

Abstract
The selective breakdown by autophagy of lipid droplet-stored lipids termed lipophagy is a lysosomal lipolytic pathway that complements the actions of cytosolic neutral lipases. The physiological importance of lipophagy has been demonstrated in multiple mammalian cell types, as well as in lower organisms, and this pathway has many functions in addition to supplying free fatty acids to maintain cellular energy stores. Recent studies have begun to delineate the molecular mechanisms of the selective recognition of lipid droplets by the autophagic machinery, and the intricate crosstalk that exists among the forms of autophagy and neutral lipases. These studies have led to increased interest in the role of lipophagy in both human disease pathogenesis and therapy.
Keywords: autophagy, lipid droplet, lipophagy, liver, macrophage, perilipin
Cellular lipid breakdown and autophagy
A critical mechanism of cellular energy storage is the sequestration of neutral lipids in a cellular organelle, the lipid droplet (LD) (see Glossary) [1]. LDs exist in most organisms and cell types but in varying amounts [2]. The LD structure consists of a core of neutral lipids that are predominantly triglycerides (TGs) and steryl esters, surrounded by a phospholipid monolayer and a group of heterogeneous proteins referred to as LD-associated proteins [3]. Prominent among these proteins are the five members of the perilipin family. LDs are not simply passive lipid storage vessels [4], but participants in a variety of complex cellular functions that range from the proteasomal degradation of apolipoprotein B [5] to the replication of the hepatitis C virus [6]. The breakdown of stored lipids from LDs into free fatty acids (FFAs) not only provides energy to the cell, but also modulates other cellular processes as FFAs can for example activate cell signaling pathways or trigger cellular injury. The process of lipid breakdown from LDs is therefore a critical cellular pathway that requires both robust and precise regulation.
Autophagy is one of the two major degradative pathways in the cell together with the proteasomal pathway. The nature of the autophagic machinery and pathways has been well summarized in other reviews [7,8]. Three forms of autophagy are recognized: macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy. In macroautophagy cytosolic organelles and proteins are sequestered in a double-membrane autophagosome that then fuses with a lysosome to form an autolysosome in which the autophagosome cargo is mixed with and degraded by the lysosomal enzymes for release back into the cytosol. This process is regulated by over 30 autophagy-related (atg) genes. These genes provide a source of genetic inhibition of autophagy, but it must be remembered that the protein products of these genes can have autophagy independent functions, although none have as yet been reported for lipid metabolism. CMA targets proteins with a specific peptide motif by association to the chaperone heat-shock cognate protein 70 (Hsc70) for uptake into lysosomes by binding to lysosome-associated membrane protein (Lamp) 2. Micoautophagy involves the removal of cell components within lysosomal invaginations but has not been implicated in lipolysis. Although lysosomes have long been known to have acidic lipases and degrade lipid membranes and exogenous lipoproteins, the ability of macroautophagy to remove and degrade TGs and cholesterol from LDs was only recently identified [9].
Lipolysis has been classically recognized to occur through the actions of cytosolic neutral lipases that hydrolyze LD-stored TGs into FFAs and glycerol. Recognition of similarities in regulation (inhibition by insulin and stimulation by glucagon) and function (induction by nutrient deprivation to supply energy to the cell) of lipolysis and autophagy led to identification of a specific form of autophagy termed lipophagy that degrades LD-sequestered lipids [9]. Studies in cultured hepatocytes and mouse liver have demonstrated the selective uptake of LDs by autophagosomes, the trafficking of lipids and perilipins through the autophagic pathway, and the increased accumulation of lipid and LDs and a resultant decrease in FFA p-oxidation with a genetic or chemical inhibition of autophagy [9]. The combined inhibition of neutral lipases and autophagy led to an additive increase in cellular lipid content indicating that the cytosolic and lysosomal pathways of lipolysis are complementary [9]. Although originally described in hepatocytes, lipophagy functions to metabolize lipids in probably all cells as this pathway has been described subsequently in a variety of mammalian cells including neurons [10], immune cells [11], intestinal epithelium [12] and cancer cells [13], as well as in lower organisms such as yeast [14] and fungi [15]. Recent investigations have increased our understanding of the lipophagic machinery, demonstrated crosstalk among the autophagic pathways and neutral lipases, and suggested potential roles for the lipolytic function of autophagy in disease states.
Targeting LDs for selective autophagy
The preferential sequestration of LDs over other cytosolic components as autophagosome cargo in the livers of starved mice indicates that lipid degradation by macroautophagy is a selective form of autophagy [9]. The cellular autophagic machinery is therefore able to identify and distinguish LDs from other potential cytosolic substrates. Other types of selective autophagy, such as the mitochondrial form mitophagy, rely on the recognition of organelle-specific proteins [16]. Proteomic analyses of LDs in a variety of organisms have identified many LD-associated proteins that are involved in membrane trafficking [17-19], and thus represent potential components of the lipophagic pathway.
The association of the membrane GTPase Rab7 with LDs from different organisms [20,21], suggests that this protein might represent a universal regulator of LD docking and degradation. In addition, GTPases regulate membrane trafficking events [22], and are involved in endosome and autophagosome interactions [23], which prompted investigations into Rab7 as a component in autophagic LD degradation [24]. Increased Rab7-LD association was found in starved hepatocytes, whereas a Rab7 dominant-negative mutant defective in GTP-binding failed to interact with LDs. Rab7 activity increased with starvation in both cell lysates and purified LDs indicating that nutrient deprivation induces Rab7 activation. Consistent with a critical role for Rab7 in lipophagy is that Rab7 depletion results in reduced LD breakdown during starvation that is restored by wild-type or constitutively active Rab7, but not by expression of the dominant-negative mutant. Studies of LD interactions with endocytic components involved in autophagy demonstrated that starvation-activated Rab7 recruits multivesicular bodies (MVBs) to the LD-autophagosome interface to promote amphisome formation. Rab7 also regulates the subsequent step of LD/amphisome-lysosome docking, as demonstrated by increased LD interactions with constitutively active versus wild-type Rab7. Interestingly, while LDs were surrounded by MVBs, their interaction with lysosomes appeared to be a transient one that may allow LD sampling or the removal of a portion of the droplet's stores [24]. It is likely that an interaction between MVBs and LDs creates a contact point for subsequent lysosome LD targeting. This is in agreement with the concept that LDs do not fuse directly with lysosomes [9], however whether amphisome/autophagosome formation on LDs is essential for lipophagy to occur remains unknown. These findings in hepatocytes are consistent with observations in adipocytes where Rab7 is essential for autolysosome-mediated LD degradation during hormone-stimulated lipolysis [25].
Consistent with a conserved regulatory role of Rab7 in lipophagy is that the yeast Rab7-like protein Ypt7p regulates LD dynamics and membrane trafficking in S. Cerevisiae [26]. Microscopic and proteomic analysis demonstrated that Ypt7p is associated with LDs in yeast grown in poor medium. Defective Ypt7p strains have increased FFA content and numbers of LDs in low nutrient conditions [26]. Together with the previous mammalian findings, these studies indicate that Rab7 may be a conserved regulator of lipophagy.
Although further studies are needed to delineate the complete machinery modulating lipophagy, additional regulatory elements have been reported in both yeast and mammals. In yeast, Ypt7p partners have been identified by co-immunoprecipitation studies, among which the H subunit of the V-ATPase proton pump, Vma13p, is involved in LD dynamics. Selective mutagenesis studies showed that another component of the V-ATPase is required for lipophagy, the homotypic fusion and protein sorting (HOPS)-tethering complex [26], which mediates autophagosome-lysosome fusion [27]. In mammals, Rab-interacting lysosomal protein (RILP) protein has been reported to be essential for Rab7 function in starvation-induced LD breakdown in hepatocytes [24]. In fat cells, LD-associated structural protein perilipin 1 (PLIN1) does not directly interact with Rab7, however PLIN1 protects LDs from autophagic degradation by blocking Rab7 access to the LD surface [25].
A novel protein recently implicated in the regulation of autophagic lipolysis is the membrane curvature protein Bif-1 [28]. In adipocytes, autophagy induces Bif-1-dependent PLIN1 degradation and Bif-1 deficiency leads to decreased TG hydrolysis, suggesting that PLIN1 degradation is necessary for autophagy-driven LD breakdown. Bif-1 deficiency resulted in adipose tissue hypertrophy, obesity and hyperinsulinemia with high fat diet feeding or aging. However, Bif-1 deficiency did not inhibit adipose tissue lipolysis upon fasting [28], suggesting that lipophagy is not essential to hormone-stimulated adipocyte lipolysis. Cell-type differences may therefore exist in lipophagy, but interpretation of the role of lipophagy in adipose tissue is complicated by effects of autophagy on adipocyte differentiation [29,30], although loss of Bif-1 was reported not to have this effect [28]. Decreased levels of the autophagy related proteins Atg9a and Lamp1 in Bif-1 knockout mice upon dietary challenge or aging suggest that defective autophagy might contribute to the LD accumulation in these mice. Membrane curvature varies with LD size and little is known about the effects of LD size on utilization. The Bif-1 findings raise the interesting question of whether the size of LDs affects their targeting by lipophagy, but this area has not yet been explored.
Cytosolic lipase-autophagy crosstalk
Recent investigations indicate that lysosomal lipid breakdown by lipophagy and cytosolic-mediated lipolysis are not completely independent pathways but actually regulate each other (Figure 1). In addition to directly targeting LD-stored lipids to the lysosome for degradation by acid lipases, autophagy may promote lipolysis by cytosolic neutral lipases. LD-associated proteins shield the core lipids from lipases. CMA targets specific proteins for lysosomal proteolysis suggesting that CMA may initiate the lipolytic process by promoting the degradation of LD-associated proteins to expose the LD core to neutral lipases. The LD proteins PLIN2 and PLIN3 were found to undergo CMA-mediated degradation prior to the onset of lipolysis [31]. CMA promoted LD association of adipose triglyceride lipase and macroautophagy proteins to trigger lipolysis, and CMA inhibition decreased lipid oxidation and promoted LD accumulation. Lipolysis is associated with PLIN2 phosphorylation which has been shown to be AMPK-mediated after the interaction of PLIN2 with the CMA chaperone HSPA8/Hsc70 [32]. These studies were conducted in fibroblasts and the findings need to be confirmed in a cell type with more significant fat stores. In addition, the overall contribution of decreased CMA to metabolism is multifactorial, as mice with an inhibition of CMA due to a knockout of Lamp-2A develop abnormalities in carbohydrate and lipid metabolism secondary to reduced degradation of a number of metabolic enzymes [33]. All of these findings do suggest that that a decrease in CMA has important detrimental metabolic effects. In addition, it is apparent that all of the effects of autophagy on LD degradation cannot be attributed simply to the mechanism of lipophagy.
Figure 1. Cellular crosstalk among autophagic pathways and neutral lipases.
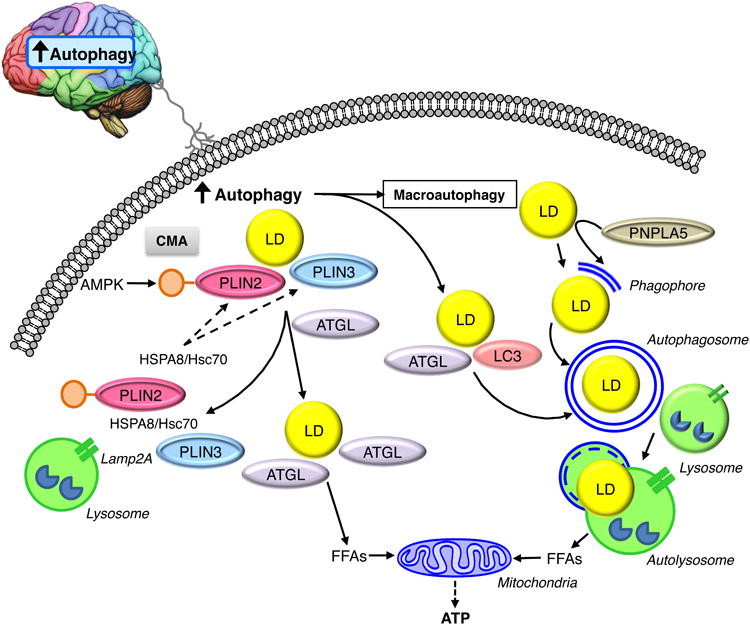
Autophagy in the brain sends signals that induce lipophagy by promoting the interactions of LC3 and ATGL on the LD surface, and by increasing the direct removal of LDs by macroautophagy. PNPLA5 may promote autophagy by supplying lipids from LDs necessary for the formation of the pre-autophagosome membrane structure the phagophore. CMA promotes neutral lipolysis by degrading AMPK-phosphorylated PLIN2 and PLIN3, thereby allowing ATGL access to the lipid core of the LD. Together lipophagy and neutral lipolysis generate FFAs that drive mitochondrial FFA β-oxidation to supply the cell with ATP.
A complex function for macroautophagy in brown adipose tissue (BAT) lipid breakdown has been described that involves both central nervous system (CNS) functions of autophagy as well as local effects on cytosolic lipases [34]. Cold-induced autophagy in CNS proopiomelanocortin neurons induced autophagy and lipid utilization in BAT and liver that was blocked by denervation. These studies also demonstrated that the autophagy protein LC3 recruited adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) to LDs indicating a coordinated regulation of lipolysis by both CNS effects and peripheral protein-protein interactions. Whether LC3 has this function in other tissues, or in response to non-CNS stimuli, needs to be further investigated.
Levels of autophagy are regulated by LDs and lipases
Recent studies have established that a reverse relationship exists between lipolysis and autophagic function in which neutral lipase-mediated LD breakdown regulates autophagosome biogenesis and levels of autophagy. In HeLa cells, dynamic interactions occur between LDs and early autophagosomes, and LDs are required for autophagy [35]. LD-mediated enhancement of autophagy in these cells is dependent on patatin-like phospholipase domain-containing protein 5 (PNPLA5), a presumed neutral TG lipase, together with other lipid biosynthetic and remodeling enzymes. These findings suggest that neutral lipase-mobilized lipids from LDs provide critical lipid building blocks for autophagosomal membranes. These findings extend the possible cellular sources of autophagosome membrane components beyond the already implicated ER, Golgi apparatus, mitochondrial and plasma membranes [36]. However, it remains possible that the cellular requirement of LDs for autophagy is related to energy needs or lipid effects on signaling pathways rather than as simply a supplier of structural lipids.
Additional evidence of the interrelationship of LDs to autophagosomes has been provided by studies in yeast [37]. With nitrogen starvation, chemical or genetic inhibition of FFA synthesis prevented Atg8-measured autophagosome formation, which was restored by the addition of FFAs. Yeast defective in LDs had a similar autophagy defect that was not reversed by FFAs, indicating that the droplets themselves and not the FFAs from their breakdown are critical. Investigations in a variety of yeast mutants suggested that lipolysis of both LD-stored TGs and steryl esters for supply to the ER through direct contact between the two organelles was required for autophagosome membrane formation. These findings are consistent with those of Dupont et al. [35]. However, other studies have shown that with starvation there is impaired autophagy in yeast lacking LDs, but the autophagic machinery is intact, as autophagy was still inducible by rapamycin even in the absence of LDs [38]. In these studies, the defect in autophagy was secondary to an altered phospholipid composition and failure to buffer external FFAs in LDs, leading to ER stress and an altered ER lipid composition that resulted in decreased autophagy. Thus, although these findings are all consistent with the existence of a bidirectional regulation of LD lipolysis and levels of autophagy, and identify a new function for lipolysis, the underlying mechanism for the regulation of autophagy by lipolysis remains to be confirmed.
Role of lipophagy in disease states
Lipophagy in fatty liver disease
Considering the important role of lipid metabolism in liver homeostasis, impaired lipophagy may be a contributing factor in the development of excessive hepatic lipid accumulation or steatosis that underlies both alcoholic and nonalcoholic fatty liver disease (NAFLD) [39,40]. In addition, lipophagy regulates cell death and inflammation which are other important features of these diseases (Figure 2) [40]. In the initial description of lipophagy, mice deficient in hepatocyte autophagy had excessive hepatic TG and cholesterol accumulation [9]. Consistent with defective autophagy promoting fatty liver disease is that several conditions that predispose to nonalcoholic steatohepatitis (NASH), which is steatosis combined with liver injury and inflammation, including aging and obesity, also impair hepatic autophagy [40]. Consistent with this concept is the finding that inhibition or activation of autophagy increased or decreased respectively, methionine choline diet-induced steatosis in mice [41]. Hepatic steatosis was also attenuated in this diet by the autophagy activator resveratrol, suggesting that this agent may be a possible treatment for NASH [42].
Figure 2. Potential contributions of lipophagy among the various cell types in the liver with obesity-induced fatty liver disease.

Genetic and diet-induced obesity in mice has been demonstrated to decrease overall levels of macroautophagy in the liver. Hepatocytes have decreased autophagy, which may promote both lipid over-accumulation and cell injury from oxidant stress, due to compromised energy homeostasis. These hepatocyte effects promote liver steatosis and cell death. Obesity also decreases autophagy in macrophages leading to increased inflammation through effects on M1 and M2 polarization. The effect of obesity on hepatic stellate cells has not been determined, but a decrease in autophagy in this cell type would decrease activation into myofibroblasts leading to an anti-fibrotic effect.
The effects of autophagy in fatty liver disease may also be mediated through changes in lipids other than TGs. The bioactive sphingolipid ceramide is increased in Atg7 knockout mouse liver [43]. Autophagy increased when sphingolipid de novo synthesis was upregulated, indicating that lipid degradation was activated to prevent excessive sphingolipid accumulation. Considering that ceramide is elevated in a variety of diseases, including obesity [44] and diabetes [45] which are both related to the development of steatosis, dysfunctional autophagy may lead to altered sphingolipid content and related pathologies.
In agreement with the essential role of autophagy in liver LD clearance are genetic studies in which hepatic TGs were increased when autophagy was inhibited by a knockdown of Atg14 [46], or knockout of the positive transcriptional regulator of autophagy transcription factor EB (Tfeb) [47]. The converse approach of adenoviral overexpression of Atg7 or Atg14 to increase hepatic autophagy reversed liver steatosis in obese mice [46,48]. Although a large amount of data indicates that lipophagy protects against hepatic lipid accumulation, some mouse models with autophagy gene knockouts have shown unchanged or even reduced steatosis [49,50]. This discrepancy might be due to differences in experimental conditions and animal models (Table 1). One in vitro study has suggested that lipophagy functions to replenish LDs with FAs, whereas neutral lipolysis removes lipids from LDs [51]. However, these findings were from fibroblasts with limited lipid stores, and occurred only with prolonged culture in a saline solution but not in serum-free medium. Thus, the physiological significance of these findings remains unclear. Further in vivo studies are necessary to confirm whether lipophagy protects against the development of steatosis and liver injury in NAFLD.
Table 1.
Summary of studies of different genetic mouse models of autophagy and the effects on hepatic lipid accumulation.
| Animal model | Genetic alteration | Effect on autophagy | Diet | Effect | Ref. |
|---|---|---|---|---|---|
| Atg7F/F-Alb-Cre mouse | Hepatocyte-specific Atg7 knockout | Decrease | Normal diet | Increased steatosis, hepatic TGs and cholesterol | [9] |
| Atg14 shRNA adenovirus-infected mouse | Atg14 hepatic knockout | Decrease | Normal diet | Increased hepatic TGs | [46] |
| Atg14 expressing adenovirus-infected mouse | Atg14 hepatic overexpression | Increase | High fat diet | Decreased hepatic TGs | [46] |
| Atg7 adenovirus-infected ob/ob mouse | Atg7 hepatic overexpression | Increase | Normal diet | Decreased steatosis and liver TGs | [48] |
| Tcfeb-LiKO mouse | Hepatocyte Tfeb knockout | Decrease | Starvation or high fat diet | Increased steatosis and hepatic TGs | [47] |
| HDAd-TFEB mouse | Adenoviral TFEB overexpression | Increase | High fat diet | Decreased hepatic TGs | [47] |
| FILKO mouse | FIP200 hepatocyte knockout | Decrease | Normal diet with or without starvation | Decreased liver TGs with starvation | [49] |
| Cre adenovirus-infected FIP200F/F mouse | FIP200 hepatocyte knockout | Decrease | High fat diet | Decreased liver TGs | [49] |
| Atg7Δhep mouse | Atg7 hepatocyte knockout | Decrease | High fat diet or starvation | Decreased steatosis | [50] |
Abbreviations: Tcfeb-LiKO, Tcfeb liver-specific knockout; FILKO, liver-specific FIP200 knockout
Autophagy also mediates lipid degradation in in vitro and in vivo models of acute alcohol-induced hepatic steatosis and injury [52]. Lipophagy is induced by acute ethanol ingestion from the generation of reactive oxygen intermediates [52]. Similar findings in mice that overexpress the pro-oxidant enzyme CYP2E1, which is induced by sustained alcohol ingestion, were found under chronic ethanol-fed conditions [53]. The livers of chronic alcohol-fed mice showed decreased levels of the principal hepatic antioxidant glutathione, and inhibition of hepatocyte autophagy further reduced glutathione levels, suggesting that autophagy protects against chronic alcohol-induced oxidative stress. These findings strongly indicate that lipid degradation by autophagy has a protective function against oxidative stress and liver damage induced by both acute and chronic ethanol treatment. This effect may be due to removal of the excess lipids that serve as the substrate for oxidative stress, or through the supply of FFA-generated ATP by lipophagy, which is a mechanism of hepatocyte resistance to oxidant injury [54].
Macrophage autophagy regulates inflammation
Macrophage foam cells contribute to atherosclerosis, and the removal of excessive cholesterol from these cells is a potential therapeutic strategy for this disease [55]. LD-stored cholesterol esters are degraded through macroautophagy by lysosomal acid lipases (LALs) in addition to neutral lipases [11]. It is unknown whether a defect in autophagy underlies foam cell development or progression, so the significance of this finding to the pathogenesis of atherosclerosis is not clear. Studies of the tumor suppressor gene programmed cell death 4 (PDCD4) have shown that this protein inhibits autophagy in macrophages [56]. Pdcd4 null mice have increased atherosclerosis demonstrating a link of autophagy, although not specifically lipophagy, to atherogenesis [56].
Macrophage activation is energy dependent and anti-inflammatory M2 macrophage polarization relies on FFA oxidation, suggesting a potential role for autophagy in this process. M2 polarization in response to parasitic infection is mediated by LALs, although the involvement of autophagy was not examined [57]. With obesity, macrophage autophagy is decreased, and an inhibition of autophagy in bone marrow-derived and hepatic macrophages causes these cells to assume a more proinflammatory phenotype due to both increased M1 and decreased M2 polarization [58]. However, it was not determined whether lipophagy was the mechanism of this effect. Adipose tissue macrophages from obese mice have been shown to increase autophagy to metabolize lipids without changes in activation, leading to the speculation that macrophage autophagy in this tissue functions to buffer the increase in lipids with obesity [59]. Another report failed to demonstrate any effect of an inhibition of autophagy on adipose tissue macrophage lipid stores, however this conclusion was based strictly on fluorescence imaging in the absence of any biochemical analysis [60]. This study also found increased rather than decreased autophagy in adipose tissue macrophages from obese mice [60]. Thus, controversy exists over the function of lipophagy in macrophages, which may just reflect macrophage heterogeneity that depends on environment and inflammatory stimulus. However, this possibility needs to be confirmed by direct comparisons of macrophages from different tissues.
Lipophagy mediates stellate cell activation and fibrosis
Liver fibrosis results from the activation of hepatic stellate cells into matrix-producing myofibroblasts through a process mediated by lipophagy. Quiescent liver stellate cells have large lipid stores, primarily in the form of vitamin A, that are metabolized during activation. Macroautophagy is increased in hepatic stellate cells in response to a fibrotic stimulus [61,62]. Knockdown of stellate cell autophagy blocked their activation, which was restored by supplementation with the FFA oleate, indicating that lipophagy drove transdifferentiation by supplying substrates for the energy needed for activation [61]. Stellate cell-specific autophagy knockout mice developed decreased fibrosis in vivo [61]. Whether lipophagy mediates fibrosis in organs other than the liver remains to be determined, but stellate cell activation is central to fibrogenesis among tissues suggesting that lipophagy in this cell type may be critical in a variety of fibrotic diseases. Together with the previously discussed involvement of lipophagy in hepatic steatosis, injury and the innate immune response, the role in fibrosis suggests that autophagy may mediate many of the hepatic features of NAFLD through effects on multiple cell types (Figure 2).
Concluding remarks and future perspectives
The breakdown of cellular lipid stores in LDs occurs through the actions of both cytosolic neutral lipases and LALs via the autophagic pathway of lipophagy. Autophagy not only regulates cell lipid content but also supplies the cell with FFAs for cellular functions, such as resistance to oxidant stress. Whether the basic lipolytic function of autophagy is the same in all cell types remains to be further clarified. Lipophagy also promotes specific cell processes such as macrophage polarization and stellate cell transdifferentiation, and other functions will undoubtedly be defined. The mechanisms for the selective targeting of LDs by the autophagic pathway have begun to be elucidated through the identification of proteins such as Rab7 that mediate lipophagy, but this process is likely far more complex than our current understanding, and requires further investigation. Although initially thought to be distinct pathways, neutral lipolysis and lipophagy are intertwined and the signals that mediate crosstalk between the two pathways have begun to be defined. The ability to stimulate lipophagy without affecting other forms of autophagy could be a potential therapy for metabolic diseases such as obesity. Lipophagy is a promising therapeutic target, but the role of lipophagy in various lipid overload states must be further examined, as for example controversy exists on the involvement of lipophagy in liver steatosis and macrophage foam cell formation.
Outstanding Questions.
Are the functions of autophagy in lipid homeostasis constant among different cells or cell type specific? Does the function of lipophagy vary among cells with different amounts of stored lipid and LDs.
What are the mechanisms by which components of the autophagic machinery sense and locate LDs for degradation? Do LDs contribute to or serve as a site for autophagosome formation? How are signals emanating from nutrient deprivation transduced into increases in lipophagy?
Are the pathways of neutral lipolysis and lipophagy unrelated or coordinated pathways that act in concert to maintain cellular lipid and energy homeostasis? What is the nature of this crosstalk?
Is autophagic function impaired in human diseases such as the metabolic syndrome and in what cell types? Do defects in autophagy in cells such as hepatocytes and macrophages mediate diseases such as nonalcoholic fatty liver disease and atherosclerosis?
Trends.
Cellular lipid breakdown from LD stores depends on the direct actions of cytosolic neutral lipases on LDs and on acidic lysosomal lipases that act on LDs delivered to lysosomes by macroautophagy.
Lipophagy (a selective form of macroautophagy) relies on the sensing of LD-associated proteins such as Rab7. This selectivity allows autophagy to specifically break down LD lipid stores in times of prolonged nutrient deprivation.
Lipolysis by neutral lipases and lipophagy are not independent pathways but have significant crosstalk such as the degradation of perilipins by autophagy to facilitate the actions of ATGL or the ability of neutral lipases to supply lipids for autophagosome biogenesis.
The functions of lipophagy in diverse processes ranging from steatosis to macrophage activation, and the fact that autophagy levels are often altered in disease, suggest that lipophagy is important in the pathogenesis of metabolic diseases.
Acknowledgments
This work was supported by National Institutes of Health Grants R01DK61498 and R01AG031782.
Glossary
- Amphisome
an autophagic vacuole formed by the fusion of an autophagosome and an endosome to deliver autophagic cargo to the lysosome as an alternative to direct autophagosome-lysosome fusion
- Autolysosome
cellular organelle resulting from the fusion of an autophagosome and a lysosome. In this structure, the cargo from the autophagosome mixes with the hydrolytic enzymes of the lysosome for degradation and release into the cytosol
- Autophagosome
double membrane vesicle in the cytosol of the cell that forms around cellular components for translocation to a lysosome as part of the process of macroautophagy. The origin of the membrane remains unclear with the ER, mitochondria, Golgi apparatus, mitochondrial and plasma membranes and LDs all implicated as sources
- Chaperone-mediated autophagy (CMA)
one type of autophagy that targets cytosolic proteins containing a pentapeptide motif that is recognized by the chaperone Hsc70. This complex is translocated to the lysosomal membrane where binding to the Lamp-2A receptor leads to internalization and degradation
- Lipid droplet (LD)
cellular organelle in which TGs and cholesterol are stored in a central core surrounded by a phospholipid monolayer and a variety of proteins
- Lipophagy
a selective form of macroautophagy that specifically targets LD-contained lipids for degradation by lysosomal acid lipases
- Macroautophagy
the major type of autophagy in which cytosolic organelles and proteins are sequestered in a double-membrane structure termed the autophagosome which fuses with a lysosome to form an autolysosome in which the cellular components are degraded and the products released into the cytosol
- Macrophage foam cells
fat-laden macrophages that accumulate in the walls of arterial blood vessels as part of the process of atherosclerosis
- Nonalcoholic fatty liver disease (NAFLD)
a liver disorder with histological similarity to alcoholic liver disease that occurs in the absence of excessive alcohol intake. NAFLD is a continuum from simple lipid over accumulation or steatosis to steatosis together with hepatocyte injury and inflammation and eventual fibrosis. The mechanisms of this liver disease are unclear but it is strongly associated with insulin insensitivity and is a component of the metabolic syndrome
- Nonalcoholic steatohepatitis (NASH)
the more serious form of NAFLD in which steatosis is accompanied by hepatocyte injury and inflammation. Whereas simple steatosis is considered benign, NASH can progress to chronic liver disease and its complications
- Perilipin
a member of a family of proteins that coat LDs and function to regulate the breakdown of lipids by both cytosolic lipases and lipophagy in part by controlling the exposure of proteins in these pathways to the lipid core of the LD
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hashemi HF, Goodman JM. The life cycle of lipid droplets. Curr Opin Cell Biol. 2015;33:119–124. doi: 10.1016/j.ceb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilfling F, et al. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29:39–45. doi: 10.1016/j.ceb.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kory N, et al. Targeting fat: mechanisms of protein localization to lipid droplets. Trends Cell Biol. 2016 doi: 10.1016/j.tcb.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welte MA. Expanding roles for lipid droplets. Curr Biol. 2015;25:R470–R481. doi: 10.1016/j.cub.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki M, et al. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salloum S, et al. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013;9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi AM, et al. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaushik S, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13:258–265. doi: 10.1038/embor.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouimet M, et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khaldoun SA, et al. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol Biol Cell. 2014;25:118–132. doi: 10.1091/mbc.E13-06-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaini RR, et al. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate. 2012;72:1412–1422. doi: 10.1002/pros.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Zutphen T, et al. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2014;25:290–301. doi: 10.1091/mbc.E13-08-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen LN, et al. Autophagy-related lipase FgATG15 of Fusarium graminearum is important for lipid turnover and plant infection. Fungal Genet Biol. 2011;48:217–224. doi: 10.1016/j.fgb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 17.Gannon J, et al. ARFGAP1 is dynamically associated with lipid droplets in hepatocytes. PLoS One. 2014;9:e111309. doi: 10.1371/journal.pone.0111309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, et al. The proteomics of lipid droplets: structure, dynamics, and functions of the organelle conserved from bacteria to humans. J Lipid Res. 2012;53:1245–1253. doi: 10.1194/jlr.R024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang P, et al. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol Cell Proteomics. 2012;11:317–328. doi: 10.1074/mcp.M111.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brasaemle DL, et al. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 21.Liu P, et al. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279:3787–3792. doi: 10.1074/jbc.M311945200. [DOI] [PubMed] [Google Scholar]
- 22.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 23.Gutierrez MG, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 24.Schroeder B, et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61:1896–1907. doi: 10.1002/hep.27667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lizaso A, et al. β-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy. 2013;9:1228–1243. doi: 10.4161/auto.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouchez I, et al. Regulation of lipid droplet dynamics in Saccharomyces cerevisiae depends on the Rab7-like Ypt7p, HOPS complex and V1-ATPase. Biol Open. 2015;4:764–775. doi: 10.1242/bio.20148615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang P, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25:1327–1337. doi: 10.1091/mbc.E13-08-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, et al. Bif-1 deficiency impairs lipid homeostasis and causes obesity accompanied by insulin resistance. Sci Rep. 2016;6:20453. doi: 10.1038/srep20453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baerga R, et al. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy. 2009;5:1118–1130. doi: 10.4161/auto.5.8.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh R, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015;17:759–770. doi: 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy. 2016;12:432–438. doi: 10.1080/15548627.2015.1124226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider JL, et al. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014;20:417–432. doi: 10.1016/j.cmet.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Lopez N, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupont N, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014;24:609–620. doi: 10.1016/j.cub.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res. 2014;24:58–68. doi: 10.1038/cr.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shpilka T, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015;34:2117–2131. doi: 10.15252/embj.201490315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Velazquez AP, et al. Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J Cell Biol. 2016;212:621–631. doi: 10.1083/jcb.201508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolganiuc A, et al. Autophagy in alcohol-induced liver diseases. Alcohol Clin Exp Res. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci. 2016 doi: 10.1007/s10620-015-4025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen R, et al. Protective role of autophagy in methionine-choline deficient diet-induced advanced nonalcoholic steatohepatitis in mice. Eur J Pharmacol. 2016;770:126–133. doi: 10.1016/j.ejphar.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 42.Ji G, et al. Resveratrol ameliorates hepatic steatosis and inflammation in methionine/choline-deficient diet-induced steatohepatitis through regulating autophagy. Lipids Health Dis. 2015;14:134. doi: 10.1186/s12944-015-0139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexaki A, et al. Autophagy regulates sphingolipid levels in the liver. J Lipid Res. 2014;55:2521–2531. doi: 10.1194/jlr.M051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Xiong X, et al. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma D, et al. Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol. 2013;27:1643–1654. doi: 10.1210/me.2013-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim KH, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 51.Rambold AS, et al. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding WX, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Y, Cederbaum AI. Autophagy protects against CYP2E1/chronic ethanol-induced hepatotoxicity. Biomolecules. 2015;5:2659–2674. doi: 10.3390/biom5042659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, et al. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52:266–277. doi: 10.1002/hep.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khera AV, Rader DJ. Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep. 2010;12:73–81. doi: 10.1007/s11883-009-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, et al. Pdcd4 deficiency enhances macrophage lipoautophagy and attenuates foam cell formation and atherosclerosis in mice. Cell Death Dis. 2016;7:e2055. doi: 10.1038/cddis.2015.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang SC, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu K, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu X, et al. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–830. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grijalva A, et al. Autophagy is dispensable for macrophage mediated lipid homeostasis in adipose tissue. Diabetes. 2016 doi: 10.2337/db15-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez-Gea V, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thoen LF, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]