

Abstract
Introduction
Heat shock proteins (HSP) protect cells under adverse conditions such as infection, inflammation, and disease. The differential expression of HSPs in human periapical granulomas suggests a potential role for these proteins in periapical lesion development, which may contribute to different clinical outcomes. Therefore, we hypothesize that polymorphisms in HSP genes leading to perturbed gene expression and protein function may contribute to an individual’s susceptibility to periapical lesion development.
Methods
Subjects with deep carious lesions, with or without periapical lesions (≥ 3 mm) were recruited at the University of Texas School of Dentistry at Houston and at the University of Pittsburgh. Genomic DNA samples of 400 patients were sorted into 2 groups: 183 cases with deep carious lesions and periapical lesions (cases), and 217 cases with deep carious lesions but without periapical lesions (controls). Eight single nucleotide polymorphisms in HSPA4, HSPA6, HSPA1L, HSPA4L and HSPA9 genes were selected for genotyping. Genotypes were generated by endpoint analysis using Taqman chemistry in a real-time polymerase chain reaction assay. Allele and genotype frequencies were compared among cases and controls using chi-square and Fisher Exact tests as implemented in PLINK v.1.07. In silico analysis of SNP function was performed using Polymorphism Phenotyping V2 and MirSNP softwares.
Results
Overall, SNPs in HSPA1L and HSPA6 showed significant allelic association with cases of deep caries and periapical lesions (P<0.05). We also observed altered transmission of HSPA1L SNP haplotypes (P=0.03). In silico analysis of HSPA1L rs2075800 function showed that this SNP results in a glutamine to lysine substitution at position 602 of the protein and might affect the stability and function of the final protein.
Conclusions
Variations in HSPA1L and HSPA6 may be associated with periapical lesion formation in individuals with untreated deep carious lesions. Future studies could help predict host susceptibility to developing apical periodontitis.
Keywords: Heat shock proteins, apical periodontitis, polymorphism
INTRODUCTION
Apical periodontitis is an inflammatory disorder of periapical tissues caused by continuous microbial infections within the root canal system (1). The inflammatory response offers a unique study of many facets of pathogenesis, including bacterial ecology and pathogenicity, innate and acquired immune responses to infection, the regulation of such responses and their effects on host tissues, particularly the periapical region (1). It is viewed as a dynamic encounter between local microbial factors and host defenses at the interface between infected radicular pulp and periodontal ligament that results in local inflammation, resorption of hard tissues, destruction of other periapical tissues, and eventual formation of various histopathological categories of apical periodontitis (1). Equilibrium of cytokines, endotoxins, and cell-to-cell contacts may influence the defense and inflammatory responses and the balance between bone resorption and regeneration, resulting in lesion expansion or healing of apical periodontitis (2–4). In addition to local factors, genetic predisposition has been suggested as a differential etiologic factor for apical periodontitis development (5–11). Moreover, genetic susceptibility may influence host response to endodontic infection (9).
The most primitive mechanism of cellular protection involves the expression of a polypeptide family called heat shock proteins (HSPs) (12). HSPs have essential roles in the synthesis, transport, and folding of proteins and are often referred to as molecular chaperones. However, HSPs are characteristically induced by stress signals such as elevated temperature, reduced oxygen supply, infectious agents, and inflammatory mediators (13, 14). Stressors that cause protein unfolding, misfolding or aggregation trigger a protective response that leads to the induction of gene transcription for proteins with the capacity to stabilize and re-fold proteins, thereby re-establishing the balance between protein synthesis, assembly and degradation (15). Therefore, HSPs also exert a protective role against harmful environment (12).
HSPs are subdivided based on their molecular weight (HSPH [HSP110], HSPC [HSP90], HSPA [HSP70], DNAJ [HSP40], HSPD [HSP60], and HSPB [HSP27], and are especially effective in triggering the innate immune response by activating macrophages and macrophage-like cells (16–18). HSPs can increase the cellular response to lipopolysaccharide (LPS) to stimulate the production of prototypic inflammatory cytokines such as tumor necrosis factor alpha (TNF-a) (19).
HSP70 proteins are found in all species and are located in numerous cellular components, (i.e., mitochondria, the endoplasmic reticulum, lysosomes, cytosol and nucleus) and are subdivided into HSPA1A, HSPA1B, HSPA1L, HSPA2, HSPA4, HSPA5, HSPA6, HSPA7, HSPA8, HSPA9, HSPA12A, HSPA12B, HSPA13 and HSPA14 (20). These proteins are involved in prevention of aggregation of unfolded polypeptides and the disassembly of multimeric protein complexes using a mechanism of protein trafficking between cellular compartments, protein folding, and regulation of heat shock response (16, 17). Interestingly, HSPs, such as HSP70, can play dual roles in the modulation of host inflammatory immune reaction. It has been shown that the induction of proinflammatory cytokines by HSP70 may contribute to the pathogenesis of autoimmune disease and chronic inflammation (19, 20). Conversely, HSP70 has shown to down regulate toll like receptors (19), thus inducing LPS tolerance and preventing augmentation of pro-inflammatory cytokines levels following LPS stimulation (20).
We have previously shown the differential expression patterns of HSPs in periapical granulomas, with a marked increase in the expression of four HSP genes, DNAJC3, HSPA4, HSPA6 and HSPB1, when compared to control tissues (21). We observed that the HSP expression in response to LPS was concentration-dependent, and that an increase in LPS concentration was positively correlated with HSP expression. These observations support the hypothesis that HSPs have a role in periapical lesion development and their expression patterns may be related to different clinical outcomes (21). Therefore, we hypothesize that altered HSP expression due to HSP gene polymorphisms may contribute to an individual’s susceptibility to periapical lesion development.
MATERIAL AND METHODS
Sample Population
Two distinct population datasets were used in this study. The first dataset was obtained through the University of Pittsburgh Dental Registry and DNA Repository (DRDR), which gathers clinical information and DNA samples from saliva from patients seeking treatment at the School of Dental Medicine and that agree to participate in the registry. Data is extracted from electronic patient records and linked to the DNA samples for genetic/clinical research and provided to investigators in de-identified format upon approved protocols. The second dataset was ascertained at the Endodontic clinic of the University of Texas Health Science Center School of Dentistry at Houston. In brief, all patients receiving treatment in the Endodontic clinic were invited to participate, and dental/medical history as well as a saliva sample was collected upon written informed consent. The University of Pittsburgh Institutional Review Board and the University of Texas Health Science Center at Houston Committee for the Protection of Human Subjects approved this study.
For this study, samples were collected over a period of five years. Individuals from both datasets were selected for inclusion based on their radiographic records showing deep carious lesions, involving at least 2/3 of the dentin depth, and periapical lesions ≥3mm in diameter (cases), and individuals showing deep carious lesions but no periapical lesions (controls). Endodontic diagnostic tests to assess pulpal and periapical status were performed in all individuals. In brief, pulpal testing was performed using Endo-Ice® and the EPT (electric pulp tester) using established protocols. Periapical status was assessed using routine palpation and percussion tests. Individuals with diagnosis of pulp necrosis and apical periodontitis were included in the ‘case’ group; individuals with diagnosis of vital pulps and normal apical tissues (no apical periodontitis) were included as controls. Patients with any systemic conditions such as diabetes or other hormonal alterations that are related to exacerbated or uncontrolled inflammatory responses, patients with medical conditions requiring the use of systemic modifiers of bone metabolism or other assisted drug therapy (i.e., systemic antibiotics, anti-inflammatory, hormonal therapy) during the last 6 months before the study were excluded. At last, our sample population consisted of 400 Caucasian individuals with deep carious lesions: 183 case individuals with deep carious lesions and periapical lesions, and 217 control individuals with deep carious lesions and no periapical lesions.
Selection of Candidate Genes and Single Nucleotide Polymorphisms
We selected eight single nucleotide polymorphisms spanning the HSPA1L, HSPA4, HSPA4L, HSPA6 and HSPA9 genes. Some of the SNPs were selected based on published reports and/or their locations within the genes. Additional SNPs were selected based on their likelihood to have functional consequences (i.e., located in the promoters, exons, or near exon/intron boundaries), or considered tag-SNPs as surrogates for the linkage disequilibrium blocks surrounding the candidate gene. We used information available at the NCBI dbSNP (http://www.ncbi.nlm.gov/SNP/) and HapMap Project (http://www.hapmap.org) databases to select polymorphisms. Details of studied genes and polymorphisms are presented in Table 1.
Table 1.
Details of SNPs investigated
| Gene | SNP Id.* | Chr.: Base Position* | SNP function | Alleles# |
|---|---|---|---|---|
| HSPA1L | rs2075800 | 6:31777946 | Missense | C/T |
| rs2227956 | 6:31778272 | C/T | ||
| rs2227955 | 6:31778077 | G/T | ||
| HSPA4 | rs14355 | 5:132440285 | 3′ UTR | C/G |
| HSPA4L | rs1380154 | 4:128723042 | Missense | C/T |
| HSPA6 | rs1042881 | 1:161496530 | 3′ UTR | C/G |
| HSPA9 | rs1042665 | 5:137902339 | Missense | C/T |
| rs10117 | 5:137892170 | A/G |
Chr. = Chromosome
according to NCBI GRCh37.p10 assembly
ancestral allele in bold
Genotyping
Genomic DNA was extracted from saliva using established protocols. Genotypes were generated using Taqman chemistry (22). Reactions were carried out in 5-μL volumes in a ViiA7 Sequence Detection System (Applied Biosystems, Foster City, CA). Assays and reagents were supplied by Applied Biosystems. The results were analyzed using EDS v.1.2.3 software (Applied Biosystems). In order to ensure quality control of genotyping reactions, a non-template control (water instead of DNA) as negative control and a DNA sample of known genotype as positive control.
Association analyses
Allele and genotype frequencies for each polymorphism were compared among cases and controls using PLINK software version 1.07 (23). We used Bonferroni correction as implemented in PLINK to adjust for multiple testing. Haplotype analyses were performed for SNPs in HSPA1L and HSPA9 using PLINK. P-values ≤ 0.05 were considered statistically significant.
In silico analysis of SNP function
We performed in silico analysis of SNP function to predict the effects of the associated HSPA1L and HSPA6 SNPs. We used Polymorphism Phenotyping V2 (24) and MirSNP (25) prediction softwares.
RESULTS
There was no evidence of deviation from Hardy-Weinberg equilibrium for any of the investigated polymorphisms (data not shown).
We assessed genotypic and allelic associations between the investigated HSP SNPs and individuals affected with deep caries and periapical disease. A SNP in HSPA6 (rs1042881) showed allelic association in our combined Pittsburgh and Houston datasets (P=0.04). HSPA1L rs2075800 and rs2227956 showed significant association with the presence of deep caries and periapical disease in both Pittsburgh (P=0.03) and Houston (P=0.02) data sets.
Haplotype analyses showed altered transmission of HSPA1L SNP alleles in cases with deep caries and periapical disease. More specifically, the combination of C-T-T alleles for SNPs rs2075800-rs2227956-rs2227955 was overrepresented in cases (P=0.03) (Table 2). No additional individual or haplotype associations were found with the remaining SNPs.
Table 2.
Haplotype Results
| SNPs | Chr. | Gene | Haplotype | Frequency cases | Frequency controls | P-value | Dataset |
|---|---|---|---|---|---|---|---|
| rs2075800|rs2227956|rs2227955 | 6 | HSPA1L | CTT | 0.05 | 0.13 | 0.03 | Houston |
| TCT | 0.27 | 0.36 | 0.04 | Pittsburgh | |||
| rs14355|rs1042665|rs10117 | 5 | HSPA9 | GTA | 0.09 | 0.05 | 0.05 | Combined |
In silico analysis of HSPA1L rs2075800 function showed that this SNP results in a glutamine to lysine substitution at position 602 of the protein (E602K) that may affect the stability and function of the final protein, hence considered potentially damaging (Figure 2). Interestingly, while the additional HSPA1L SNPs (rs2227955 and rs2227956) results in glutamine to alanine (E558A) and threonine to methionine (T493M) changes, these SNPs were predicted as benign with no damaging effects. The 3′ UTR SNP in HSPA6 rs1042881 was predicted to have different binding affinity to the microRNA hsa-miR-4290 with an effect on mRNA expression levels of its target gene (Figure 1).
Figure 1.
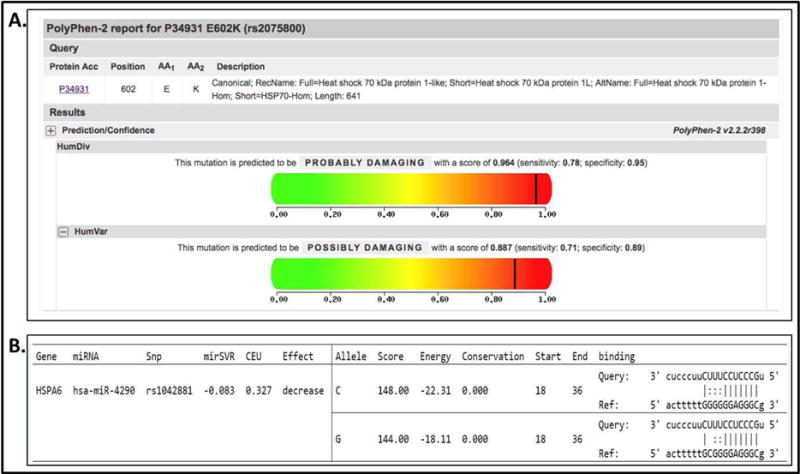
Results of HSPA1L and HSPA6 in silico analysis of SNP function. A, HSPA1L rs2075800 results in a glutamine to lysine substitution at position 602 of the protein (E602K) that is considered potentially damaging; B, HSPA6 rs1042881 located in the 3′ UTR is predicted to have different binding affinity to the microRNA hsa-miR-4290. Albeit not significant, the C allele appears to have increased affinity to hsa-miR-4290.
DISCUSSION
In this study, we selected eight single nucleotide polymorphisms spanning HSP70 family genes HSPA1L, HSPA4, HSPA4L, HSPA6, and HSPA9 to test for association with the presence of deep carious lesions and periapical lesions. Our hypothesis was that the presence of HSP gene polymorphisms could lead to altered HSP expression and contribute to an individual’s increased susceptibility to apical periodontitis.
Microbial infections of the dental pulp trigger inflammatory responses and ultimately cause bone destruction in the periradicular tissues (26). A network of cytokines, endotoxins, and cell-to-cell contacts may influence the defense and inflammatory responses and the balance between bone resorption and regeneration, resulting in a periapical lesion (1–4). Heat shock proteins or stress proteins are produced by cells in response to a variety of insults (12). Previously, our group has shown the differential expression patterns of HSPs in periapical granulomas, and observed a marked increase in the expression of four HSP genes, DNAJC3, HSPA4, HSPA6 and HSPB1 when compared to control tissues. We also observed that HSP expression in response to LPS was concentration-dependent, and that an increase in LPS concentration was positively correlated with increased HSP expression (21). These observations support a role for HSPs as potential candidate genes for apical periodontitis.
Our results showed that SNPs in HSPA6 (rs1042881) and HSPA1L (rs2075800 and rs2227956) were associated with the presence of deep carious lesions and periapical lesions in cases, and could play a role in individual predisposition to periapical disease in these individuals. There was no evidence of deviation from Hardy-Weinberg equilibrium for any of the investigated polymorphisms between the groups suggesting that our sample dataset was representative of a general population. Hardy-Weinberg Equilibrium (HWE) is used to describe the genotype distribution of a population when it is large, self-contained, and randomly mating (27). Testing for HWE in the control group is commonly used to detect genotyping errors in genetic association studies. We also observed the combination of specific alleles (C-T-T) for HSPA1L SNPs rs2075800-rs2227956-rs2227955 more frequently in cases, suggesting that this gene, and not a specific polymorphic variant, may have an important role in the periapical disease process.
The associated HSPA1L SNPs tested reflect missense mutations, resulting in amino acid substitutions at their respective locations, with potential downstream effects on the structure of their final proteins. More specifically, HSPA1L SNP (rs2075800, E602K) results in a glutamine to lysine substitution at position 602 of the protein, that is predicted as probably damaging and has been previously associated with other inflammatory diseases in humans (28–31). While the presence of the T allele has been associated with rheumatoid arthritis (28), it has been shown to play a protective role in sudden sensorineural hearing loss (29). Moreover, the C allele has been associated with increased susceptibility to sarcoidosis and Lofgren’s syndrome (30) and is a risk factor for Sarcoidosis-related uveitis (31). Interestingly, HSPA1L rs2227955 and rs2227956, also missense substitutions, resulting in threonine-to-methionine and in glycine-to-alanine changes, respectively, do not appear to have a direct damaging effect on the HSPA1L protein. The associated HSPA6 SNP rs1042881 is located at the 3′ untranslated region (UTR) of the gene, often characterized by the presence of microRNA (miRNA) binding sites that contribute to regulation of gene expression. In silico analysis of SNP function revealed that the miRNA hsa-miR-4290 is predicted to bind to the 3′UTR of HSPA6, and it is possible that this binding may contribute to downregulation of HSPA6.
Both HSPA1L and HSPA6 have important roles in other organ systems. HSPA1L is clinically significant in the context of trauma since it has been shown to contribute to upregulation of cytokine expression in patients with otherwise comparable trauma load (32). The HSPA1L C2437T polymorphism was significantly associated with multiple organ failure in trauma patients, suggesting that the CT genotype may be related to an induction of proinflammatory cytokine expression by HSPA1L (32). In contrast, it has been suggested that HSPA1L does not influence the risk to infectious morbidities nor specific organ failure or increased mortality risk in surgical care unit patients (33). Also, the presence of this protein has been suggested as an early predictor for chronic graft versus host disease at the mRNA expression level (34). HSPA6 is expressed in basal and stress-inducible human keratinocytes and is essential to increasing survival of cells exposed to increased temperatures and chemicals; it was also shown to provide critical benefits to buffering neuronal migration from cellular stress in the human brain (35). Other studies have suggested that HSPA6 is a secondary regulator of stress at low expression levels possibly depending on the cell type and growth conditions under non-stressed conditions (36). Members of the HSP70 gene family play a crucial role in the maintenance of infection and subsequent tissue destruction in autoimmune diseases and cancer (37), as well as in apical periodontitis (21). Further, HSP70 proteins have known roles in the induction of proinflammatory cytokines and thereby may contribute to the pathogenesis of autoimmune disease and chronic inflammation (38). Corroborating with these findings, our association findings with HSPA1L and HSPA6 may suggest a protective role for these genes under stressful conditions such as in a periapical lesion scenario.
Genetic predisposition has been suggested as an etiologic factor for apical periodontitis development and additional gene polymorphisms have been recently reported in association with apical periodontitis (5–11). Previous studies assessing the role of polymorphisms of matrix metalloproteinase (MMP) and interleukin genes, for which an increased expression has been shown to correlate with the development of pulpitis and apical periodontitis cases, and found strong associations with MMP2, MMP3 and IL1B genes in cases with apical periodontitis (9, 10). Given the complexity of the inflammatory response observed in an apical periodontitis scenario, it is expected that perturbations of expression in critically responsive genes, such as HSPs (and also MMPs or interleukins), could contribute to the underlying etiology of the condition. While additional studies in other populations will further support a role for HSP genetic polymorphisms in endodontic pathologies, the present study provide insights into the development of potential markers to predict host susceptibility for the development of apical periodontitis.
Acknowledgments
The authors deny any conflicts of interest related to this study. We are grateful for the participation of the many individuals in this study. This research was supported in part by a Research Grant from the American Association of Endodontists Foundation (to AL and RMS), by NIH RO1-DE18914 (to ARV), FAPESP (to GPG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stashenko P, Teles R, D’Souza R. Periapical inflammatory responses and their modulation. Crit Rev Oral Biol Med. 1998;9:498–521. doi: 10.1177/10454411980090040701. [DOI] [PubMed] [Google Scholar]
- 2.Nair PN. Pathogenesis of apical periodontitis and the causes of endodontic failures. Crit Rev Oral Biol Med. 2004;15:348–381. doi: 10.1177/154411130401500604. [DOI] [PubMed] [Google Scholar]
- 3.Marton IJ, Kiss C. Overlapping protective and destructive regulatory pathways in apical periodontitis. J Endod. 2014;40:155–163. doi: 10.1016/j.joen.2013.10.036. [DOI] [PubMed] [Google Scholar]
- 4.Graves DT, Oates T, Garlet GP. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol. 2011;3:5304. doi: 10.3402/jom.v3i0.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amaya MP, Criado L, Blanco B, et al. Polymorphisms of pro-inflammatory cytokine genes and the risk for acute suppurative or chronic nonsuppurative apical periodontitis in a Colombian population. Int Endod J. 2013;46:71–78. doi: 10.1111/j.1365-2591.2012.02097.x. [DOI] [PubMed] [Google Scholar]
- 6.Rocas IN, Siqueira JF, Jr, Del Aguila CA, et al. Polymorphism of the CD14 and TLR4 genes and post-treatment apical periodontitis. J Endod. 2014;40(2):168–172. doi: 10.1016/j.joen.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Siqueira JF, Jr, Rocas IN, Provenzano JC, et al. Relationship between Fcgamma receptor and interleukin-1 gene polymorphisms and post-treatment apical periodontitis. J Endod. 2009;35:1186–1192. doi: 10.1016/j.joen.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Morsani JM, Aminoshariae A, Han YW, et al. Genetic predisposition to persistent apical periodontitis. J Endod. 2011;37:455–459. doi: 10.1016/j.joen.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menezes-Silva R, Khaliq S, Deeley K, et al. Genetic susceptibility to periapical disease: conditional contribution of MMP2 and MMP3 genes to the development of periapical lesions and healing response. J Endod. 2012;38:604–607. doi: 10.1016/j.joen.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dill A, Letra A, Chaves de Souza L, et al. Analysis of multiple cytokine polymorphisms in individuals with untreated deep carious lesions reveals IL1B (rs1143643) as a susceptibility factor for periapical lesion development. J Endod. 2015;41:197–200. doi: 10.1016/j.joen.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aminoshariae A, Kulild JC. Association of Functional Gene Polymorphism with Apical Periodontitis. J Endod. 2015;41:999–1007. doi: 10.1016/j.joen.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 12.De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock. 1999;11:1–12. doi: 10.1097/00024382-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 14.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 15.Pockley AG. Heat shock proteins in health and disease: therapeutic targets or therapeutic agents? Expert Rev Mol Med. 2001;3:1–21. doi: 10.1017/S1462399401003556. [DOI] [PubMed] [Google Scholar]
- 16.Gething M-J. Guidebook to molecular chaperones and protein-folding catalysts. Oxford University Press; 1997. [Google Scholar]
- 17.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 18.Kampinga HH, Hageman J, Vos MJ, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferat-Osorio E, Sanchez-Anaya A, Gutierrez-Mendoza M, et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J Inflamm (Lond) 2014;11:19. doi: 10.1186/1476-9255-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aneja R, Odoms K, Dunsmore K, et al. Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J Immunol. 2006;177:7184–7192. doi: 10.4049/jimmunol.177.10.7184. [DOI] [PubMed] [Google Scholar]
- 21.Goodman SC, Letra A, Dorn S, et al. Expression of heat shock proteins in periapical granulomas. J Endod. 2014;40:830–836. doi: 10.1016/j.joen.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Ranade K, Chang M-S, Ting C-T, et al. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:1262–1268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 Jan; doi: 10.1002/0471142905.hg0720s76. Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C, Zhang F, Li T, et al. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genomics. 2012;13:661. doi: 10.1186/1471-2164-13-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K. Microbiological, pathological, inflammatory, immunological and molecular biological aspects of periradicular disease. Int Endod J. 1998;31:311–325. doi: 10.1046/j.1365-2591.1998.00171.x. [DOI] [PubMed] [Google Scholar]
- 27.Yu C, Zhang S, Zhou C, et al. A likelihood ratio test of population Hardy-Weinberg equilibrium for case-control studies. Gen Epidemiol. 2009;33:275–280. doi: 10.1002/gepi.20381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenkins SC, March RE, Campbell RD, et al. A novel variant of the MHC-linked hsp70, hsp70-hom, is associated with rheumatoid arthritis. Tissue antigens. 2000;56:38–44. doi: 10.1034/j.1399-0039.2000.560105.x. [DOI] [PubMed] [Google Scholar]
- 29.Chien CY, Chang NC, Tai SY, et al. Heat shock protein 70 gene polymorphisms in sudden sensorineural hearing loss. Audiol Neurootol. 2012;17:381–5. doi: 10.1159/000341815. [DOI] [PubMed] [Google Scholar]
- 30.Bogunia-Kubik K, Koscinska K, Suchnicki K, et al. HSP70-hom gene single nucleotide (+2763 G/A and +2437 C/T) polymorphisms in sarcoidosis. Int J Immunogenet. 2006;33:135–140. doi: 10.1111/j.1744-313X.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- 31.Spagnolo P, Sato H, Marshall SE, et al. Association between heat shock protein 70/Hom genetic polymorphisms and uveitis in patients with sarcoidosis. Invest Ophthalmol Vis Sci. 2007;48:3019–3025. doi: 10.1167/iovs.06-1485. [DOI] [PubMed] [Google Scholar]
- 32.Schröder O, Schulte K-M, Ostermann P, et al. Heat shock protein 70 genotypes HSPA1B and HSPA1L influence cytokine concentrations and interfere with outcome after major injury. Crit Care Med. 2003;31:73–79. doi: 10.1097/00003246-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Bowers DJ, Calvano JE, Alvarez SM, et al. Polymorphisms of heat shock protein-70 (HSPA1B and HSPA1L loci) do not influence infection or outcome risk in critically ill surgical patients. Shock. 2006;25:117–22. doi: 10.1097/01.shk.0000190826.36406.27. [DOI] [PubMed] [Google Scholar]
- 34.Atarod S, Turner B, Pearce KF, et al. Elevated level of HSPA1L mRNA correlates with graft-versus-host disease. Transpl Immunol. 2015;32:188–194. doi: 10.1016/j.trim.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Ramirez VP, Stamatis M, Shmukler A, et al. Basal and stress-inducible expression of HSPA6 in human keratinocytes is regulated by negative and positive promoter regions. Cell Stress Chaperones. 2015;20:95–107. doi: 10.1007/s12192-014-0529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noonan EJ, Place RF, Giardina C, et al. Hsp70B regulation and function. Cell Stress Chaperones. 2007;12:393. doi: 10.1379/CSC-278e.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calderwood SK, Stevenson MA, Murshid A. Heat shock proteins, autoimmunity, and cancer treatment. Autoimmune Dis. 2012;2012:486069. doi: 10.1155/2012/486069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70 role of Toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]