

Abstract
The endoplasmic reticulum (EnR) stress sensor, the unfolded protein response (UPR), plays a key role in regulating intracellular protein homeostasis. The extensively studied reactive mode of UPR activation is characterized by unfolded protein, or other EnR stress, triggering UPR activation. Here we focus on the emerging anticipatory mode of UPR activation in which mitogenic steroid and peptide hormones and other effectors pre-activate the UPR and anticipate a future need for increased protein folding capacity. Mild UPR activation in breast cancer can be protective and contributes to antiestrogen resistance. Hyperactivation of the anticipatory UPR pathway in cancer cells with a small molecule converts it from cytoprotective to cytotoxic, highlighting its potential as a therapeutic target in estrogen receptor positive breast cancer.
Keywords: Estrogen, Cancer, Unfolded Protein Response, BHPI
The Reactive UPR Pathway
Protein folding homeostasis and quality control is maintained by the sensor system for endoplasmic reticulum (EnR) stress (see Glossary), the unfolded protein response (UPR) [1–4]. The UPR consists of three main branches that together balance the synthesis of new proteins, with the availability of chaperones and other proteins to help fold and transport proteins within cells. EnR stress activates the three main arms of the UPR (Figure 1). The transmembrane kinase protein kinase RNA-like endoplasmic reticulum kinase (PERK) is activated by autophosphorylation. P-PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), resulting in transient inhibition of most protein synthesis [5]. UPR activation also results in proteolytic cleavage and activation of activating transcription factor 6 α (ATF6α). Activated ATF6α (p50-ATF6α) enters the nucleus and regulates expression of UPR targets. Upon activation by oligomerization and autophosphorylation, the third UPR sensor, inositol-requiring enzyme 1 α (IRE1α), alternatively splices inactive XBP1 mRNA (X-box binding protein 1) producing active spliced XBP1 (sp-XBP1). IRE1α and ATF6α activation leads to induction of the chaperone BiP/GRP78/HSPA5 (binding immunoglobulin protein/glucose regulated protein 78 kDa/heat shock protein A5) and other chaperones that increase protein-folding capacity, and to altered mRNA decay and translation. Simultaneously, degradation of misfolded protein is increased.
Fig. 1. Schematic Representation of the Cytoprotective and Cytototxic Anticipatory UPR Pathways.
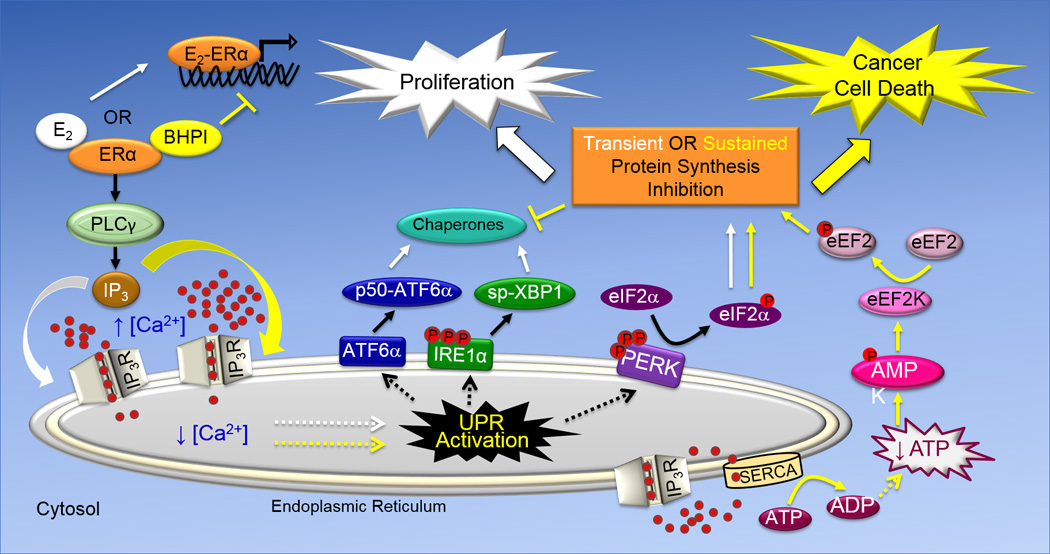
The UPR consists of three arms, ATF6α, IRE1α and PERK that are named after proteins embedded in the membrane of the endoplasmic reticulum. While activation of the ATF6α and IRE1α arms of the UPR leads to induction of chaperones that increase protein folding capacity, activation of the PERK arm of the UPR leads to reduced protein production. ATF6α activation leads to translocation of the transmembrane protein p90-ATF6α from the EnR to the Golgi apparatus, where it encounters proteases that liberate the N-terminal fragment of ATF6α (p50-ATF6α). p50-ATF6α is a transcription factor that increases the protein-folding capacity of the EnR by inducing EnR-resident chaperones, including BiP. The transmembrane protein IRE1α is a nuclease. Oligomerization and phosphorylation activates IRE1α. Activated IRE1α removes an intron from full-length XBP1 mRNA, producing spliced (sp)-XBP1 mRNA. sp-XBP1 is a transcription factor that increases the protein-folding capacity of the EnR and turnover of misfolded proteins by inducing EnR resident-chaperones. Oligomerization and autophosphorylation activate the transmembrane kinase PERK. Activated p-PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), leading to inhibition of protein synthesis and a reduction in the endoplasmic reticulum protein folding load. E2 and BHPI bind at distinct sites on ERα. In the anticipatory UPR (shown on the left), E2 and BHPI act via ERα to activate a pathway that results in opening of EnR IP3R calcium channels allowing efflux of calcium stored in the lumen of the EnR into the cytosol. This activates the UPR. In ERα containing cancer cells, strong and sustained opening of the EnR IP3R calcuim channels by BHPI sets up a futile cycle of ATP-dependent pumping of calcium into the EnR by SERCA pumps and leakage back into the cytosol through the open IP3R channels (lower right). This depletes intracellular ATP, activating the energy sensor AMPK (AMP kinase). Elevated intracellular calcium and activated AMPK phosphorylate and activate eukaryotic elongation factor 2 (eEF2) kinase. eEF2 kinase phosphorylates and inactivates eEF2. This inhibits protein synthesis at a second site, resulting in strong and sustained toxic inhibition of protein synthesis by BHPI.
In this “reactive” mode, EnR stress resulting from accumulation of unfolded or misfolded protein, or other stresses, triggers UPR activation. How are the UPR sensors activated? IRE1α contains domains that bind unfolded proteins leading to structural alterations that might result in oligomerization and autophosphorylation [6–9]. Whether the other UPR sensors are activated by binding unfolded proteins is unclear. The EnR membrane contains ATP-dependent SERCA (sarcoplasmic/endoplasmic reticulum calcium ATPase) pumps that maintain a high concentration of calcium in the lumen of the EnR. The SERCA pump inhibitor, thapsigargin, and the ionophore, ionomycin, activate the UPR by depleting EnR calcium. The calcium-dependent chaperone BiP binds to the 3 UPR sensors, inhibiting their activation. Unfolded protein or low calcium may remove BiP from UPR sensors, allowing sensor oligomerization and UPR activation [2, 10].
Reactive UPR activation is the initial response of cells to EnR stress. Recently, a different mode of UPR activation, termed “anticipatory” activation [1] has emerged as a hormone-activated pathway and a therapeutic target [11]. In this pathway, cells pre-activate the UPR in anticipation of a future requirement for increased protein folding capacity due to protein secretion (Box 1) or hormone-stimulated cell proliferation. Here we focus on the anticipatory UPR pathway, its likely role in hormone-dependent cancer and therapeutic targeting of this pathway.
BOX 1: Anticipatory Activation of the UPR Across Cell Types and Species.
B-cell Differentiation Triggers Anticipatory Activation of the UPR
Anticipatory activation of the UPR was first observed in differentiating immune cells. Following exposure to an antigen, B-cells differentiate into plasma cells, which ultimately synthesize and secrete massive amounts of immunoglobulins. Anticipating the need to expand protein-folding capacity, the UPR is activated prior to increased immunoglobulin secretion [85, 86]. Importantly, differentiating mutant B cells that cannot make immunoglobulin still activate the UPR [85]. Although the full pathway has not been elucidated, B-cell antigen receptor (BCR), acting through tyrosine kinases (Figure 2) likely initiates the pathway [87]. Thus a wide range of stimuli can activate the anticipatory UPR pathway,
The Anticipatory UPR Pathway in Insects: Ecdysone
One factor that suggests a pathway is important is its conservation across species. The steroid hormone, ecdysone, acting through the ecdysone receptor (EcR), plays an important role in insect metamorphosis. Recent studies suggest that the anticipatory UPR pathway plays an important role in ecdysone action. The anticipatory UPR pathway is activated when inositol triphosphate receptor (IP3R) calcium channels in the EnR membrane are opened; this allows high concentrations of calcium stored in the lumen of the EnR to flow into the cytosol. IP3R calcium channels open following binding of inositol triphosphate (IP3). IP3 is produced by the enzyme reaction catalyzed by activated phospholipase C γ (PLCγ) (Figure 1). Consistent with activation of the anticipatory UPR pathway, in the lepidopteran insect Helicoverpa armigera, (the cotton bollworm) 20-hydroxyecdysone-EcR acts through G-protein coupled receptors (GPCRs) and Src (proto-oncogene Sarcoma) tyrosine kinase to phosphorylate and activate PLCγ, increasing cytosol calcium. Several lines of evidence support a role in ecdysone action for the early steps in the anticipatory UPR pathway. Inhibition or knockdown of PLCγ blocked 20-hydroxyecdysone induced pupation and caused larval death. Moreover, an IP3R inhibitor blocked the increase in cytosol calcium [88], and blocking the increase in cytosol calcium inhibited 20-hydroxyecdysone induced transcription [88, 89]. These data suggest ecdysone activates an evolutionarily conserved insect anticipatory UPR pathway.
A Largely Common Pathway for Anticipatory Activation of the UPR
The biological functions of steroid and peptide hormones are mediated through direct interaction with hormone-specific receptors. Mitogenic hormones, including the steroid hormones estrogen (17β-estradiol: E2), androgen (dihydrotestosterone, DHT) and ecdysone (Ec) bind to their respective receptors (ERα, AR, and EcR) and peptide hormones epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) bind to their plasma membrane receptors (EGFR, VEGFR). The mitogenic human steroid and peptide hormones E2, DHT, EGF and VEGF contribute to the pathology of several types of cancer and are important therapeutic targets. Estrogens and androgens act via their receptors to play pivotal roles in breast and prostate cancer, respectively [12, 13]. Endocrine therapies based on inhibitors that target the synthesis of estrogens and androgens and antagonists that inhibit hormone binding to their respective receptors are mainstays in the treatment of breast and prostate cancer. EGF activates progrowth and antapoptotic signaling pathways in several types of cancer [14]. Tumor growth and metastases depends on growth of a new vascular network. VEGF acting through VEGF receptors, promotes cell viability and angiogenesis, the formation of new blood vessels [15]. The EGF and VEGF pathways are major therapeutic targets in many types of cancer.
Steroid hormones act in the nucleus to regulate gene expression. While activation of genomic steroid hormone regulated gene expression programs initiates quickly, they play out over many hours. In addition, a diverse set of rapid extranuclear actions of steroid receptors, often initiated at or near the plasma membrane both modulate the genomic programs and influence many cell functions [16]. In contrast to the well-studied effects of steroid and peptide hormones on established signaling pathways, until recently rapid anticipatory activation of the UPR by steroid and peptide hormones was largely unexplored.
Several lines of evidence indicate that rapid hormone activation of the UPR occurs in the absence of EnR stress and accumulation of unfolded protein. VEGF did not alter EnR processing of a GFP-tagged secreted protein [17] and EGF activates the UPR in cells in which EGF does not induce cell proliferation [18]. How do hormones elicit rapid anticipatory activation of the UPR? The EnR is a closed membrane and the EnR lumen contains a very high concentration of calcium relative to the cell body. As discussed above for the reactive UPR pathway, loss of EnR calcium activates the UPR. Since the concentration of EnR calcium is high relative to the cell body, opening calcium channels in the EnR membrane results in rapid efflux of calcium from the lumen of the EnR into the cell body. Although there are several classes of calcium channels in the EnR membrane, all the known activators of the anticipatory UPR pathway use a common pathway that results in opening the EnR inositol triphophoshate receptor (IP3R) calcium channels. The IP3R calcium channels open on binding of inositol triphosphate (IP3). IP3 is produced enzymatically by activated phosphorylated phospholipase C γ (PLCγ). All the hormones that elicit anticipatory activation of the UPR activate PLCγ (Figure 2) [17–20]. The receptors for EGF and VEGF are tyrosine kinases that directly phosphorylate and activate PLCγ. The receptors for E2 and ecdysone lack tyrosine kinase activity. Binding of E2 to ERα and of ecdysone to EcR induces a conformational change that ultimately alters the activity of a tyrosine kinase that in turn activates PLCγ. Importantly, BHPI, a recently described ERα biomodulator with anticancer activity, binds ERα at a different site than estrogens and induces a conformational change in ERα that leads to strong and sustained activation of the anticipatory UPR pathway [20]. While the mitogens E2, EGF and VEGF and the small molecule BHPI all activate the rapid anticipatory UPR pathway, DHT does not [21]. Instead, DHT activates the UPR after many hours [22]. Supporting the proposed pathway, inhibition and knockdown of PLCγ blocks UPR activation by all four rapid activators [17–20]. Moreover, E2, EGF and BHPI all increased IP3 levels [18–20]. For each activator, inhibition of the IP3Rs by 2-aminoethoxydiphenyl borate (2-APB) abolishes increases in cytosol calcium [17–20]. Moreover, for EGF, E2 and BHPI simultaneous knockdown of the three EnR IP3R calcium channels blocked the increase in cytosol calcium and abolished UPR activation [18–20]. In contrast, other findings indicate that VEGF mediated UPR activation does not involve calcium depletion; the UPR remained activated after co-treatment with VEGF and the IP3R inhibitor 2-APB. Instead, a complex between VEGF-activated PLCγ and mTORC was proposed as triggering UPR activation [17]. This raises the possibility of multiple pathways for hormone-mediated anticipatory activation of the UPR.
Fig. 2. The Diverse Activators of PLCγ.
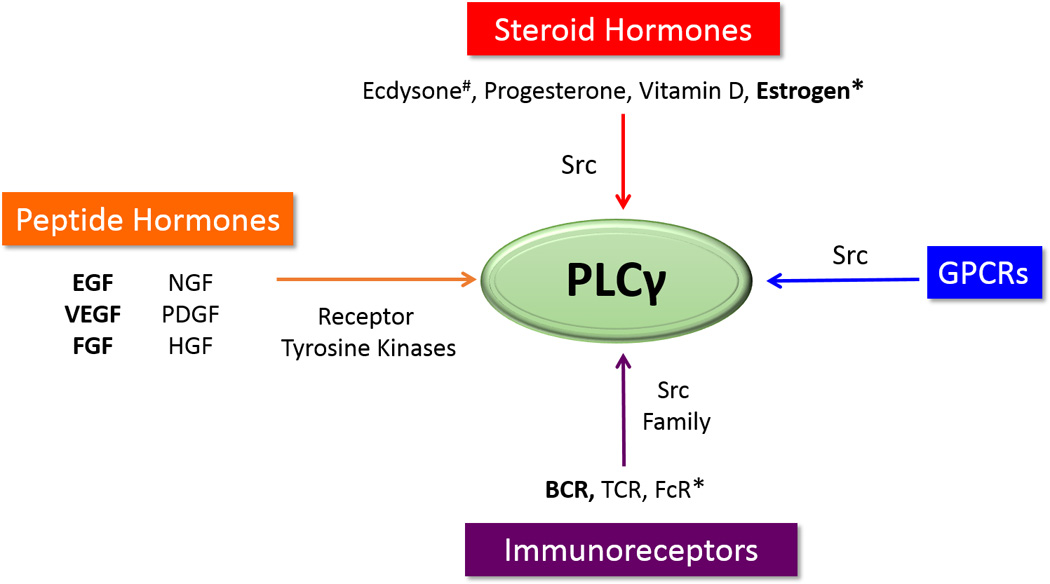
Activators of PLCγ known to activate the UPR are in bold. * indicates the activating tyrosine kinase has not been identified. #Ecdysone acts with a GPCR to open the IP3R calcium channels, but activation of the UPR arms has not been investigated. Tyrosine kinases that activate PLCγ are expected to activate the anticipatory UPR pathway. Peptide hormone receptors that are membrane tyrosine kinases that activate PLCγ are shown on the left. Src kinase has also been shown to activate PLCγ. Several steroid hormones activate Src kinase and are likely activators of the anticipatory UPR pathway (center). Immunoreceptors, shown on the bottom, working through GPCRs and Src family kinases are not mitogenic hormones and instead activate the UPR to antcipate the increased protein folding load that accompanies antibody production.
Additional experiments show that VEGF, EGF, E2 and BHPI induce PLCγ-dependent anticipatory activation of two UPR arms via p50-ATF6α and p-IRE1α (Figure 1), resulting in increased activated sp-XBP1 and induction of BiP mRNA and protein [17–20]. Is the protein synthesis inhibiting PERK arm activated as part of these mitogenic programs? VEGF, EGF and E2 each induce a mild and transient activation of the PERK arm of the UPR and transient appearance of p-eIF2α [17–19]. Detailed studies show E2 induces a transient increase in phosphorylation of PERK and eIF2α and a transient and mild inhibition of protein synthesis [19]. These studies suggest mitogenic hormones initiate anticipatory activation of the chaperone-inducing, pro-proliferation arms of the UPR without impacting the supply of new protein needed for cell proliferation.
Rapid Anticipatory Activation of the UPR is Important for Hormone Induction of Cell Proliferation
The observations discussed above raise several questions. Does rapid UPR activation by VEGF, EGF and E2 enhance their ability to induce cell proliferation? If so, what UPR-related molecules and signals facilitate cell proliferation? For all three hormones, PLCγ knockdown strongly inhibited hormone-induced cell proliferation [17–19]. Upon activation of the PLCγ-UPR pathway, intracellular calcium levels increase very rapidly. Several hours later BiP chaperone levels increase. Elevated intracellular calcium is a proliferation signal and is associated with aggressive tumors [23, 24]. Elevated BiP is associated with cancer cell proliferation, a poor prognosis and therapy resistance [25–28]. Knockdown experiments were employed to test the role of different pathway components in cell proliferation. Knockdown of eIF2α or ATF6α reduced VEGF-mediated endothelial cell survival by ~50% [17]; knockdown of ATF6α and XBP1 reduced EGF-stimulated cell proliferation 30–40% [18]. IP3R knockdown strongly inhibited E2-ERα induced proliferation of breast cancer cells, while XBP1 and ATF6α knockdown moderately inhibited proliferation. As expected, since estrogen had minimal effects on the PERK arm of the UPR, PERK knockdown had no effect [19]. Since XBP1 and ATF6α act after the increase in cytosol calcium, both elevated intracellular calcium and UPR induced chaperones play a role in hormone-induced cell proliferation.
PLCγ-IP3R-mediated Elevated Intracellular Calcium is Important for Hormone-regulated Gene Expression
The role of anticipatory activation of the UPR in the mitogen-regulated signaling pathways and gene expression programs that underlie their effects on cell proliferation was explored. In addition to promoting survival of endothelial cells, activation of the UPR plays a major role in VEGF induced blood vessel formation. In a sophisticated matrigel plug angiogenesis assay, knockdown of PLCγ, eIF2α or ATF6α nearly abolished neovascularization [17]. Solid tumors are subject to hypoxia and hypoxia in turn activates a signaling pathway involving hypoxia inducible factor 1α (HIF-1α) and VEGF. UPR activation may impact induction of VEGF. Emerging data suggests the possible existence of a feed-forward regulatory loop in which VEGF activates the UPR, up-regulating XBP1. The XBP1 then works together with HIF-1α to induce VEGF [29]. In ERα negative breast cancer, elevated XBP1 is associated with a poor prognosis [30]. Tumor progression may be driven by this HIF-1α-XBP1 axis [31].
EGF binding to EGFR rapidly activates the pro-proliferation ERK and AKT pathways [32]. This is quickly followed by initiation of the immediate early gene expression program. Immediate early gene expression is important for EGF-stimulated cell proliferation [33–35]. Since the ERK inhibitor, UO126, blocks EGF induction of immediate early genes, ERK activation is essential for immediate early gene expression. Because immediate early genes Fos and EGR1 were induced in 20 minutes, the IP3R inhibitor, 2-APB, was unlikely to exhibit off-target and secondary effects. Blocking the increase in intracellular calcium with 2-APB completely blocked the rapid EGF-EGFR induction of Fos and EGR1 mRNAs [18]. Since 2-APB also blocked EGF-EGFR-mediated down regulation of gene expression, this is a specific action of EGF-EGFR mediated through the EnR IP3Rs and is not the result of global inhibition of transcription. Surprisingly, 2-APB had no effect on the rapid EGF-EGFR activation of the ERK and AKT signaling pathways [18]. Since 2-APB did not block EGF activation of the ERK pathway, the PLCγ-IP3R-mediated increase in intracellular calcium is a previously undescribed independent regulator of EGF-induced gene expression that works together with ERK activation to regulate immediate early gene expression.
ERα is an intensively studied ligand-regulated transcription factor. Blocking opening of the EnR IP3R calcium channels with 2-APB, or chelating intracellular calcium with BAPTA, strongly inhibited E2-ERα regulated gene expression [19]. A significant body of early work described a functional interaction between the calcium sensor calmodulin (CaM) and ERα in E2-ERα regulation of gene expression [36–39]. The PLCγ-IP3R pathway couples rapid extranuclear E2-ERα regulation of calcium levels to nuclear regulation of gene expression. Future studies will doubtless explore mechanisms by which these increased calcium levels selectively regulate the E2-ERα gene expression program.
The Anticipatory UPR Pathway in Therapy Resistant Breast Cancer
Activation of the UPR protects cells against stress. The important role of the UPR in protecting cancer cells against stress has been the subject of excellent recent reviews [3, 4, 40, 41], as has the more focused subject of the UPR in therapy-resistant ERα positive breast cancer [40]. Here, we focus primarily on anticipatory activation of the UPR in cancer with limited consideration of the widely studied role of the reactive UPR pathway.
Endocrine therapy using antiestrogens that compete with E2 for binding to ERα and aromatase inhibitors that block estrogen synthesis is a mainstay in treatment of the ~70% of breast cancers that are ERα positive. Although most tumors respond initially, ~50% will develop acquired resistance to endocrine therapy. Resistance mechanisms are diverse and have been recently reviewed [42]. Recently, ERα mutations have been the focus of intense interest as a likely cause of resistance to endocrine therapy. In ~1/3 of patients with metastatic therapy-resistant breast cancer, integrative sequencing has revealed ERα mutations thought to be constitutively active [43–47]. The ERα mutation, K303R, is reportedly common in tumors, and is undetectable by standard DNA sequencing [48, 49]. Also, up-regulation of the UPR and cross-talk of the UPR with autophagy can play an important role in allowing these tumors to proliferate in nutrient-deprived, hypoxic environments and resist therapy [40, 42, 50]. In the reactive pathway, resistance to therapy is likely driven by selection and outgrowth of cancer cells that survive in part because they activated the UPR in response to hypoxia, nutritional deprivation and therapy-induced stress.
Since breast cancers likely undergo hypoxic stress throughout tumor progression, it is difficult to dissect out the contributions to UPR activation of the estrogen-driven anticipatory pathway and of reactive activation of the UPR in response to hypoxia and nutritional deprivation. Two factors suggest the anticipatory pathway plays a significant role in UPR activation early in tumor outgrowth. There is a strong correlation between elevated expression of a UPR gene signature, consisting of UPR sensors and downstream targets of UPR activation, and expression levels of E2-ERα regulated genes [19]. Moreover, compared to normal mammary epithelial cells, the UPR is activated in premalignant mammary cells [19]. It is likely that the extent of activation of the anticipatory UPR pathway plays an important early role in selection of tumors that grow and progress; as tumors grow, hypoxia, nutritional deprivation and therapy-induced stress probably play an increasing role in UPR activation.
The UPR gene signature has been used to explore whether UPR activation at diagnosis predicts subsequent clinical outcome. Bioinformatic analysis of data from ~1,000 ERα positive breast cancer patients showed that elevated expression of the UPR gene signature at diagnosis was tightly correlated with subsequent resistance to tamoxifen therapy, reduced time to recurrence and reduced survival [19]. This supports a role for the anticipatory UPR in selection and progression of breast cancers. UPR activation can play an important role in protection of cancer cells from subsequent therapeutic insult. Weakly activating non-toxic concentrations of the UPR activator tunicamcyin elicit an adaptive stress response that increases EnR chaperones, and renders cells resistant to subsequent exposure to an otherwise lethal concentration of tunicamycin [51, 52]. Prior exposure of breast cancer cells to E2, or to a low concentration of tunicamycin, each elicited an ~10 fold increase in the concentration of tunicamycin required to induce apoptosis [19]. This suggested that E2-induced anticipatory activation of the UPR might both facilitate tumor proliferation and protect ERα positive breast tumors against subsequent stress due to hypoxia, nutritional deprivation and therapy.
Two UPR-regulated proteins, BiP chaperone and XBP1 are strongly implicated in antiestrogen resistance [40, 53–56]. As mentioned above, elevated BiP is associated with a poor prognosis [27]. BiP overexpression confers antiestrogen resistance and BiP knockdown resensitizes ERα positive breast cancer cells to antiestrogens [28]. Similarly, XBP1 knockdown restores sensitivity to antiestrogens, and XBP1 overexpression elicits antiestrogen resistance [28, 30, 57, 58]. Similar to E2, tamoxifen and ICI 182780/fulvestrant can activate pro-survival signaling through the UPR [58]. Notably, in women more than 5 years post menopause, whose tumors grow in a low estrogen environment, which can lead to elevated ERα levels [59, 60], or after long-term treatment with SERMs, removal of SERMs, and treatment with high dose estrogens slows tumor growth and induces tumor regression [61, 62]. Both UPR activation and increased cellular reactive oxygen species (ROS) appear to play a role in high dose estrogen-induced apoptosis [63].
Mild activation of the UPR that primarily results in chaperone induction and does not strongly activate the PERK arm of the UPR is protective. However, robust and sustained activation of the UPR that leads to strong activation of the PERK arm of the UPR is toxic. Supporting the idea that UPR activation is an attractive therapeutic strategy, the ability of proteasome inhibitors to induce apoptosis of cancer cells is largely based on their ability to increase levels of unfolded proteins and thereby strongly activate the UPR [64–66]. Thus, the UPR in cancer is a finely titrated system, where stress that exceeds a threshold tips the scales from protective to lethal (Figure 3). In the anticipatory UPR pathway, hormones, such as estrogen induce a moderate and transient increase in intracellular calcium that results in weak activation of the UPR while BHPI induces a much larger and more sustained increase in intracellular calcium leading to strong and toxic activation of the PERK arm of the UPR [19, 20].
Fig. 3. The UPR is a Molecular Rheostat Responding to Different Levels of UPR Activation.

The UPR is nearly off in normal non-malignant cells. In cells likely to initiate rapid proliferation, in antiestrogen-treated cancer cells, and in cells that will soon expand protein production and secretion, UPR activation is mild and protective. Strong and sustained UPR activation induced by BHPI, by proteasome inhibitors and by classical UPR activators, such as tunicamycin and thapsigargin, is toxic. Because the classical UPR activators are quite toxic, they have not found therapeutic application.
The Preclinical Anticancer Drug, BHPI, Induces Lethal Hyperactivation of the UPR
BHPI was the most effective biomodulator to emerge from an unbiased high throughput screen for small molecules that inhibit E2-ERα induction of a luciferase reporter gene [20, 67]. Surprisingly, BHPI elicited rapid, near quantitative inhibition of protein synthesis, a seemingly unlikely action for a small molecule inhibitor of E2-ERα regulated gene expression. It was therefore important to test whether BHPI works through ERα. BHPI gains the ability to inhibit protein synthesis when ERα is stably transfected into ERα negative MCF10A cells and loses the ability to inhibit protein synthesis when ERα is knocked down or degraded with ICI 182,780. Moreover, increasing levels of ERα elicit progressively increasing inhibition of protein synthesis, and BHPI only inhibits protein synthesis in ERα positive cell lines. Supporting BHPI acting through ERα, and not through the estrogen binding protein GPR30, BHPI has no effect on protein synthesis in ERα negative HepG2 cells, which contain functional GRP30 [68], and activating GPR30 with G1 did not inhibit protein synthesis. Consistent with binding to ERα, BHPI alters the intrinsic fluorescence emission pattern of ERα and alters the ERα digestion pattern in partial protease digestion studies [20]. Moreover, at early times, when inhibition of protein synthesis and UPR activation do not inhibit E2-ERα-induced gene expression, BHPI non-competitively inhibits E2-ERα induction and repression of gene expression and reduces binding of E2-ERα to gene regulatory regions [20]. Taken together, these data indicate that BHPI binds ERα and that BHPI binding induces a conformational change in ERα.
The PERK arm of the UPR was identified as the pathway responsible for rapid BHPI inhibition of protein synthesis (Figure 1). This led to the finding that BHPI works by hyperactivating the anticipatory UPR pathway. Compared with E2, BHPI more strongly activates PLCγ, producing much higher IP3 levels, calcium release from the EnR, and UPR activation. BHPI potently inhibits protein synthesis by inducing rapid and robust phosphorylation of PERK and eIF2α. Supporting the role of the PERK arm of the UPR in inhibiting protein synthesis, knockdown and inhibition of ERα, PLCγ, the IP3Rs, and PERK blocked rapid BHPI inhibition of protein synthesis [20].
How does BHPI convert the normal transient and protective activation of the UPR into a sustained toxic UPR activation? The substantial level of IP3 produced by strong BHPI activation of PLCγ binds to and opens the EnR IP3Rs, resulting in rapid efflux of calcium stored in the lumen of the EnR into the cytosol. To restore EnR calcium, the cell activates SERCA pumps, which catalyze ATP-dependent transfer of calcium from the cytosol into the lumen of the EnR. Since the IP3R calcium channels are still open, the calcium pumped into the EnR leaks back out creating a futile cycle that rapidly depletes cell ATP. Depleting intracellular ATP activates the important metabolic sensor, AMP kinase (AMPK). Supporting this model, thapsigargin, which inhibits the SERCA pumps, blocks the BHPI-mediated decline in ATP levels and AMPK activation. Together, AMPK activation and elevated intracellular calcium activate the eukaryotic elongation factor 2 kinase (CAMKIII/eEF2K). Activated eEF2K phosphorylates eEF2, inhibiting protein synthesis at a second site. Inhibiting protein synthesis at a second site prevents synthesis of BiP and other chaperones and p58IPK and GADD34 (growth arrest and DNA damage-inducible protein 34) that normally reverse PERK activation [20]. Working together, several actions of BHPI, including long-term inhibition of protein synthesis, sustained UPR activation, ATP depletion and AMPK activation likely contribute to BHPI’s ability to block proliferation and often kill ERα positive cancer cells. At nanomolar concentrations BHPI blocked growth, and killed diverse ERα positive breast and endometrial cancer cells. In a mouse xenograft model of breast cancer, BHPI stopped tumor growth and induced rapid and substantial regression of large tumors [20].
Aromatase inhibitors and competitor antiestrogens such as tamoxifen and ICI work by preventing E2-ERα action, and are therefore ineffective in ERα positive cancer cells that do not require estrogens and ERα for growth. Although ~50% of ovarian cancers are ERα positive [69], ovarian cancers do not depend on estrogens or ERα for growth and endocrine therapy is therefore ineffective [70]. In contrast, BHPI retains full effectiveness in ERα positive ovarian, breast and endometrial cancer cells that do not depend on estrogens or ERα for growth [17]. BHPI is effective in these cells because it is a noncompetitive biomodulator that uses ERα to block cell growth by hyperactivating the UPR. Moreover BHPI was highly effective in multiple breast and ovarian cell-based models for tamoxifen and ICI resistance. BHPI’s impressive effectiveness demonstrates the therapeutic potential of small molecules that target the anticipatory UPR pathway.
Concluding Remarks and Future Perspectives
It remains to be established whether all of the known activators of PLCγ (Figure 2), and perhaps activators of other PLC family members, activate the UPR. Calcium efflux through the EnR IP3R channels is regulated by interaction with multiple proteins. Consistent with IP3R modulator proteins influencing responses to E2 and BHPI, RACK1 is an E2-ERα induced gene that is overexpressed in aggressive breast tumors [71] and RACK1 increases calcium efflux through the EnR IP3R channels [72–74]. This suggests that overexpression in tumors of proteins that enhance the calcium efflux through the IP3R calcium channels may contribute to the selectivity of BHPI for cancer cells. How the diverse regulators of the IP3R channels influence the anticipatory UPR pathway is unexplored.
Whether non-hormonal activators of ERα, such as the metals calcium [75, 76] and cadmium [77], activate the UPR has not been investigated. E2 increases cytosol calcium in ~1 min. suggesting this may be the initial response of a cell to estrogen [19]. The protein complex in which E2-ERα activates a tyrosine kinase that phosphorylates and activates PLCγ is as yet unidentified. Progress in identifying the tyrosine kinase linked to E2-ERα activation of the UPR may shed light on how other PLCγ activators that are not tyrosine kinases activate the UPR.
Anticipatory activation of the UPR induces an extremely rapid increase in intracellular calcium and after several hours, increased levels of BiP and other chaperones. How these signals from activation of the UPR pathway couple to and control the EGF-EGFR, VEGF-VEGFR and E2-ERα gene expression and cell proliferation programs will be important areas for future study.
Studies to date have largely focused on effects of anticipatory activation of the UPR by VEGF, EGF and E2 on gene expression and cell proliferation in cancer. However, estrogens protect against stress in pancreatic islet cells in diabetes [78, 79] and in the nervous system [80–82] and important roles for the UPR have been described in these systems [78, 83, 84]. Whether some of the well-known protective effects of estrogen in the nervous system and in diabetes are due to anticipatory activation of the UPR remains to be explored.
Outstanding Questions.
How do ERα and other steroid receptors couple to tyrosine kinases to activate PLCγ and initiate the anticipatory UPR pathway?
Does activation of other isoforms of PLC also trigger activation of the anticipatory UPR pathwway?
What is the role of the anticipatory UPR pathway in the brain and nervous system in which estrogens are known to mitigate stress?
Increased intracellular calcium released by opening endoplasmic reticulum calcium channels is important for initiating hormone regulated gene expression programs. How is the calcium signal read at the level of transcription and how is selectivity maintained?
Using ERα to elicit cytotoxic hyperactivation of the UPR shows impressive therapeutic potential. Can new approaches be identified for selective hyperactivation of the PLCγ-IP3R anticipatory UPR pathway in ERα negative cancer cells?
Trends.
Diverse signaling molecules including peptide hormones, steroid hormones, immunoreceptors and GPCRs signal through phospholipase C γ (PLCγ) to activate the anticipatory UPR pathway.
Rapid activation of the anticipatory UPR pathway is important for subsequent hormone-regulated gene expression and cell proliferation.
The anticipatory and reactive UPR pathways work together to enable antiestrogen resistance in ERα positive breast cancer.
Targeting the anticipatory UPR, converting it from protective to toxic holds significant therapeutic promise.
Acknowledgments
This work was supported by Department of Defense Breast Cancer Research Program Grant W81XWH-14-1-0159 (to D.J.S) and by National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health Grant RO1 DK071909 (to D.J.S.), by a National Science Foundation Graduate Research Fellowship under Grant DGE-114245 (to M.L.) and by University of Illinois, Department of Biochemistry Westcott (to N.A.) and Carter (to X.Z.) Predoctoral Fellowships.
Glossary
- Aromatase inhibitor
a class of anticancer drugs that work by inhibiting the enzyme aromatase (CYP19A1), which converts precursors into estrogen.
- BHPI
a potent non-competitive small molecule ERα biomodulator that selectively blocks proliferation of ERα positive breast, ovarian and endometrial cancer cells by hyperactivating the unfolded protein response.
- Ecdysone
a steroidal pro-hormone produced by the prothoracic gland of insects that regulates molting and stimulates metamorphosis.
- Endoplasmic reticulum (EnR)
a eukaryotic membrane organelle. Protein folding and processing occur in the interior, or lumen, of the EnR. (Since estrogen receptor is abbreviated ER, to abbreviate endoplasmic reticulum, we use EnR, rather than the more common ER.)
- Epidermal growth factor (EGF)
a polypeptide growth factor with diverse biological effects including stimulating proliferation and differentiation of mesenchymal and epithelial cells.
- ER positive breast cancer
a type of breast cancer that grows in response to estrogen and contains estrogen receptor α. At diagnosis, about 70% of breast cancers are ERα positive.
- Estrogen receptor α (ERα)
a nuclear steroid hormone receptor that is activated by estrogen. Upon estrogen binding, ERα undergoes a conformational change and carries out both nuclear and extranuclear actions.
- Reactive and anticipatory activation of the unfolded protein response (UPR)
in reactive activation of the UPR, sensors respond to unfolded proteins, or other forms of stress, by activating the UPR. The anticipatory UPR is activated in the absence of endoplasmic reticulum stress, in anticipation of future increased need for protein folding capacity.
- Src
a membrane-associated non-receptor tyrosine kinase. It plays a role in diverse cellular pathways including cell survival, angiogenesis, cell proliferation and invasion.
- Tamoxifen
a prodrug used to treat ER positive breast cancer. It is a competitive inhibitor of estrogen binding to ERα and inhibits growth of breast cancer cells. In vivo, tamoxifen is converted to the more active, 4-hydroxytamoxifen.
- Unfolded protein response (UPR)
when unfolded or misfolded proteins accumulate in the lumen of the EnR, the UPR is activated, resulting in induction of molecular chaperones, decreased translation and increased degradation of misfolded proteins.
- Vascular endothelial growth factor (VEGF)
it was originally isolated from tumor cells and was initially referred to as ‘tumor angiogenesis factor’. VEGF stimulates development of new vascular networks that provide oxygen and nutrients to tumors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 5.Tsaytler P, et al. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 6.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 7.Gardner BM, et al. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Credle JJ, et al. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preissler S, et al. Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife. 2015;4:e08961. doi: 10.7554/eLife.08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hetz C, et al. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 12.Korach KS, DBJ Estrogen receptors and human disease. J Clin Inv. 2006;116:561–571. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson PA, et al. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salomon DS, et al. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 15.Kieran MW, et al. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:a006593. doi: 10.1101/cshperspect.a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin ER. Extranuclear steroid receptors are essential for steroid hormone actions. Annu Rev Med. 2015;66:271–280. doi: 10.1146/annurev-med-050913-021703. [DOI] [PubMed] [Google Scholar]
- 17.Karali E, et al. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559–572. doi: 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Yu L, et al. Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol Cell Endocrinol. 2016;422:31–41. doi: 10.1016/j.mce.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andruska N, et al. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene. 2015;34:3760–3769. doi: 10.1038/onc.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andruska ND, et al. Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A. 2015;112:4737–4742. doi: 10.1073/pnas.1403685112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng X, et al. Interplay between steroid hormone activation of the unfolded protein response and nuclear receptor action. Steroids. 2016 doi: 10.1016/j.steroids.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng X, et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol Med. 2015;7:788–801. doi: 10.15252/emmm.201404509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 24.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–375. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- 25.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong D, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 27.Lee E, et al. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–7853. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 28.Fu Y, et al. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–3740. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 29.Pereira ER, et al. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF) J Biol Chem. 2014;289:3352–3364. doi: 10.1074/jbc.M113.507194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies MP, et al. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int J Cancer. 2008;123:85–88. doi: 10.1002/ijc.23479. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda H, et al. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy LO, et al. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 34.Brown JR, et al. Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol. 1998;18:5609–5619. doi: 10.1128/mcb.18.9.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zwang Y, et al. Consistency test of the cell cycle: roles for p53 and EGR1. Cancer Res. 2012;72:1051–1054. doi: 10.1158/0008-5472.CAN-11-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, et al. Calmodulin enhances the stability of the estrogen receptor. J Biol Chem. 2001;276:17354–17360. doi: 10.1074/jbc.M010238200. [DOI] [PubMed] [Google Scholar]
- 37.Gallo D, et al. Calmodulin, a regulatory partner of the estrogen receptor alpha in breast cancer cells. Mol Cell Endocrinol. 2008;291:20–26. doi: 10.1016/j.mce.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Garcia Pedrero JM, et al. Calmodulin is a selective modulator of estrogen receptors. Mol Endocrinol. 2002;16:947–960. doi: 10.1210/mend.16.5.0830. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Structural basis for Ca2+-induced activation and dimerization of estrogen receptor alpha by calmodulin. J Biol Chem. 2012;287:9336–9344. doi: 10.1074/jbc.M111.334797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clarke R, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–1331. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajapaksa G, et al. Estrogen signaling and unfolded protein response in breast cancer. J Steroid Biochem Mol Biol. 2016 doi: 10.1016/j.jsbmb.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 42.Clarke R, et al. Endocrine resistance in breast cancer - An overview and update. Mol Cell Endocrinol. 2015 doi: 10.1016/j.mce.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toy W, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson DR, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merenbakh-Lamin K, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73:6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 46.Fanning SW, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. 2016:5. doi: 10.7554/eLife.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alluri PG, et al. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014;16:494. doi: 10.1186/s13058-014-0494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuqua SA, et al. Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat. 2014;144:11–19. doi: 10.1007/s10549-014-2847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuqua SA, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. 2000;60:4026–4029. [PubMed] [Google Scholar]
- 50.Tavassoly I, et al. Dynamic Modeling of the Interaction Between Autophagy and Apoptosis in Mammalian Cells. CPT Pharmacometrics Syst Pharmacol. 2015;4:263–272. doi: 10.1002/psp4.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rutkowski DT, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Cook KL, et al. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012;72:3337–3349. doi: 10.1158/0008-5472.CAN-12-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu R, et al. NF-kappaB signaling is required for XBP1 (unspliced and spliced)-mediated effects on antiestrogen responsiveness and cell fate decisions in breast cancer. Mol Cell Biol. 2015;35:379–390. doi: 10.1128/MCB.00847-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gomez BP, et al. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. Faseb J. 2007;21:4013–4027. doi: 10.1096/fj.06-7990com. [DOI] [PubMed] [Google Scholar]
- 56.Ming J, et al. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget. 2015;6:40692–40703. doi: 10.18632/oncotarget.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Booth L, et al. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. J Cell Physiol. 2015;230:1661–1676. doi: 10.1002/jcp.24919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cook KL, et al. Knockdown of estrogen receptor-alpha induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014;28:3891–3905. doi: 10.1096/fj.13-247353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katzenellenbogen BS, et al. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987;47:4355–4360. [PubMed] [Google Scholar]
- 60.Welshons WV, Jordan VC. Adaptation of estrogen-dependent MCF-7 cells to low estrogen (phenol red-free) culture. Eur J Cancer Clin Oncol. 1987;23:1935–1939. doi: 10.1016/0277-5379(87)90062-9. [DOI] [PubMed] [Google Scholar]
- 61.Obiorah IE, et al. Selective estrogen-induced apoptosis in breast cancer. Steroids. 2014;90:60–70. doi: 10.1016/j.steroids.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Fan P, et al. The molecular, cellular and clinical consequences of targeting the estrogen receptor following estrogen deprivation therapy. Mol Cell Endocrinol. 2015;418(Pt 3):245–263. doi: 10.1016/j.mce.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan P, et al. Integration of Downstream Signals of Insulin-like Growth Factor-1 Receptor by Endoplasmic Reticulum Stress for Estrogen-Induced Growth or Apoptosis in Breast Cancer Cells. Mol Cancer Res. 2015;13:1367–1376. doi: 10.1158/1541-7786.MCR-14-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ishii Y, et al. Bortezomib enhances the efficacy of fulvestrant by amplifying the aggregation of the estrogen receptor, which leads to a proapoptotic unfolded protein response. Clin Cancer Res. 2011;17:2292–2300. doi: 10.1158/1078-0432.CCR-10-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nawrocki ST, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 66.Lee AH, et al. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andruska N, et al. Evaluation of a luciferase-based reporter assay as a screen for inhibitors of estrogen-ERalpha-induced proliferation of breast cancer cells. J Biomol Screen. 2012;17:921–932. doi: 10.1177/1087057112442960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikeda Y, et al. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS One. 2012;7:e40465. doi: 10.1371/journal.pone.0040465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuo K, et al. Estrogen receptor expression and increased risk of lymphovascular space invasion in high-grade serous ovarian carcinoma. Gynecol Oncol. 2014;133:473–479. doi: 10.1016/j.ygyno.2014.03.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito K, et al. Inhibition of estrogen actions in human gynecological malignancies: new aspects of endocrine therapy for endometrial cancer and ovarian cancer. Mol Cell Endocrinol. 2010;340:161–167. doi: 10.1016/j.mce.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 71.Cao XX, et al. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int J Cancer. 2010;127:1172–1179. doi: 10.1002/ijc.25120. [DOI] [PubMed] [Google Scholar]
- 72.Patterson RL, et al. Phospholipase C-gamma: diverse roles in receptor-mediated calcium signaling. Trends Biochem Sci. 2005;30:688–697. doi: 10.1016/j.tibs.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Qiu Y, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells. Sci Signal. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu D, et al. IRE1-RACK1 axis orchestrates ER stress preconditioning-elicited cytoprotection from ischemia/reperfusion injury in liver. J Mol Cell Biol. 2015 doi: 10.1093/jmcb/mjv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Divekar SD, et al. The role of calcium in the activation of estrogen receptor-alpha. Cancer Res. 2011;71:1658–1668. doi: 10.1158/0008-5472.CAN-10-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leclercq G. Calcium-induced activation of estrogen receptor alpha--New insight. Steroids. 2012;77:924–927. doi: 10.1016/j.steroids.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 77.Stoica A, et al. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- 78.Kooptiwut S, et al. Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic beta-cell death. J Steroid Biochem Mol Biol. 2014;139:25–32. doi: 10.1016/j.jsbmb.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 79.Le May C, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103:9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ishihara Y, et al. Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J Steroid Biochem Mol Biol. 2015;145:85–93. doi: 10.1016/j.jsbmb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 81.Gollapudi L, Oblinger MM. Estrogen and NGF synergistically protect terminally differentiated, ERalpha-transfected PC12 cells from apoptosis. J Neurosci Res. 1999;56:471–481. doi: 10.1002/(SICI)1097-4547(19990601)56:5<471::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 82.Engler-Chiurazzi EB, et al. From the 90s to now: A brief historical perspective on more than two decades of estrogen neuroprotection. Brain Res. 2016;1633:96–100. doi: 10.1016/j.brainres.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghosh R, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu CC, et al. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009;28:1624–1636. doi: 10.1038/emboj.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Anken E, et al. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity. 2003;18:243–253. doi: 10.1016/s1074-7613(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 87.Kurosaki T, et al. Regulation of the phospholipase C-gamma2 pathway in B cells. Immunol Rev. 2000;176:19–29. doi: 10.1034/j.1600-065x.2000.00605.x. [DOI] [PubMed] [Google Scholar]
- 88.Liu W, et al. Phospholipase Cgamma1 connects the cell membrane pathway to the nuclear receptor pathway in insect steroid hormone signaling. J Biol Chem. 2014;289:13026–13041. doi: 10.1074/jbc.M113.547018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jing YP, et al. The steroid hormone 20-hydroxyecdysone via nongenomic pathway activates Ca2+/calmodulin-dependent protein kinase II to regulate gene expression. J Biol Chem. 2015;290:8469–8481. doi: 10.1074/jbc.M114.622696. [DOI] [PMC free article] [PubMed] [Google Scholar]