

Summary
Background
A variant of von Willebrand disease (VWD) type 2A, phenotype IIC (VWD2AIIC) is characterized by recessive inheritance, low von Willebrand factor antigen (VWF:Ag), lack of VWF high molecular weight multimers, absence of VWF proteolytic fragments, and mutations in the VWF propeptide. A family with dominantly inherited VWD2AIIC but markedly elevated VWF:Ag of >2 U/L was described as VWD type IIC Miami (VWD2AIIC-Miami) in 1993, however the molecular defect remained elusive.
Objectives
To identify the molecular mechanism underlying the phenotype of the original VWD2AIIC-Miami.
Patients and Methods
We studied the original family with VWD2AIIC-Miami phenotypically and by genotyping. The identified mutation was recombinantly expressed and characterized by standard techniques, confocal imaging, and in a mouse model, respectively.
Results
By Multiplex ligation-dependent probe amplification we identified an in-frame duplication of VWF exons 9-10 (c.998_1156dup; p.Glu333_385dup) in all patients. Recombinant mutant (rm)VWF only presented as a dimer. Co-expressed with wild type VWF, the multimer pattern was indistinguishable from patients’ plasma VWF. Immunofluorescence studies indicated retention of rmVWF in unusually large intracellular granules in the Endoplasmic Reticulum. ADAMTS13 proteolysis of rmVWF under denaturing conditions was normal, however an aberrant proteolytic fragment was apparent. A decreased ratio of VWF propeptide to VWF:Ag and a DDAVP test in one patient indicated delayed VWF clearance which was supported by clearance data after infusion of rmVWF into VWF−/− mice.
Conclusion
The unique phenotype of VWD2 type IIC-Miami results from dominant impairment of multimer assembly, an aberrant structure of mutant mature VWF, and reduced clearance in vivo.
Keywords: von Willebrand disease, type 2A, von Willebrand factor, genetics, physiopathology, ADAMTS13, mouse
Introduction
Von Willebrand disease (VWD) is a common disorder of platelet dependent hemostasis, characterized by quantitative and/or qualitative deficits of von Willebrand factor (VWF), a large protein, which occurs in plasma in the form of homo-polymers of different sizes designated as VWF multimers. VWD is classified into 3 main types, characterized by either i) reduced VWF quantity and correspondingly its function (type 1), ii) virtually complete absence of VWF in plasma and platelets (type 3), or iii) functional deficits with or without reduction of VWF levels (type 2) [1]. VWD symptoms typically comprise a prolonged bleeding time, mucocutaneous bleeding symptoms and in severe cases also hemophilia like bleeding manifestations, e.g. joint, muscle and intracranial bleedings correlating with very low FVIII levels. Several distinct structural and functional defects of VWF causing VWD have been described (reviewed in [2]) Among them, VWD type 2A features functional deficits of VWF platelet GPIb binding (VWF:GPIbB) and VWF collagen binding (VWF:CB) due to the reduction or absence of VWF high molecular weight multimers (HMWM) [1] which are the most active in primary hemostasis.
VWD type 2A is rather heterogeneous in its molecular background. The most common type 2A phenotype results from enhanced proteolytic susceptibility of mutant VWF to its protease ADAMTS13 caused by mutations in the VWF A2 domain [1, 3]. Other mutations prevent proper multimerization by directly impairing either VWF dimerization at the VWF carboxy-terminal CK domain [4] or the polymerization of VWF dimers to HMWM at the amino-terminal VWFD3 domain [5]. This last post-translational step is mediated by the VWF-propeptide (VWFpp) [6, 7]. Hence, some mutations located in VWFpp also correlate with a profound multimerization deficit [8-12]. The corresponding phenotype was originally described as VWD subtype IIC (VWD2AIIC), characterized by low VWF antigen (VWF:Ag), decreased platelet dependent function (low ristocetin-cofactor [VWF:RCo]), and lack of VWF HMWM, which in medium resolution multimer analysis show a virtually complete absence of proteolytic sub-bands of individual VWF oligomers [3]. In contrast to most other type 2A subtypes, inheritance is recessive in VWD2AIIC [13].
In 1993, a family with several affected members with the typical multimer pattern of VWD2AIIC but obviously dominant inheritance was reported by Ledford and co-workers [14]. In contrast to conventional VWD2AIIC, VWF:Ag was markedly elevated to levels >2 U/L. VWF:RCo was moderately decreased, but the ratio of VWF:RCo to VWF:Ag was significantly reduced which correlated well with the bleeding tendency in the index case and the affected family members.
Previous attempts to identify the underlying molecular defect in this family applying complete sequencing of all VWF coding exons, RT-PCR and cDNA sequencing, by several groups (represented by M.L., J.E.S., R.R.M., R.S.) were not successful. In particular, in the VWFpp, where mutations causing VWD2AIIC cluster, no mutations were detected. We included patients of this family in a VWF deletion screening project by multiplex ligation-dependent probe amplification (MLPA). It was by using this technique that we have finally identified the molecular cause of VWD type IIC Miami.
Patients, Materials and Methods
Patients
The affected mother (II-3), the two affected daughters (III-3 and III-5) and the affected son (III-1) of the original family with VWD type IIC Miami were available for our study (s. supplemental Fig. 1A).
We updated the previous medical history of four of the family members described in the original report [14] in December 2010 and established bleeding scores according to the EU-VWD MCMDM type 1 study [15]. Historical data derived from a DDAVP test performed in patient II-3 were also available.
Diagnosis of VWD
Conventional diagnostic tests for VWF parameters were carried out in a reference laboratory (U.B., S.S.) with patients’ plasma (0.011 M trisodium citrate). VWF:Ag [16], VWF:RCo [17] and VWF:CB [18] by using equine collagen (Horm®, Takeda, Berlin, Germany) a mixture of 95 % collagen type I and 5 % type III) as binding substrate in comparison to a normal plasma pool were obtained as standard parameters. VWF:GPIb-binding (VWF:GPIbB) was assessed as described previously [5]. We also determined VWF:FVIII-binding properties [19] and the VWFpp by a commercial ELISA assay according to the directions of the provider (Sanquin White label, Amsterdam, The Netherlands). VWFpp normal ranges and ratios with VWF:Ag for this assay had previously been published [20].
Multimer analysis was performed by SDS-agarose gel electrophoresis [21] with luminescent visualization [22, 23]. The luminescent blot was stored on electronic media by means of photo imaging (FluorChem 8000™, Alpha Innotech Corp., San Leandro, CA, U.S.A.).
Electrophoresis of the VWFpp was carried out on NuPAGE Bis-Tris 4-12% gradient gels (Invitrogen, Darmstadt, Germany) with subsequent immunoblotting using a polyclonal rabbit IgG antibody (Mango) to detect the native VWFpp and a monoclonal antibody (mab 242.6), both provided by the laboratory of R.R.M. to detect VWFpp after reduction by ß-mercaptoethanol and luminescent visualization by photo imaging as described for multimer analysis.
Molecular studies
Genomic DNA from patients’ leukocytes was used for the amplification of VWF coding exons 2 through 52 by PCR as previously described [5]. PCR products were sequenced by the ‘ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit’ on an ABI Prism 3130 (ABI, Foster City, U.S.A.).
Total RNA was extracted from platelets with SV RNA Kit (Promega, Mannheim, Germany) and reverse-transcribed into cDNA by the use of Superscript III (Invitrogen) according to the supplier's instructions.
Overlapping fragments of cDNA were amplified by PCR using cDNA specific primer pairs covering the complete coding VWF gene sequence and finally sequenced as described above.
We applied MLPA to detect possible heterozygous deletions or gains according to directions of the provider (MRC Holland, Amsterdam, The Netherlands). The results were evaluated by the software Sequence pilot (JSI Medical Systems, Ettenheim, Germany).
Breakpoint analysis at the cDNA level was carried out by using VWF cDNA PCR primers with the sense primer in exon 10 and the antisense primer in exon 9. Genomic DNA was studied by long range PCR and primer walking to further define the genomic alterations in the region of interest.
This region was analyzed using the RepeatMasker web server version 4.0.3 to identify repetitive DNA elements located at or nearby the recombination sites (URL: http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker). In silico experiments of DNA folding and the calculations were carried out by using the MFold program (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form).
Recombinant expression studies
Methods for VWF mutagenesis, transfection and recombinant expression of wild type (wt) and mutant (m) VWF were performed as follows: Mutagenesis of full length VWF cDNA was carried out by using the Quick Change kit (Stratagene, Kassel, Germany) to introduce an artificial SgsI restriction site after nucleotide c.1156 of VWF. A PCR product of exon 9 to 10 (c.998-1156) flanked by SgsI restriction sites at their ends was introduced into the SgsI restriction site of VWF cDNA with subsequent deletion of artificial restrictions sites by mutagenesis. Transient expression of rmVWF alone (homozygous) and coexpressed with rwtVWF (heterozygous) in 293 EBNA cells was carried out as previously described [5]. To analyze intracellular VWF, transfected cells were lysed by three rounds of freezing (−80°C) and thawing in lysis buffer (0.1 M Tris/HCl pH 8.0; 0.6% (v/v) Triton X-100).
All experiments were done in triplicate. Data are presented as arithmetic mean and standard deviation.
The rmVWF was characterized in comparison to rwtVWF as described for plasma VWF. ADAMTS13-mediated proteolysis of VWF was assessed by a static assay (plasma VWF according to [24], rm and rwtVWF according to [25]).
Immunofluorescence and confocal microscopy
Immunofluorescence studies were performed with transfected HEK293 as previously described [26]. HEK293 cells were seeded in Ibidi treat 8-well μ-slides (Ibidi, Martinsried, Germany) 24h prior transfection with Lipofectamine LTX (Invitrogen) and VWF-plasmid-constructs in vector pcDNA3 [5]. Antibodies used were: rabbit anti-VWF (DAKO, Hamburg, Germany, 1:1,000), mouse anti-Protein Disulfide Isomerase (PDI) (Abcam, Cambridge, UK, 1:500), goat anti-rabbit AF488 (Invitrogen, 1:5,000), goat anti-mouse AF 546 (Invitrogen, 1:5,000). Images were captured at RT with a confocal microscope (TCS SP5, Leica, Wetzlar, Germany). For settings, please refer to legend of Fig. 4.
Fig. 4.
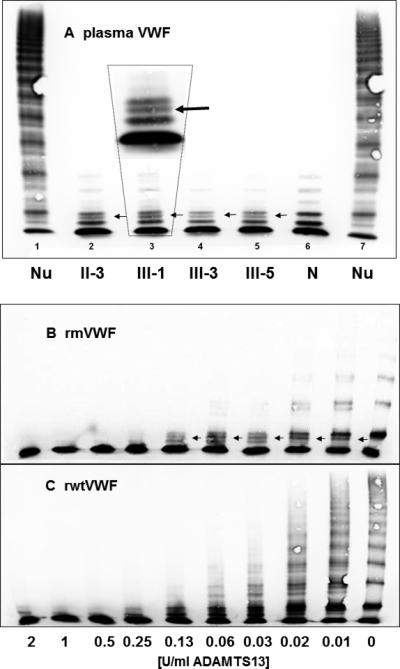
ADAMTS13 proteolysis of mutant compared to wtVWF. No evident resistance to proteolysis of patients’ plasma (A) and rmVWF (B) compared to normal plasma (N) and rwtVWF (C), respectively. Nu = normal undigested plasma VWF. Arrows point to an extra band (magnified in panel A) both in the banding pattern of patients’ and of rmVWF.
Animal studies
To assess a possible effect of mutations on the clearance of VWF, VWF−/−C57Bl/6 mice were injected by tail vein puncture with rmVWF or rwtVWF (200 or 300 μl culture medium corresponding to either 6.6 μg VWF for the mutant protein and 5.95 μg VWF for the wt protein). At different time points after injection (3, 15, 30, 60, 90, 120, 240, 480 and 1440 min), mice were anesthetized using isoflurane, blood was collected by retro-orbital venous puncture and plasma was prepared. Three mice were used for each time-point and each mouse was bled once or twice. Residual VWF at the different time points was quantified using an ELISA.
Analysis of pharmacokinetics was performed using the GraphPad Prism program as described [27]. Clearance data were fitted to the biexponential equation Cp = Ae-αt + Be-βt to obtain A, α, B, and β. Cp refers to the amount of residual VWF antigen in plasma relative to the amount injected. These parameters were used to calculate mean residence time (MRT) employing the equation MRT = (A/α2 + B/β2)/(A/α + B/β). Furthermore, α and β were used to calculate half-lives of the initial and terminal phase, respectively, employing t1/2αα = ln 2/α and t1/2β = ln 2/β.
Results
Clinical data and VWF laboratory parameters
All studied patients from this family suffer from a significant bleeding tendency with typical mucocutaneous bleeding symptoms. Comprehensive cumulative bleeding scores of 21 (II-3), 9 (III-1) and 14 (III-3) were recorded for three family members. Bleeding symptoms were frequent epistaxis, hematoma, bleeding from minor wounds, oral cavity bleeding, bleeding at tooth extraction in all three patients, post-surgery bleeding in patients II-3 and III-1. Both women suffered from menorrhagia and patient II-3 suffered from post-partum bleeding finally requiring hysterectomy.
Previous hemostasis parameters were confirmed by analyzing patient samples obtained in December 2010. We additionally noticed a low ratio of plasma FVIII:C to VWF:Ag, corresponding to decreased VWF:FVIII binding in the patients.
Patients’ VWF multimers showed the characteristic pattern of a VWD2AIIC subtype with decreased VWF HMWM and absence of a VWF triplet structure, which usually results from ADAMTS13 mediated proteolysis (s. supplemental. Fig. 1B). VWFpp/VWF:Ag ratio was decreased in all patients (0.32-0.71; 2.5-97.5 percentiles 0.8-2.2). Patients’ clinical presentation and laboratory parameters are summarized in Table 1.
Table 1.
Clinical and laboratory data of patients and a normal control.
| ID | Age | BS | PFA-E | PFA-A | Ag | pp* | ppr* | CB | CBr | RC | RCr | GPIb | GPIbr | FVIII | FVIIIr | F8Br |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nor | y | <4 | ≤165 | ≤118 | ≥0.5-≤1.6 | 0.82-1.73 | 0.8-2.2 | ≥0.5 | ≥0.8 | ≥0.5 | ≥0.8 | ≥0.5 | nd | ≥0.6 | nd | ≥0.6 |
| II-3 | 67 | 21 | >300 | >300 | 2.50 | 1.78 | 0.71 | 0.46 | 0.18 | 0.54 | 0.22 | 0.74 | 0.30 | 1.08 | 0.43 | 0.61 |
| III-1 | 43 | 9 | >300 | >300 | 2.72 | 0.88 | 0.32 | 0.37 | 0.14 | 0.40 | 0.18 | 0.55 | 0.20 | 0.99 | 0.36 | 0.49 |
| III-3 | 41 | 14 | >300 | >284 | 1.78 | 1.01 | 0.57 | 0.22 | 0.12 | 0.27 | 0.15 | 0.35 | 0.20 | 0.75 | 0.42 | 0.50 |
| III-5 | 37 | nd | >267 | >278 | 2.01 | 1.22 | 0.61 | 0.29 | 0.14 | 0.30 | 0.15 | 0.39 | 0.19 | 0.85 | 0.42 | 0.44 |
| C | 60 | 0 | nd | nd | 0.87 | 1.39 | 1.60 | 0.86 | 0.99 | 1.11 | 1.28 | 0.92 | 1.06 | 0.99 | 1.14 | 1.14 |
Patient numbers correspond to those in the pedigree (Fig. 1), C=normal person. y=years, BS=Bleeding score, PFA-E=PFA 100 (Epinephrine) [s], PFA-A=PFA 100 (ADP) [s], Ag=VWF:Ag [U/L], pp=VWF propeptide:Ag [U/L], CB=VWF:CB [U/L], RC=VWF:RCo [U/L], GPIb=VWF:GPIbα binding [U/L], F8B=VWF:FVIII binding, r = ratios of parameters to VWF:Ag
nor=2.5-97.5 percentiles of 387 healthy controls [20]
Molecular studies
Previous partial [14] and subsequent complete PCR-based mutation screening even with alternative primers and sequencing of all VWF coding exons (M.L., J.E.S., R.R.M., R.S.) revealed no candidate mutation in this family. However, the final application of MLPA identified signal gains of exon 9 and 10 in all four investigated family members, suggesting a duplication of these 2 exons (supplemental Fig. 2). We designed duplication specific primers for the putative fusion region at the cDNA level with the sense primer located in exon 10 and the antisense primer in exon 9. The RT-PCR only amplified the fusion site and was negative for wtVWF cDNA. RT-PCR using primers located in exons 8 and 11 followed by sequencing confirmed an inframe duplication of VWF exons 9 and 10: c.998_1156dup (Ref Seq. NM_000552.3), (supplemental Fig. 3 and 4). The identified mutation predicts a 53 amino acids duplication (p.Glu333_Pro385dup) in the D1 domain of VWF.
By PCR primer walking on genomic DNA and sequencing we analyzed the complete region of interest. A complex genetic rearrangement caused a duplication of VWF intron 8 to 10, including exons 9 and 10 (Fig. 1A). However, the duplicated genomic region of 5047 bp also contains 2 deletions of 1,348 and 2,301 bp, in intron 10, separated from each other by a small residual sequence of 40 bp.
Fig. 1.

Partial gene structure of the VWF gene harboring the duplicated region. (A) Exons 1-11 are shown schematically. The area of VWF intron 8 to 10 is highlighted below. A gene fragment of 5,047 bp - spanning intron 8, exon 9, intron 9, exon 10 and a portion of intron 10 - is duplicated leading to a particular exon repetition of VWF exons 9/10, resulting in an in-frame fusion at the translational level. A comparison of the recombination sites is shown below.
(B) The duplicated area also exhibits 2 partial deletions of 1,348 and 2301 nucleotides, separated by residual 40 bp located in VWF intron 10. Folding this area resulted in a complex structure that has been depicted. The main base-paired stretch of this structure is due to the ALU3 and ALU4 elements that are inversely orientated and which display a very high sequence identity of about 62%. See also supplemental Fig. 3
To analyze the particular recombination events, RepeatMasker was used to identify repetitive DNA sequences. An ALU element was identified 5' to the recombination site, while intron 10 harbored 3 ALU elements that were affected by the deletion event. Two of these ALU elements (ALU2 and ALU3) were located in direction of the VWF gene transcript, while the third element (ALU4) was oriented in the opposite direction.
When investigating the recombination sites, only a small homologous DNA sequence of 4-6 bp (5'-TCTGTA/TG-3') was present at the borders of the duplication event. However, neither homologous nucleotides nor mini-direct repeats, reminiscent of Non Homologous End-Joining-mediated DNA repair were identified at the deletion sites. Also potential RAG1/2 recombination signal sequences were absent. However, our in silico DNA folding experiments demonstrated that Alu3 and Alu4 are able to form a highly stable DNA structure under physiological conditions with a free energy ΔG of −321.62 kcal/mol (Fig. 1B). This DNA structure is based on a nearly perfect match between the two DNA sequences of the inversely oriented Alu3 and Alu4 elements (sequence identity of 62%). While the identified homologous DNA sequence at the duplication sites is indicative for a “replication slippage” mechanism that might have caused the identified duplication, we propose that the very stable structure of the inverse ALU repeat 3 and 4 may have caused the deletion event.
Characterization of the mutant protein
By transient “homozygous” expression of mVWF and by co-expression of mVWF with wtVWF, we tried to assess the molecular pathomechanism and to reproduce the patients’ laboratory phenotype of VWF2AIIC Miami, respectively.
After co-transfection, mVWF:Ag secreted into the medium was only 0.38 in comparison to wtVWF, arbitrarily set to 1.0. Similar results were obtained for VWF in cell lysate, suggesting that the elevated VWF:Ag values in vivo were not the result of increased synthesis and/or secretion. As expected, functional parameters - VWF:CB, VWF:GPIbB but also VWF:FVIII binding - of the mutant derived from the homozygous expression were severely reduced, whereas co-expression attenuated these deficiencies (Table 2). Comparative multimer analysis of plasma VWF (Fig. 2A) and recombinant VWF (Fig. 2B) could convincingly confirm the causative nature of the duplication: “homozygous” mVWF did not show multimers beyond the size of a dimer, and the co-expression experiment resulted in multimers that could not be distinguished from patients’ plasma VWF and thereby exactly reproduced the phenotype.
Table 2.
Concentration and functional properties of rwt and rmVWF respectively.
| Ag Med | Ag Lys | pp | ppr | CB | CBr | GPIb | GPIbr | F8Br | |
|---|---|---|---|---|---|---|---|---|---|
| wt | 1.00 ± 0.03 | 1.00 ± 0.02 | 1.00 ± 0.02 | 1.00 | 1.00 ± 0.04 | 1.00 | 1.00 ± 0.04 | 1.00 | 1.00 ± 0.06 |
| ho | 0.19 ± 0.02 | 1.02 ± 0.06 | 0.08 ± 0.00 | 0.42 | 0.02 ± 0.01 | 0.11 | 0.02 ± 0.00 | 0.08 | 0.20 ± 0.02 |
| co | 0.38 ± 0.01 | 0.85 ± 0.03 | 0.29 ± 0.04 | 0.76 | 0.08 ± 0.01 | 0.21 | 0.13 ± 0.01 | 0.34 | 0.69 ± 0.02 |
Parameters are in relation to rwtVWF, ho=“homozygous” expression of duplication mutant, co=co-expression of mut and wtVWF, Ag Med=VWF:Ag in cell culture medium, Ag Lys=VWF:Ag in cell lysate, pp=VWFpp, CB=VWF:CB, GPIb=VWF:GPIbα binding in medium, r=ratios of parameters to VWF:Ag in medium,.
Fig. 2.
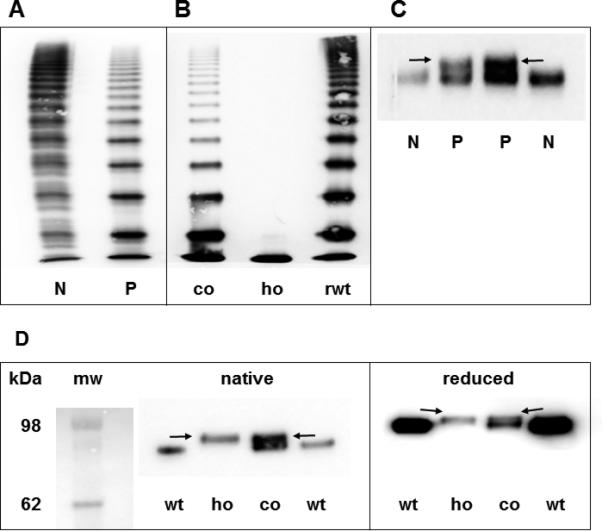
(A) Multimer analysis of VWF from patient's plasma. (B) Multimer analysis of rmVWF. (C) Western Blot of VWFpp from patients’ plasma. (D) Western Blot of recombinant VWFpp under native and reducing conditions. Arrows point to the mutant propeptide band.
N = normal plasma, P = patient, co = co-expression of wt and mVWF, ho = “homozygous” expression of mVWF, rwt = expression of rwtVWF, mw = molecular weight marker, r = recombinant, wt = wild type, m = mutant.
By gradient PAGE and immunoblotting using a VWFpp antibody we could visualize the expected larger mVWFpp in patients’ plasma (Fig. 2C). This could also be reproduced by a Western blot of rmVWFpp (Fig. 2D). Reduction of recombinant VWFpp by ß-mercaptoethanol resulted in reduced electrophoretic mobility, indicating a more expanded structure of reduced VWFpp, presumably due to loss of intramolecular disulfide bonds [28] (Fig. 2D).
Intracellular localization of the mutant protein
Since HEK293 cells secreted only mutant dimers after homozygous expression, we investigated the mutant's intracellular localization, to gain insight into the underlying processing defect. We observed an enrichment of the mutant protein within large intracellular granules (Fig. 3), strongly co-localizing with PDI, thereby indicating retention in the endoplasmic reticulum (ER) (Fig. 3D-F).
Fig. 3.
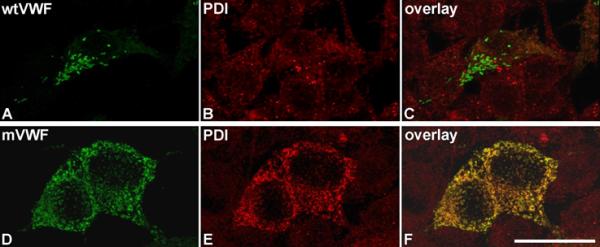
Intracellular localization of mVWF.
Wt (A) and mVWF (D) were transiently expressed in HEK293 cells. 48 h after transfection, cells were fixed and VWF proteins (A,D) and PDI (B,E) were detected by indirect immunofluorescence employing rabbit anti-VWF and mouse anti-PDI antibodies, respectively. Z-stacks were recorded with a confocal microscope using an HC PL APO CS2 63.0x1.40 OIL UV objective and the following settings: image size of 512 × 512, laser power of the 543 and 488 lasers was set to 9 % and 20 %, respectively. Overlays are shown in (C,F). 3D reconstruction was performed using the LAS software (Leica). Scale bar represents 25 μm. For movies of the rotating complete 3D reconstruction please refer to supplemental Movies S1 (rwtVWF) and S2 (rmVWF).
ADAMTS13 mediated proteolysis
To assess the influence of ADAMTS13 proteolysis on the mutant protein we exposed plasma VWF and recombinant VWF, respectively, to proteolysis by recombinant ADAMTS13. In our static assay, multimers of patients’ VWF and co-expressed wt/rmVWF were readily proteolyzed with no sign of resistance to ADAMTS13. Even more, by multimer analysis in a medium resolution gel we could identify an additional proteolytic band, both in the electrophoretic banding pattern of rmVWF and patients’ VWF (Fig. 4).
Clearance of rmVWF from the circulation
To investigate whether decreased clearance could explain the increased antigen levels in patients, VWF-deficient mice were injected with either wt or mVWF and the clearance of the protein was studied using this in vivo model. Recovery (calculated in samples obtained 3 min after injection) appeared to be similar between the two proteins (Table 3). Both wt and mutant were cleared from the circulation in a biphasic manner, characterized by a rapid initial phase and a slow secondary phase. Pharmacokinetic analysis allowed the calculation of the corresponding apparent half-lives that were significantly prolonged for the VWFpp mutant (Table 3). Mean residence time (MRT) was 1.90±0.30h for wt and 3.15±0.44h for the mutant (p=0.015) (Table 3 and Fig. 5).
Table 3.
Recovery and half-life of rmVWF compared to rwtVWF infused into VWF−/− mice.
| Recovery | MRT (h) | T1/2α (min) | T1/2β (h) | |
|---|---|---|---|---|
| rwtVWF:Ag | 69.7 ± 17.6 | 1.90 ± 0.30 | 7.0 ± 0.7 | 2.31 ± 0.17 |
| rmVWF:Ag | 93.1 ± 29.1 | 3.15 ± 0.44 | 10.1 ± 1.1 | 3.85 ± 0.38 |
| p-value | NS | 0.0153 | 0.0146 | 0.003 |
Recovery in % of injected VWF:Ag, MRT = mean residence time, T1/2α and T1/2β = half-life of rVWF
Fig. 5.

Kinetic study of rmVWF compared to rwtVWF infused into VWF−/− mice. VWF-deficient mice were injected intravenously with rmVWF (closed circles) or rwtVWF (open circles). Blood samples were taken at the indicated time points, and the amount of residual VWF antigen in plasma was quantified in an immunosorbent assay. Plotted is the percentage of residual VWF antigen in plasma relative to the amount injected versus time after injection. Data represent mean values ±SD of 3-9 mice for each time point.
Discussion
No VWF gene mutation could be identified by the original study on VWD2AIIC Miami, nevertheless, intragenic haplotype analysis suggested linkage to the VWF locus and not the contribution of a modifier gene [14]. After several previous attempts to detect a causative mutation in this family, we could finally identify by MLPA a heterozygous complex duplication/deletion event, including an in-frame-duplication of exons 9 and 10 in all studied family members. The identified duplication/deletion could be the result of “replication slippage” which subsequently caused the duplication and stalled replication at the inverted ALU3/4 elements causing the deletion, respectively. The mutation predicts the duplication of 53 amino acids in the D1 domain of VWF, which corresponds to a slower migrating electrophoretic band of the mutant VWFpp.
Characterization of co-expressed rwt/rmVWF convincingly confirmed the causative nature of the novel mutation by showing the typical multimer pattern of the heterozygous patients and by results of the functional assays with very low ratios of VWF:CB and VWF:GPIbα binding to VWF:Ag. This additionally confirmed the first example of dominant inheritance of a VWD subtype 2AIIC due to a mutation in the VWFpp.
After cleavage of pro-VWF at its furin cleavage site, the VWFpp is still bound non-covalently to mature VWF, mediating multimerization of VWF dimers and directing high molecular weight VWF multimers to their storage organelles, the Weibel-Palade Bodies (WPB) [6, 7, 29]. VWF with its propeptide deleted, remains at the stage of the dimer, emphasizing the role of the propeptide for the multimerization process [29].
How the duplication of 53 amino acids in the VWFD1 domain encoded by exons 9 and 10 could interfere with this process is not evident at first sight. Certainly, it can be assumed that the duplication which contains 8 cysteine residues has an impact on the structure of the propeptide which is largely shaped by intra-molecular cysteine bonds [28]. Possible scenarios also include a negative impact of the duplication on a presumably concerted action of the D1 and D2 CGLC sequences on multimerization, by increasing the distance between these two CGLCs, which are encoded on exons 5 and 14, respectively. Since exons 9 and 10 contain several cysteine codons, the duplicated amino acids may also directly interfere with multimerization. Three cysteine mutations in the VWFD2 domain causing VWD2AIIC [8, 11, 12] and several examples of cysteine mutations in the VWFD3 domain causing multimerization deficits [5], support this idea. Similar to other mutations in the D1 and D2 domains [26, 30], the intracellular transport via the Golgi and Trans-Golgi compartments into WPB, a prerequisite for the formation of VWF HMWM, seems to be severely impaired.
Mutant VWF is only detected in the ER and does not seem to initiate WPB formation, the natural storage organelles for VWF HMWM prior to their stimulated secretion into the blood stream. But in contrast to these previously described mutants the Miami mutant seems to induce formation of PDI granules in which PDI and the mutant strongly co-localize (Fig. 3D-F). Because PDI is a protein disulfide isomerase that catalyzes the disulfide bond formation between cysteine residues, we additionally investigated the intracellular localization of the VWD2AIIC mutant p.Cys410Ser. For this mutant, we indeed found the same phenotype (supplemental Fig. 5) as for VWF2AIIC-Miami. It thus seems that cysteine mutations can alter VWF-PDI-interaction, which should only serve VWF dimerization [32], and induce a prolonged and strongly increased interaction with PDI, thereby trapping these mutants in the ER. In heterozygous patients and in co-expression experiments, multimers may escape ER retention, depending on the contribution of wtVWF monomers to the individual multimer, whereas dimers may be secreted by the constitutive pathway. This would be in accordance with the observed multimer pattern of both, patients’ and co-expressed rwt/rmVWF.
We also focused on the absent ADAMTS13 proteolysis of mutant VWF seen in all patients with the VWD2AIIC subtype [3, 8, 9] by incubation of patient's plasma VWF and rmVWF with rADAMTS13. Mutant VWF from either source was readily digested by ADAMTS13. This prompted us to question if the lack of mVWF proteolysis was due to reduced accessibility of the VWFA2 domain cleavage site for ADAMTS13 under normal shear forces of the blood stream. Under physiological conditions, this cleavage site becomes only accessible when VWF is stretched under shear flow, which requires prior binding to its receptors at the vessel wall and platelets [31-33]. It then seems logical to assume that reduced VWF function would also reduce VWF proteolysis by ADAMTS13 in vivo.
This suggests that the reduced in vivo proteolytic susceptibility of VWF2AIIC Miami is not an intrinsic property of the mutant protein but rather the result of its functional deficits. In this model, lack of function due to absence of VWF HMWM would prevent VWF binding to sub-endothelial collagen and platelet receptors, resulting in reduced shear stress on the molecule under flow conditions, as a prerequisite for sufficient VWF unfolding [34] and subsequently in reduced accessibility of the VWF ADAMTS13 cleavage site in the A2 domain. Similar observations were made for another subtype of VWF2A with reduced proteolysis, namely the phenotype IIE [5].
In addition or alternatively, the aberrant proteolytic sub-band, experimentally produced by ADAMTS13 proteolysis of mutant plasma VWF and of the rmVWF, indicating an aberrant primary structure of mature mVWF, may confer ADAMTS13 resistance under shear flow conditions in vivo.
Elevated VWF:Ag is present in all family members with VWD type 2AIIC Miami, which might reflect either increased secretion or decreased clearance. However, our first experiments did not explain the elevated VWF:Ag. VWF:Ag of rmVWF was reduced rather than elevated in the cell culture medium and normal in the cell lysate, speaking against increased synthesis and/or secretion. VWF clearance seems to be independent from ADAMTS13 mediated proteolysis [35]. Therefore, the lack of VWF proteolysis by ADAMTS13 should not prolong the half-life of VWF in the circulation. However, the elevated ratio of mature mVWF to its propeptide strongly suggests decreased clearance, and historical data of a DDAVP test in one of the patients is consistent with this hypothesis (s. supplemental Fig. 6). Finally, infusion studies of rmVWF into VWF−/− mice provided further confirmation. These results indicate that the clearance of the mVWF is indeed decreased in vivo, which will ultimately result in elevated VWF levels in VWD2AIIC Miami.
Mutations influencing presumable receptor mediated clearance are located in mature VWF [27, 35]. In contrast, the VWFpp-mutation should not directly affect receptor dependent clearance since it is located in the VWFpp and no longer attached to mature VWF. However, due to the physiological role of the propeptide to catalyze multimerization by disulfide bond formation in the VWFD3 domain, the aberrant structure of the mutant VWFpp could cause aberrant disulfide bond formation in mature VWF, a hypothesis that is supported by our detection of an aberrant VWF proteolytic band after ADAMTS13 digestion and in addition by the significantly reduced VWF:FVIII binding property of the recombinant mutant protein. This seems reasonable since the VWF:FVIII binding property is located in the D’ [36] and presumably also in the D3 domain and cysteine mutations in both domains have been shown to affect FVIII binding [37-39].
In conclusion, more than 20 years after the original report in “Blood” we have finally identified and studied in detail an in frame duplication of VWF exons 9 and 10 as the elusive dominant mutation causing VWD2AIIC-Miami in the original family. The mutation interferes with proper multimerization due to ER retention, and does not cause enhanced synthesis of VWF in vitro. The elevated VWF:Ag in vivo is due to decreased clearance, resulting in prolonged half-life of mutant VWF. In contrast to the decreased proteolysis of mutant VWF by ADAMTS13 in the circulation, ADAMTS13 susceptibility of mutant VWF under static conditions in vitro is not decreased. Therefore, the lack of VWF proteolysis by ADAMTS13 in the patients’ circulation probably results from the functional deficits of the mutant protein, which does not sufficiently unfold under the shear forces in a physiological environment, a prerequisite for the VWF/ADAMTS13 interaction in vivo. The aberrant proteolytic band, experimentally generated by ADAMTS13 proteolysis of plasma and rmVWF, respectively, points to an influence of the mutation on the primary structure of mature mutant VWF, which would explain the observed dominant inheritance of VWD2AIIC-Miami.
Supplementary Material
Essentials.
Von Willebrand disease (VWD) IIC Miami features high von Willebrand factor (VWF) with reduced function.
We aimed to identify and characterize the elusive underlying mutation in the original family.
An inframe duplication of VWF exons 9-10 was identified and characterized.
The mutation causes a defect in VWF multimerization and decreased VWF clearance from the circulation.
Acknowledgements
This work has been made possible by DFG grants DFG Schn 325-4, DFG Research Group FOR 1543 (SHENC), and by grants from the German National Genome Research Net, NGFN2 (NHK-S17T22) to R.S., NIH grant HL081588 to R.R.M. and NIH grants HL072917 and HL112303 to J.E.S., respectively.
We thank the UKE Microscopy Imaging Facility for technical support and providing the Leica SP5 microscope.
The article is dedicated to the original family for their extraordinary support of our study, and their enduring interest in the scientific issues over two decades.
Footnotes
Authorship
Contribution: T. Obser mapped and characterized the duplication, designed and carried out the expression studies, functionally characterized plasma and rmVWF, carried out proteolysis studies with recombinant ADAMTS13, and wrote part of the manuscript. M. L. Kraemer did the first sequencing studies, recruited the patients, assessed their clinical phenotype, co-wrote and critically reviewed the manuscript, F. Oyen carried out genotyping, RNA analysis and the MLPA. M. A. Brehm carried out studies on VWF intracellular trafficking and wrote part of the manuscript. C. V. Denis carried out the animal studies, interpreted the pharmacokinetic data and wrote part of the manuscript. R. Marschalek analyzed and described the complex duplication to identify the recombination mechanism. R. R. Montgomery and J. E. Sadler did mutation screening and critically reviewed the manuscript. S. Schneppenheim and U. Budde characterized the patients’ and recombinant VWF by basic VWF-parameters and multimer analysis and reviewed the manuscript. R. Schneppenheim designed the study, collected, evaluated and interpreted the phenotypic, genotypic and expression data and wrote the paper.
Conflict-of interest-disclosure: The authors declare no competing financial interests.
Disclosure J. E. Sadler reports personal fees from Baxter HealthCare, BioMarin, XO1 Limited, Band Therapeutics, Ablynx and 23andMe, outside the submitted work.
Other authors have nothing to disclose.
References
- 1.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, Federici AB, Working Party on von Willebrand Disease C Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 2.Schneppenheim R, Budde U. von Willebrand factor: the complex molecular genetics of a multidomain and multifunctional protein. J Thromb Haemost. 2011;9(Suppl 1):209–15. doi: 10.1111/j.1538-7836.2011.04324.x. [DOI] [PubMed] [Google Scholar]
- 3.Zimmerman TS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor. Cleavage is present in normal individuals, increased in IIA and IIB von Willebrand disease, but minimal in variants with aberrant structure of individual oligomers (types IIC, IID, and IIE). J Clin Invest. 1986;77:947–51. doi: 10.1172/JCI112394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneppenheim R, Brassard J, Krey S, Budde U, Kunicki TJ, Holmberg L, Ware J, Ruggeri ZM. Defective dimerization of von Willebrand factor subunits due to a Cys-> Arg mutation in type IID von Willebrand disease. Proc Natl Acad Sci U S A. 1996;93:3581–6. doi: 10.1073/pnas.93.8.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneppenheim R, Michiels JJ, Obser T, Oyen F, Pieconka A, Schneppenheim S, Will K, Zieger B, Budde U. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115:4894–901. doi: 10.1182/blood-2009-07-226324. [DOI] [PubMed] [Google Scholar]
- 6.Verweij CL, Hart M, Pannekoek H. Expression of variant von Willebrand factor (vWF) cDNA in heterologous cells: requirement of the pro-polypeptide in vWF multimer formation. EMBO J. 1987;6:2885–90. doi: 10.1002/j.1460-2075.1987.tb02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–46. doi: 10.1146/annurev.cb.06.110190.001245. [DOI] [PubMed] [Google Scholar]
- 8.Gaucher C, Dieval J, Mazurier C. Characterization of von Willebrand factor gene defects in two unrelated patients with type IIC von Willebrand disease. Blood. 1994;84:1024–30. [PubMed] [Google Scholar]
- 9.Schneppenheim R, Thomas KB, Krey S, Budde U, Jessat U, Sutor AH, Zieger B. Identification of a candidate missense mutation in a family with von Willebrand disease type IIC. Hum Genet. 1995;95:681–6. doi: 10.1007/BF00209487. [DOI] [PubMed] [Google Scholar]
- 10.Holmberg L, Karpman D, Isaksson C, Kristoffersson AC, Lethagen S, Schneppenheim R. Ins405AsnPro mutation in the von Willebrand factor propeptide in recessive type 2A (IIC) von Willebrand's disease. Thromb Haemost. 1998;79:718–22. [PubMed] [Google Scholar]
- 11.Enayat MS, Ravanbod S, Rassoulzadegan M, Jazebi M, Ala F, Budde U, Schneppenheim S, Obser T, Schneppenheim R. Identification of a homozygous Cys410Ser mutation in the von Willebrand factor D2 domain causing type 2A(IIC) von Willebrand disease phenotype in an Iranian patient. Haemophilia. 2013;19:e261–4. doi: 10.1111/hae.12161. [DOI] [PubMed] [Google Scholar]
- 12.Lanke E, Kristoffersson AC, Philips M, Holmberg L, Lethagen S. Characterization of a novel mutation in the von Willebrand factor propeptide in a distinct subtype of recessive von Willebrand disease. Thromb Haemost. 2008;100:211–6. [PubMed] [Google Scholar]
- 13.Ruggeri ZM, Nilsson IM, Lombardi R, Holmberg L, Zimmerman TS. Aberrant multimeric structure of von Willebrand factor in a new variant of von Willebrand's disease (type IIC). J Clin Invest. 1982;70:1124–7. doi: 10.1172/JCI110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ledford MR, Rabinowitz I, Sadler JE, Kent JW, Civantos F. New variant of von Willebrand disease type II with markedly increased levels of von Willebrand factor antigen and dominant mode of inheritance: von Willebrand disease type IIC Miami. Blood. 1993;82:169–75. [PubMed] [Google Scholar]
- 15.Tosetto A, Rodeghiero F, Castaman G, Goodeve A, Federici AB, Batlle J, Meyer D, Fressinaud E, Mazurier C, Goudemand J, Eikenboom J, Schneppenheim R, Budde U, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost. 2006;4:766–73. doi: 10.1111/j.1538-7836.2006.01847.x. [DOI] [PubMed] [Google Scholar]
- 16.Mazurier C, Parquet-Gernez A, Goudemand M. [Enzyme-linked immunoabsorbent assay of factor VIII-related antigen. Interest in study of Von Willerbrand's disease (author's transl)]. Pathol Biol (Paris) 1977;25(Suppl):18–24. [PubMed] [Google Scholar]
- 17.Weiss HJ, Hoyer LW, Rickles FR, Varma A, Rogers J. Quantitative assay of a plasma factor deficient in von Willebrand's disease that is necessary for platelet aggregation. Relationship to factor VIII procoagulant activity and antigen content. J Clin Invest. 1973;52:2708–16. doi: 10.1172/JCI107465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown JE, Bosak JO. An ELISA test for the binding of von Willebrand antigen to collagen. Thromb Res. 1986;43:303–11. doi: 10.1016/0049-3848(86)90150-7. [DOI] [PubMed] [Google Scholar]
- 19.Schneppenheim R, Budde U, Krey S, Drewke E, Bergmann F, Lechler E, Oldenburg J, Schwaab R. Results of a screening for von Willebrand disease type 2N in patients with suspected haemophilia A or von Willebrand disease type 1. Thromb Haemost. 1996;76:598–602. [PubMed] [Google Scholar]
- 20.Eikenboom J, Federici AB, Dirven RJ, Castaman G, Rodeghiero F, Budde U, Schneppenheim R, Batlle J, Canciani MT, Goudemand J, Peake I, Goodeve A, Group M-VS. VWF propeptide and ratios between VWF, VWF propeptide, and FVIII in the characterization of type 1 von Willebrand disease. Blood. 2013;121:2336–9. doi: 10.1182/blood-2012-09-455089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruggeri ZM, Zimmerman TS. The complex multimeric composition of factor VIII/von Willebrand factor. Blood. 1981;57:1140–3. [PubMed] [Google Scholar]
- 22.Schneppenheim R, Plendl H, Budde U. Luminography--an alternative assay for detection of von Willebrand factor multimers. Thromb Haemost. 1988;60:133–6. [PubMed] [Google Scholar]
- 23.Budde U, Schneppenheim R, Eikenboom J, Goodeve A, Will K, Drewke E, Castaman G, Rodeghiero F, Federici AB, Batlle J, Perez A, Meyer D, Mazurier C, Goudemand J, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1VWD). J Thromb Haemost. 2008;6:762–71. doi: 10.1111/j.1538-7836.2008.02945.x. [DOI] [PubMed] [Google Scholar]
- 24.Gerritsen HE, Turecek PL, Schwarz HP, Lammle B, Furlan M. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP). Thromb Haemost. 1999;82:1386–9. [PubMed] [Google Scholar]
- 25.Hassenpflug WA, Budde U, Obser T, Angerhaus D, Drewke E, Schneppenheim S, Schneppenheim R. Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood. 2006;107:2339–45. doi: 10.1182/blood-2005-04-1758. [DOI] [PubMed] [Google Scholar]
- 26.Brehm MA, Huck V, Aponte-Santamaria C, Obser T, Grassle S, Oyen F, Budde U, Schneppenheim S, Baldauf C, Grater F, Schneider SW, Schneppenheim R. von Willebrand disease type 2A phenotypes IIC, IID and IIE: A day in the life of shear-stressed mutant von Willebrand factor. Thromb Haemost. 2014;112:96–108. doi: 10.1160/TH13-11-0902. [DOI] [PubMed] [Google Scholar]
- 27.Lenting PJ, Westein E, Terraube V, Ribba AS, Huizinga EG, Meyer D, de Groot PG, Denis CV. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279:12102–9. doi: 10.1074/jbc.M310436200. [DOI] [PubMed] [Google Scholar]
- 28.McCarroll DR, Levin EG, Montgomery RR. Endothelial cell synthesis of von Willebrand antigen II, von Willebrand factor, and von Willebrand factor/von Willebrand antigen II complex. J Clin Invest. 1985;75:1089–95. doi: 10.1172/JCI111802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wise RJ, Pittman DD, Handin RI, Kaufman RJ, Orkin SH. The propeptide of von Willebrand factor independently mediates the assembly of von Willebrand multimers. Cell. 1988;52:229–36. doi: 10.1016/0092-8674(88)90511-9. [DOI] [PubMed] [Google Scholar]
- 30.Haberichter SL, Budde U, Obser T, Schneppenheim S, Wermes C, Schneppenheim R. The mutation N528S in the von Willebrand factor (VWF) propeptide causes defective multimerization and storage of VWF. Blood. 2010;115:4580–7. doi: 10.1182/blood-2009-09-244327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–4. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baldauf C, Schneppenheim R, Stacklies W, Obser T, Pieconka A, Schneppenheim S, Budde U, Zhou J, Grater F. Shear-induced unfolding activates von Willebrand factor A2 domain for proteolysis. J Thromb Haemost. 2009;7:2096–105. doi: 10.1111/j.1538-7836.2009.03640.x. [DOI] [PubMed] [Google Scholar]
- 33.Gao W, Anderson PJ, Majerus EM, Tuley EA, Sadler JE. Exosite interactions contribute to tension-induced cleavage of von Willebrand factor by the antithrombotic ADAMTS13 metalloprotease. Proc Natl Acad Sci U S A. 2006;103:19099–104. doi: 10.1073/pnas.0607264104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao W, Anderson PJ, Sadler JE. Extensive contacts between ADAMTS13 exosites and von Willebrand factor domain A2 contribute to substrate specificity. Blood. 2008;112:1713–9. doi: 10.1182/blood-2008-04-148759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schooten CJ, Tjernberg P, Westein E, Terraube V, Castaman G, Mourik JA, Hollestelle MJ, Vos HL, Bertina RM, Berg HM, Eikenboom JC, Lenting PJ, Denis CV. Cysteine-mutations in von Willebrand factor associated with increased clearance. J Thromb Haemost. 2005;3:2228–37. doi: 10.1111/j.1538-7836.2005.01571.x. [DOI] [PubMed] [Google Scholar]
- 36.Foster PA, Fulcher CA, Marti T, Titani K, Zimmerman TS. A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J Biol Chem. 1987;262:8443–6. [PubMed] [Google Scholar]
- 37.Allen S, Abuzenadah AM, Blagg JL, Hinks J, Nesbitt IM, Goodeve AC, Gursel T, Ingerslev J, Peake IR, Daly ME. Two novel type 2N von Willebrand disease-causing mutations that result in defective factor VIII binding, multimerization, and secretion of von Willebrand factor. Blood. 2000;95:2000–7. [PubMed] [Google Scholar]
- 38.Hilbert L, Jorieux S, Proulle V, Favier R, Goudemand J, Parquet A, Meyer D, Fressinaud E, Mazurier C, Disease INoMAivW Two novel mutations, Q1053H and C1060R, located in the D3 domain of von Willebrand factor, are responsible for decreased FVIII-binding capacity. Br J Haematol. 2003;120:627–32. doi: 10.1046/j.1365-2141.2003.04163.x. [DOI] [PubMed] [Google Scholar]
- 39.Schneppenheim R, Lenk H, Obser T, Oldenburg J, Oyen F, Schneppenheim S, Schwaab R, Will K, Budde U. Recombinant expression of mutations causing von Willebrand disease type Normandy: characterization of a combined defect of factor VIII binding and multimerization. Thromb Haemost. 2004;92:36–41. doi: 10.1160/TH04-02-0084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.