

Abstract
Chimeric antigen receptor (CAR) T cell therapy has shown promise in CD19 expressing hematologic malignancies, but how to translate this success to solid malignancies remains elusive. Effective translation of CAR T cells to solid tumors will require an understanding of potential therapeutic barriers, including factors that regulate CAR T cells expansion, persistence, trafficking, and fate within tumors. Herein, we describe the current state of CAR T cells in solid tumors; define key barriers to CAR T cell efficacy and mechanisms underlying these barriers, outline potential avenues for overcoming these therapeutic obstacles, and discuss the future of translating CAR T cells for the treatment of patients with solid malignancies.
Keywords: CAR T cells, Chimeric antigen receptor, Solid malignancies, Tumor microenvironment, Cellular immunity
1. Introduction
Adoptive cell therapy using gene-modified T cells has emerged as an exciting therapeutic approach for the treatment of cancer. Engineering T cells to express a chimeric antigen receptor (CAR) is the most common gene-modifying strategy that is being investigated. This personalized medicine approach has produced promising results for many patients across a range of hematological malignancies (Porter et al., 2011; Kochenderfer et al., 2012; Brentjens et al., 2013; Gill & June, 2015). Based on this success, there is now mounting interest around how to use CAR T cells for the treatment of solid malignancies. However, the translation of CAR T cells to solid tumors has been fraught with obstacles including issues of safety, T cell persistence and expansion, T cell trafficking into tumors, and immune resistance mechanisms established within tumors that may define the ultimate fate of CAR T cells. Many of these obstacles are unique to solid tumors given their distinct microenvironments and anatomical locations. In this review, we detail the current state of CAR T cells in solid malignancies, discuss barriers that may regulate CAR T cell efficacy in vivo, and propose next steps toward advancing CAR T cells for the treatment of solid tumors.
2. Strategies to genetically modify T cells for cancer therapy
Two main approaches, namely, T cell receptors (TCRs) and chimeric antigen receptors (CARs), have been studied to genetically modify T cells to recognize cancer. Both of these approaches involve the introduction of genetic material encoding a receptor (i.e. TCR or CAR) into T cells. Each strategy is distinct and thus, associated with unique advantages and disadvantages (Table 1).
Table 1.
Defining characteristics of CARs and TCRs
| Chimeric antigen receptors (CARs) | T cell receptors (TCRs) | |
|---|---|---|
| Signal amplification | Determined by synthetic biology | Determined by evolution |
| Affinity | High and can be engineered | Low but can be enhanced |
| Target | Cell surface proteome | Intracellular proteome |
| MHC dependency | Independent | Required |
| Safety | Toxicities possible due to antigen expression on normal tissues | Toxicities may be difficult to predict |
| Mechanisms of resistance | Antigen loss | Antigen loss, MHC loss/downregulation, defects in antigen presentation |
The distinct biology of TCR- and CAR-modified T cells is based on their structure. For example, a TCR recognizes tumor-specific peptides presented by a major histocompatibility (MHC) molecule. In contrast, a CAR is a modular protein composed of a defined extracellular component that recognizes a cell surface protein and an intracellular component made up of specific signaling domains from proteins (e.g. CD3ζ, CD28, and 4-1BB) involved in T cell activation (Fig. 1). The extracellular component commonly incorporates light and heavy chain regions of a single chain variable fragment (scFv) that are combined in series using a polypeptide linker. This extracellular component acts as the target-binding domain of a CAR and is connected to the intracellular component by a hinge region. Optimization of the hinge region is important as it has been found to significantly influence the quality of a CAR construct (Hudecek et al., 2013). For example, the length of the hinge region may impact the elasticity of the extracellular domain and, thus, its capacity to bind the target ligand (Guest et al., 2005; James et al., 2008). Most commonly, the hinge region has been derived from CD8 or IgG4 molecules and serves to connect the extracellular domain with an intracellular component that contains signaling domains designed to drive cellular activation. “First generation” CARs utilize only a single intracellular signaling domain, typically the CD3 ζ chain. Other groups have also studied the use of signaling domains derived from the Fcγ receptor for triggering T cell activation (Kershaw et al., 2006). Incorporating additional co-stimulatory molecules into the intracellular component can enhance T cell proliferation and persistence induced by CAR engagement of its target protein (Gill & June, 2015). CAR constructs that include one or two co-stimulatory domains have been termed “second generation” and “third generation” CARs, respectively. Overall, CAR-modified T cells are designed to interrogate a cell’s surface proteome which contrasts TCR-modified T cells which sample the intracellular proteome of a cell.
Fig. 1. Schematic comparing the structure of T cell receptors (TCRs) and chimeric antigen receptors (CARs).

The TCR complex is composed of TCR-α and -β chains that are assembled together with invariant CD3 molecules. In contrast, a CAR is a modular protein that comprises an extracellular domain (ligand binding region), a hinge region, and an intracellular domain (signaling components). TCR: T cell receptor; CD3: cluster of differentiation 3; scFv: single-chain variable fragment.
The unique mechanisms by which TCR- and CAR-modified T cells recognize a target cell are important in defining potential mechanisms of resistance and in understanding issues of toxicity. Whereas loss of antigen expression, downregulation of MHC molecules, or disruption of antigen processing and presentation are all potential mechanisms that cancer cells can exploit to evade recognition by TCR-modified T cells, defects in antigen processing and presentation are not sufficient for evading recognition by CAR T cells which rely on surface expression of the target protein. However, due to this reliance on cell surface protein recognition, the selection of a target protein for engineering a CAR can be quite challenging. For example, if the target protein is expressed on normal tissues, there is a possibility for off-tumor but on-target toxicities to occur. In contrast, TCRs can be engineered with high affinity and specificity for tumor-specific peptides, although their clinical application is then limited to patients with a particular MHC allele, such as HLA-A*02–01 (Schumacher, 2002; Robbins et al., 2011). However, while this selectivity of TCRs might suggest a reduced likelihood for toxicities against normal tissues, affinity-enhanced TCR-modified T cells have been found to produce off-target and organ-specific toxicities that are not always readily predictable (Johnson et al., 2009; Cameron et al., 2013; Linette et al., 2013). These properties of TCRs are again in contrast to CARs, which are MHC-independent and, therefore, can be applied more universally to patients as they depend only on expression of the target antigen by tumor cells. Thus, gene-modified T cells, while capable of potentially invoking potent anti-tumor activity, come with particular challenges in their design and application that must be reconciled during their development.
3. Current state of CAR T cells in solid malignancies
Gene-modified T cells are actively being investigated in both hematological and solid malignancies. As of March 2016, there are 17 active TCR- and 97 active CAR-modified T cell studies registered through clinicaltrials.gov (Fig. 2). Whereas the majority of TCR-modified T cell studies are focused on solid malignancies, CAR-modified T cells have most extensively been evaluated in B cell malignancies (Table 2). This focus of CAR T cells on lymphoma, leukemia, and multiple myeloma reflects (i) a tolerable safety profile with targeting proteins expressed by B lymphocytes, (ii) the ease of monitoring responses through peripheral blood and bone marrow sampling in hematological malignancies, and (iii) the natural trafficking of adoptively transferred T cells to sites (e.g. peripheral blood, bone marrow, and lymph nodes) where malignant cells naturally reside.
Fig. 2. Clinical trials using TCR and CAR therapy in cancer.
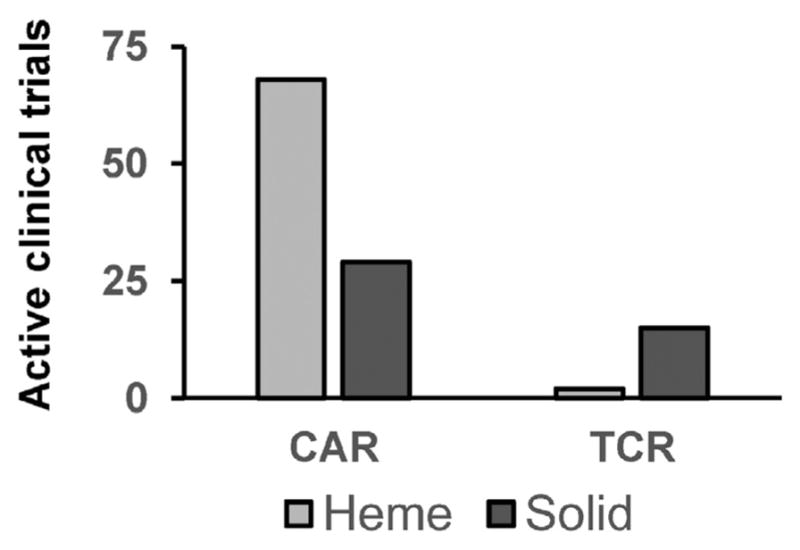
Both CAR- and TCR-based T cell immunotherapies are being investigated in solid and hematologic malignancies. Shown are active clinical trials using CAR and TCR adoptive cell therapy in solid and hematologic malignancies. Active studies were identified on clinicaltrials.gov as of March 31, 2016, using keywords “CAR,” “chimeric antigen,” and “TCR”.
Table 2.
Active clinical trials with CAR T cell therapy
| Cancer type 1 | Active clinical trials |
|---|---|
| Skin | 1 |
| Other | 2 |
| Neural | 2 |
| Stomach | 3 |
| Liver | 3 |
| Soft tissue | 4 |
| Colon | 4 |
| Multiple myeloma | 4 |
| T cell leukemia/lymphoma | 4 |
| Myeloid leukema | 5 |
| Ovary | 5 |
| Lung | 6 |
| Breast | 6 |
| Brain | 6 |
| Pancreas | 8 |
| B cell leukemia/lymphoma | 59 |
Tissue of origin for cancers being targeted in active clinical trials with CAR T cell therapy.
More than half of all CAR-modified T cell studies in hematological malignancies have focused on CD19, a protein that is expressed by many B cell malignancies (Table 3). CD19-specific CAR T cells have demonstrated potent activity in B cell acute lymphoblastic leukemia as well as B cell lymphomas (e.g., chronic lymphocytic leukemia and non-Hodgkin lymphoma) (Porter et al., 2011; Grupp et al., 2013; Davila et al., 2014; Maude et al., 2014; Lee et al., 2015; Brudno et al., 2016). However, normal B cell aplasia is a common adverse event occurring with CD19-redirected CAR T cells due to CD19 expression on normal B cells (Porter et al., 2011). This finding illustrates the potential of CAR T cells to break tolerance against normal cells that may express the CAR target protein.
Table 3.
Proteins targeted by CAR T cell therapies in active clinical trials
| CAR target 1 | Active clinical trials | CAR target | Active clinical trials |
|---|---|---|---|
| CD33 | 1 | CD123 | 2 |
| CD133 | 1 | CD20 | 2 |
| CD138 | 1 | CD22 | 2 |
| CD171 | 1 | CEA | 2 |
| cMet | 1 | GPC3 | 2 |
| EphA2 | 1 | MUC1 | 2 |
| FAP | 1 | GD2 | 3 |
| IL13Ra2 | 1 | CD30 | 4 |
| LewisY | 1 | EGFRvIII | 4 |
| NKG2D ligands | 1 | HER2 | 4 |
| ROR1 | 1 | Mesothelin | 6 |
| BCMA | 2 | CD19 | 51 |
Shown is the protein targeted by CAR T cell therapies in active trials of solid and hematologic malignancies.
CD: cluster of differentiation; FAP: fibroblast activation protein; ROR: receptor tyrosine kinase-like orphan receptor; BCMA: B-cell maturation antigen; CEA: carcinoembryonic antigen; GPC3: glypican 3; HER2: human epidermal growth factor 2; MUC1: mucin 1; EGFRvIII: epidermal growth factor receptor variant III.
Early studies attempting to translate CAR-modified T cells to solid malignancies have been hampered by expression of the CAR target on normal tissues. For example, a patient with ERBB2 (Her-2/neu)-positive metastatic colorectal carcinoma, who was treated with ERBB2-redirected CAR T cells, developed respiratory distress within 15 min of treatment followed by cardiac arrest and death. Postmortem analysis suggested localization of CAR T cells to the lung with activation triggered by low levels of ERBB2 on lung epithelial cells (Morgan et al., 2010). In a second report, a CAR specific for carbonic anhydrase IX (CAIX) was used to engineer autologous T cells for the treatment of patients with CAIX-expressing metastatic renal cell carcinoma. Here, CAR T cell infusions produced grade 2–4 liver enzyme disturbances in each of the three patients treated (Lamers et al., 2006). Liver biopsies revealed CAIX expression on bile duct epithelium and the presence of infiltrating T cells. Moreover, pre-treatment with a CAIX monoclonal antibody inhibited CAR T cell induced liver toxicity demonstrating antigen-specificity (Lamers et al., 2013). Together, these two reports illustrate the capacity of CAR T cells to induce antigen-specific toxicities directed against normal tissues, and thus, these reports strongly support the incorporation of safety measures for evaluating CAR T cells that are engineered to recognize target proteins that may also be expressed on normal tissues.
Despite promising findings with CAR T cells directed against CD19+ B cell hematological malignancies, the use of CAR T cells in solid malignancies has not yet reliably produced major or durable clinical responses. The most significant results reported to date have been in patients with neuroblastoma treated with GD2-specific CAR T cells where infusions were well-tolerated and produced complete responses in 3 of 4 patients with relapsed or refractory bone disease (Pule et al., 2008; Louis et al., 2011). However, in this study, responses in 7 additional neuroblastoma patients with relapsed bulky disease were modest with 5 patients showing disease progression and only one patient each demonstrating a partial response and stable disease. This limited efficacy of CAR T cells against malignant cells residing in solid tissues is consistent with many other reports investigating CAR T cells in sarcoma (Ahmed et al., 2015), glioblastoma (O’Rourke et al., 2015), ovarian (Kershaw et al., 2006), pancreatic (Beatty et al., 2014a; Beatty et al., 2015a), mesothelioma (Maus et al., 2013; Beatty et al., 2014a), colon (Morgan et al., 2010; Katz et al., 2015), and renal cell carcinoma (Lamers et al., 2006; Lamers et al., 2013) where stable disease is typically the best overall response observed. Many factors likely contribute to this limited potency of CAR T cells in solid malignancies including (i) poor expansion and persistence of CAR T cells in vivo, (ii) insufficient CAR T cell trafficking to tumors, and (iii) CAR T cell susceptibility to mechanisms of immunosuppression in the tumor microenvironment. Below, we discuss clinical trials that highlight each of these potential barriers in translating CAR T cells to solid tumors.
3.1. Barrier 1: Expansion and persistence
In hematological malignancies, robust in vivo expansion and persistence of CAR T cells is a critical determinant of therapeutic efficacy (Porter et al., 2015). However, in nearly all studies investigating CAR T cells for solid malignancies, expansion and persistence of CAR T cells has been a major obstacle. For example, autologous T cells engineered with a CAR specific for CD171 were detected in the peripheral blood of patients with neuroblastoma for only 1–7 days after infusion (Park et al., 2007). Similarly, CAR T cells specific for mesothelin or alpha folate receptor have largely been found to persist in patients for only several weeks (Kershaw et al., 2006; Tanyi et al., 2015). On the other hand, CAR T cells specific for GD2 and Her-2 have, in some patients, demonstrated persistence for almost a year or longer, although these cells did not appear to undergo in vivo expansion (Louis et al., 2011). The importance of CAR T cell persistence in solid malignancies is suggested by a direct correlation between persistence and longer time to progression seen in neuroblastoma patients treated with GD2-specific CAR T cells (Louis et al., 2011). The biology that determines this persistence of CAR T cells in vivo, though, remains poorly understood and could be influenced by a number of factors including the differentiation state and/or functional competence of adoptively transferred CAR T cells, the CAR target, CAR affinity for the target, CAR immunogenicity, and host-derived factors. Certainly, strategies that improve CAR T cell persistence and expansion in solid malignancies will likely be necessary for producing durable clinical responses in patients.
3.2. Barrier 2: Trafficking
Another major obstacle for CAR T cells in solid malignancies is the requirement for T cells to extravasate from the peripheral blood into solid tissues in order to mediate anti-tumor activity. This is in contrast to hematological malignancies where CAR T cells encounter malignant cells within the blood, lymph nodes, and bone marrow because of their natural tendency to migrate to these lymphoid organs. For many solid malignancies, though, tumor-infiltrating lymphocytes are often sparse and thus, mechanisms of T cell exclusion orchestrated by tumors may need to be circumvented to direct CAR T cells into tumor tissue. Furthermore, demonstrating CAR T cell infiltration into solid tissues is also challenging as it requires detection of not only T cells, but also their expression of the CAR. Several approaches have been used to monitor CAR T cell localization in vivo, including radiolabeling CAR T cells prior to adoptive cell transfer and detection of the CAR transgene by quantitative polymerase chain reaction. For example, 111In has been used to label CAR T cells specific for alpha folate receptor (Kershaw et al., 2006). When these radiolabeled gene-modified T cells were then infused in combination with high-dose interleukin-2, adoptively transferred cells were detected in the lungs, spleen, and liver, but detection in tumor tissue was limited, suggesting poor trafficking to tumors. In a second study, HER-2-specific CAR T cell infiltration into sarcoma tissue was implied by detection of CAR transgene by quantitative polymerase chain reaction in two patients who underwent tumor resection (Ahmed et al., 2015). Similar findings have been seen in biopsies obtained from patients with pancreatic cancer, ovarian cancer, and mesothelioma treated with CAR T cells specific for mesothelin (Beatty et al., 2014a; Tanyi et al., 2015). However, neither imaging nor transgene detection can adequately address issues of cellular density and localization of CAR T cells within the tumor microenvironment. This is a particularly important consideration as several clinically relevant patterns of infiltration by endogenous tumor-specific T cells into tumor tissue have been observed (Galon et al., 2013; Beatty & Gladney, 2015) including (i) diffuse infiltration, (ii) infiltration restricted to the tumor margin, and (iii) limited infiltration by only specific T cell subsets (e.g. Foxp3+ regulatory T cells). Strategies to enhance T cell trafficking into tumor tissues by direct intratumoral injection (Beatty et al., 2014a) or by hepatic artery infusion (Katz et al., 2015) to treat liver metastases have been studied but responses have been limited, suggesting that CAR T cell trafficking to the tumor tissue, while important, may be only one element regulating efficacy.
3.3. Barrier 3: Tumor microenvironment
The ultimate fate of tumor-infiltrating T cells in solid malignancies is determined by a complex network of signals orchestrated by both malignant and non-malignant cells present within the tumor microenvironment. Within solid tumors, T cell activity can be inhibited by immunosuppressive molecules (e.g. PD-L1) presented by tumor-infiltrating myeloid cells and tumor cells (Gajewski et al., 2013; Beatty & Gladney, 2015; Zou et al., 2016). In addition, solid tumors frequently demonstrate metabolic aberrations that can impact T cell biology including (i) enhanced metabolism and subsequent depletion of amino acids (e.g. arginine and tryptophan) that are important for T cell function (Bronte & Zanovello, 2005; Munn & Mellor, 2007) and (ii) hypoxia and a resultant extracellular matrix acidification due to insufficient vascular supply that is hostile to T cell survival (Palazon et al., 2011). Finally, an accumulation of immunosuppressive factors (e.g. TGF-β) and a lack of immunostimulatory factors (e.g. IL-15) may contribute to the fate of tumor-infiltrating T cells (Gajewski et al., 2013; Mlecnik et al., 2014).
Knowledge about the functional competence of CAR T cells as they penetrate the tumor microenvironment in patients is currently lacking. Preclinical models have suggested that CAR T cells, upon encountering the tumor microenvironment, rapidly lose their cytolytic and cytokine secretion capacity and enter a state of hyporesponsiveness (Moon et al., 2014). The factors that regulate this cellular fate, though, remain ill-defined. Thus, understanding mechanisms that regulate the activity of CAR T cells within human tumors may help in the rational design of combination therapies for enhancing CAR T cell efficacy in solid malignancies.
4. Challenges to CAR T cell efficacy in solid tumors
CAR T cells, when evaluated in vitro, generally demonstrate potent cytolytic, proliferative, and cytokine secretory activity. However, their anti-tumor potential upon infusion into patients with solid malignancies has, to date, been limited. For CAR T cells to be effective in vivo, they must be able to persist, undergo cellular expansion, infiltrate tumor tissues, engage their target antigen expressed on tumor cells, and finally, exert their cytolytic, proliferative, and cytokine secretory activities within the tumor microenvironment to eliminate malignant cells (Fig. 3). Thus, unleashing the full potential of CAR T cells in solid tumors will require an understanding of each of these steps. Below, we discuss several key determinants for successful translation of CAR T cells to solid malignancies.
Fig. 3. CAR T cell efficacy in solid malignancies is dependent on multiple steps.

CAR T cell therapy is a multi-step process involving (i) pheresis to collect peripheral blood leukocytes (i.e. leukapheresis) and elutriation to isolate lymphocytes from the peripheral blood of patients (Lymphocyte Collection); (ii) manufacturing of CAR T cells including gene modification of T cells to express the CAR, stimulation/expansion of gene-modified T cells in vitro, and quality control measures to evaluate CAR expression levels, T cell quality, and infectious contamination (CAR T cell Manufacturing); and (iii) the adoptive transfer of T cells to patients which then must expand, persist, traffick to tumors, and mediate effector anti-tumor activity within the tumor microenvironment (Adoptive Cell Transfer). Each of these steps is critical to the efficacy of CAR T cell therapy in solid malignancies.
4.1. CAR target selection
Early attempts at translating CAR T cells to solid malignancies demonstrated the need for caution in CAR target selection due to the potential of CAR T cells to recognize their target expressed on normal tissues and produce toxicity, so-called “off-tumor on-target” activity (Lamers et al., 2006; Morgan et al., 2010). Several approaches have now been developed to address this safety concern including (i) incorporating suicide genes into the engineered T cells, (ii) selecting CAR targets that are unique to tumor cells, and (iii) expressing a CAR transiently or designing “smart CARs” in which the CAR requires a co-signal for activation
The use of “suicide” genes, such as herpes simplex thymidine kinase (HSV-tk) and cleaved caspase 9, has been studied as strategies for selectively eliminating gene-modified T cells in vivo in the event of treatment-related toxicities. For example, T cells engineered to express HSV-tk undergo apoptosis when exposed to the anti-viral drug ganciclovir (Bonini et al., 1997; Frank et al., 2004; Berger et al., 2006). Similarly, T cells engineered to express human caspase 9 fused to a modified FK-binding domain undergo apoptosis when exposed to AP1903, a small-molecule dimerizing drug (Straathof et al., 2005; Di Stasi et al., 2011). Both strategies have demonstrated the capacity to induce “on demand” elimination of gene-modified T cells in vivo. However, not all gene-modified T cells appear to be effectively eliminated and in the case of HSV-tk, the incorporated transgene can be immunogenic thereby causing depletion of adoptively transferred cells and limiting their potential to even be efficacious (Berger et al., 2006). As a result, alternative strategies to selectively eliminate CAR T cells are also being examined. One such approach involves introducing proteins, such as CD20 or a truncated version of the epidermal growth factor receptor (EGFR). CD20- and EGFR-expressing gene-modified T cells can then be eliminated by cell-depleting antibodies rituximab and cetuximab, respectively (Kieback et al., 2008; Vogler et al., 2010; X. Wang et al., 2011).
Perhaps, the most desirable strategy for preventing on-target off-tumor toxicities with CAR T cells is to identify a target that is only expressed by malignant cells. One such example is the development of a CAR against epidermal growth factor receptor variant III (EGFRvIII), a common variant of EGFR that results from an in-frame deletion of multiple exons and produces a modified EGFR protein that is only found in human tumors and is immunogenic (Sampson et al., 2009; Mukasa et al., 2010). Single-chain variable fragments (scFvs) derived from monoclonal antibodies specific for EGFRvIII have been used to develop a humanized CAR that is being evaluated in patients with glioblastoma (Johnson et al., 2015; O’Rourke et al., 2015) (NCT02209376, NCT02664363). Preliminary results using this strategy in 6 patients with glioblastoma have been reported and suggest safety without cross-reactivity against wild-type EGFR (O’Rourke et al., 2015). Another tumor-specific target being evaluated clinically is MUC1, which undergoes altered glycosylation and loss of polarity in many human cancers, allowing for development of tumor-specific MUC1 CARs (Maher & Wilkie, 2009). Ongoing clinical investigations are evaluating MUC1-specific CAR T cells for the treatment of multiple MUC1 expressing solid tumors (NCT02587689, NCT02617134).
The most common CAR target being evaluated in the clinic in solid tumors is mesothelin, a normal protein present on healthy serosal tissue lining the peritoneum, pericardium, and pleura (Table 3). Mesothelin is also overexpressed in many solid malignancies and for this reason has attracted attention as a CAR target (Morello et al., 2016). However, healthy tissue expression of mesothelin raises the possibility for on-target off-tumor toxicities. To address this concern, the first-in-human studies with CAR T cells recognizing mesothelin used mRNA to transiently express the CAR transgene in T cells (Beatty et al., 2014a; Beatty et al., 2015a). This strategy contrasts more common approaches for engineering T cells that use retroviral and lentiviral vectors to permanently integrate the CAR transgene into the T cell genome. With RNA CAR T cells, the CAR is detected in the peripheral blood only transiently and, due to rapid loss of CAR expression on the cell surface during proliferation and activation, toxicities against normal tissues are expected to be short-lived (Zhao et al., 2010; Barrett et al., 2013).
With rapid development of gene-modifying strategies, the possibility to redesign cell behavior has become a reality. As an approach to improve the therapeutic safety of CAR T cells recognizing target antigens, T cells can now be engineered with logic circuits that require two distinct signals for activation. For example, engagement of one receptor by antigen will induce the expression of a CAR that recognizes a second antigen (Morsut et al., 2016). This strategy has been used in preclinical models to create dual-receptor AND-gate T cells. It uses a synthetic Notch receptor (synNotch), a modular receptor combining an extracellular tumor antigen recognition domain with a Notch cleavage domain fused to an intracellular transcriptional factor. Upon antigen binding to the synNotch receptor, the transcription factor is released and drives expression of a CAR recognizing a second antigen. In proof-of-concept studies, the synNotch-CAR system was shown to guide gene-modified T cells to selectively eliminate tumors that co-expressed both target antigens, leaving intact cells that expressed only one of the two target proteins (Morsut et al., 2016).
Dual-specific CAR T cells can also be designed using a trans-signaling strategy where one CAR is linked to a T cell activation signal (i.e. CD3ζ) and the second CAR is engineered with a co-stimulatory signaling domain (e.g. CD28) (Lanitis et al., 2013). In this approach, the T cell activation and co-stimulatory signals are physically dissociated from each other. As a result, full T cell activity requires target engagement of both CARs. This strategy can produce equivalent activity to second-generation CAR T cells that incorporate a single CAR fused to both CD3ζ and CD28 signaling domains. However, the trans-signaling approach provides an opportunity to minimize the potential for toxicity against normal tissues that express only one of the two CAR-specific target antigens. Despite this potential for improved safety, though, dual-specific CAR T cells may be more susceptible to antigen loss as a mechanism of immune evasion in which loss of either CAR target would be sufficient to evade elimination by CAR T cells. In contrast, bi-specific CAR T cells which incorporate two second-generation CARs may be more effective at preventing antigen escape (Hegde et al., 2013). This added potency, though, is again limited by the potential for toxicity now directed against two distinct target antigens. Thus, major challenges facing CAR T cell design in solid malignancies is not only with CAR target selection but also in how to balance safety without compromising potential efficacy.
4.2. CAR T cell manufacturing feasibility
While selection of a CAR for engineering T cells is an “off-the-shelf” approach, engineering and manufacturing CAR T cells is personalized and involves a multi-step process: (i) leukapheresis to collect leukocytes, (ii) elutriation to isolate lymphocytes based on cell size, (iii) genetic modification of isolated lymphocytes, (iv) in vitro cell expansion of T cells, and (v) quality control to assess CAR expression, T cell purity, and sterility (Fig. 3). With this process, manufacturing failures do occur. For example, the manufacturing failure rate among patients treated with CD19-redirected CAR T cells is approximately 5–10% (Davila et al., 2014; Lee et al., 2015; Porter et al., 2015). The reason for manufacturing failures is usually an inability to achieve the target cell number of CAR T cells required for treatment. This can be the result of insufficient numbers of T cells in the initial leukapheresis product or poor T cell expansion during the stimulation culture. This observation raises the possibility that T cell “fitness” could be a key factor in CAR T cell manufacturing. Consistent with this premise, recent work has suggested that interleukin-7 (IL-7) and IL-15 can enrich for T cells with enhanced expansion capacity (Singh et al., 2016). Thus, modifications to the T cell culture method may improve manufacturing feasibility.
4.3. Expansion and persistence of CAR T cells
The expansion and persistence of CAR T cells is fundamental to their therapeutic potential. In patients with CLL treated with CD19-redirected CAR T cells, expansion and persistence of the adoptively transferred cells is a clear determinant of clinical benefit (Porter et al., 2015). The potential of CAR T cells to expand and persist in vivo may be due, at least in part, to their intrinsic cellular “fitness.” However, several additional factors can influence CAR T cell expansion and persistence including (i) conditioning regimens, (ii) the intracellular activation domains of the CAR, and (iii) CAR T cell immunogenicity.
To improve the engraftment of engineered T cells, conditioning regimens including lymphodepleting agents such as cyclophosphamide and fludarabine as well as total body irradiation have been incorporated prior to T cell infusion (Dudley et al., 2005). These approaches enhance T cell engraftment likely through a variety of mechanisms that act to modify the host macroenvironment to provide “space” for T cell expansion and to induce the systemic release of cytokines (e.g., IL-7 and IL-15) involved in T cell homeostasis (Rosenberg et al., 2008; Bot et al., 2015). Alternative strategies may also be capable of promoting T cell engraftment. For example, the BTK inhibitor ibrutinib has recently been shown to improve CAR T cell engraftment and enhance CAR T cell activity in xenograft models (Fraietta et al., 2016). These studies have largely, though, been conducted in hematological malignancies, and thus, the role of conditioning regimens for improving CAR T cell persistence and expansion in solid malignancies remains poorly understood.
The intracellular signaling domain of a CAR can also be a key determinant of CAR T cell biology. For example, whereas incorporating a CD28 signaling domain into a CAR can drive a metabolic state dependent on glycolysis, CARs utilizing a 4-1BB signaling domain appear to rely on oxidative phosphorylation (Kawalekar et al., 2016). This distinct biology seen between CD28 and 4-1BB can impact the cellular fate of CAR T cells—CD28 CARs promote effector memory T cells and drive terminal differentiation/exhaustion while 4-1BB CARs reduce T cell exhaustion, promote proliferation, and drive the development of a central memory pool of T cells (Long et al., 2015). Together, these findings may provide mechanistic insight into the distinct persistence patterns seen with CAR T cells expressing 4-1BB or CD28 signaling domains. In addition, these data suggest the importance of the intracellular region of a CAR for defining CAR T cell persistence and expansion in vivo.
As CARs represent novel synthetic proteins, they can also be potentially immunogenic. As a result, endogenous immunity against the CAR may promote elimination of CAR T cells in vivo and limit persistence. Both humoral- and cellular-mediated anti-CAR responses have been observed in patients treated with CAR T cells (Riddell et al., 1996; Berger et al., 2006; Kershaw et al., 2006; Maus et al., 2013).
4.4. Trafficking of CAR T cells to solid malignancies
The exclusion of T cells from physically interacting with malignant cells is a common feature of many solid malignancies that can be mediated by innumerous mechanisms (Gajewski et al., 2013; Beatty & Gladney, 2015; Joyce & Fearon, 2015). Overcoming this barrier and facilitating the infiltration and accumulation of CAR T cells into solid tumors is fundamental to their therapeutic potential. Tumors can be classified broadly based on their degree of T cell infiltration and to date, those tumors demonstrating active T cell penetration appear more likely to respond to immunotherapy (Tumeh et al., 2014). The trafficking of T cells to solid tumors is dependent on multiple factors including (i) chemokines that act to lure T cells into tumors, (ii) endothelium that must be permissive to T cell extravasation into tumor tissue, and (iii) stroma, including extracellular matrix proteins and non-malignant cells that may restrict T cell access to tumors. Each of these factors may also be involved in regulating CAR T cell trafficking to solid tumors. Thus, devising strategies to circumvent these potential barriers to CAR T cell entry is of great interest.
Leukocyte recruitment to solid malignancies is dependent on chemokines that can be secreted by both malignant and non-malignant cells residing within the tumor microenvironment (Bonecchi et al., 2011). For example, CCL2 is commonly produced within tumors and acts to recruit myeloid cells to the tumor microenvironment (Qian et al., 2011). To improve CAR T cell infiltration into tumors, gene-modified T cells can be engineered to express a chemokine receptor that matches the chemokine profile of a tumor. For example, engineering CAR T cells with CCR2, the receptor for CCL2, was found to enhance CAR T cell trafficking to tumors in a xenograft model of human mesothelioma (Moon et al., 2011). Improved trafficking was associated with enhanced anti-tumor efficacy, thereby demonstrating that proper recruitment of CAR T cells to tumors can produce robust anti-tumor activity even against large established solid tumors.
Extravasation of T cells into solid tissues is a tightly regulated process. Aberrant expression of immune regulatory molecules, such as endothelin B receptor, on tumor vasculature, can restrict T cell entry into solid tumors (Buckanovich et al., 2008). In addition, the vasculature that feeds tumors is commonly dysregulated with poorly arranged and tortuous vessels that ineffectively supply the tumor bed with nutrients thereby leading to regional hypoxia and acidosis (Huang et al., 2013; Jain, 2014). Disruption of VEGF/VEGFR2 signaling can induce vascular normalization in tumors (Huang et al., 2012). Moreover, in preclinical models VEGF/VEGFR2 inhibition improves lymphocyte infiltration into tumors and enhances the efficacy of adoptive cell therapy (Shrimali et al., 2012). Thus, conditioning the vasculature that supplies solid tumors may be necessary in some cases to improve CAR T cell trafficking, although this hypothesis remains to be tested.
Dense fibrosis that surrounds tumors and separates malignant cells from the endothelium may be a physical barrier to CAR T cell infiltration in solid malignancies. Penetration of this stromal matrix is dependent at least in part on the capacity of T cells to produce enzymes, such as heparanase, which degrade heparin sulfate proteoglycans present within the extracellular matrix of solid tumors (Caruana et al., 2015). However, during in vitro culture conditions, T cells can lose their capacity to produce heparanase leading to an inability to penetrate the tumor microenvironment. Engineering T cells to express heparanase to allow for degradation of heparin sulfate proteoglycans can improve T cell infiltration into tumors leading to enhanced efficacy. Thus, some matrix components within solid tumors may restrict CAR T cell access.
Finally, soluble factors and non-malignant cells within tumors may be able to repel T cells from the tumor microenvironment and thereby establish a site of “immune privilege” (Mellor & Munn, 2008). Immature myeloid cells recruited to solid malignancies have been shown to suppress T cell activation, and their elimination can restore T cell accumulation within tumors (Bayne et al., 2012; Stromnes et al., 2014). Similarly, some chemokines, such as CXCL12, may be capable of excluding T cells from solid malignancies (Feig et al., 2013). In addition, tumor genetics can be a critical determinant of the chemokine profile of tumors and, thus, may determine the permissiveness of the tumor microenvironment to T cell recruitment. For example, activation of Kras induces the secretion of chemokines, including IL-8 (Sparmann & Bar-Sagi, 2004), which attracts myeloid cells to tumors, restricting T cell entry. Similarly, loss of PTEN by tumor cells can lead to release of CCL2 and subsequent recruitment of T cell suppressive monocytes and macrophages (Peng et al., 2016). Activation of the β-catenin pathway can also modulate the chemokine profile of tumors by preventing expression of CCL4, which appears to be important for dendritic cell recruitment and subsequent T cell priming and activation in draining lymph nodes (Spranger et al., 2015). In addition, hyperactivation of Hippo-YAP signaling in malignant cells can induce CXCL5-dependent recruitment of immature myeloid cells and in doing so, restrict T cell entry into tumors (Wang et al., 2016). However, it remains unclear whether these mechanisms act to mainly prohibit T cell infiltration or are primarily involved in regulating the priming and activation of tumor-specific T cells which once activated can infiltrate tumor tissue. For example, induction of tumor-specific T cells using chemoimmunotherapy (Beatty et al., 2015b) or adoptive transfer of gene-modified TCR-specific T cells (Stromnes et al., 2015) leads to T cell recruitment in models of pancreatic cancer despite dense fibrosis, a strong presence by immunosuppressive myeloid cells, and high levels of CXCL12 and CCL2. However, each of these studies has addressed the trafficking of T cells that are dependent on TCR recognition for activation. Thus, whether mechanisms regulating CAR T cell trafficking into solid malignancies will be the same as for TCR-modified T cells still remains unclear. Nonetheless, the cellular fate of tumor-infiltrating T cells will ultimately be defined by the tumor microenvironment and thus, strategies to condition tumors for improved receptiveness to gene-modified T cells will likely be important for their effective translation to solid tumors.
4.5. Circumventing immune suppressive mechanisms within the tumor microenvironment
The microenvironment that surrounds tumors often comprises a dense fibrotic matrix with an abundance of immunosuppressive myeloid cells that together may limit the efficacy of tumor-infiltrating CAR T cells. This requirement to extravasate from the peripheral blood into solid tissues is in stark contrast to hematological malignancies in which T cells routinely migrate through lymphoid organs (e.g. spleen, lymph nodes, and bone marrow) where malignant cells reside. The microenvironment that surrounds solid tumors can be hypoxic, acidic, and scarce in key amino acids (e.g. arginine and tryptophan) critical for T cell activity. CAR T cells have been shown to enter a state of hyporesponsiveness within tumor tissues (Moon et al., 2014) and thus, modulation of this microenvironment may be necessary to improve outcomes. Multiple signaling networks can regulate T cell efficacy within tissues including (i) checkpoint molecules (e.g. PD-1, CTLA-4, TIM3, LAG3, etc), (ii) non-malignant stromal cells (e.g. macrophages, granulocytes, regulatory T cells, B cells, and fibroblasts), and (iii) malignant cells which may adapt to immune pressure through a process termed “immune editing” in which they become poorly immunogenic such as by losing antigen expression.
To maintain peripheral immune tolerance, T cells are hardwired to upregulate immune checkpoint molecules (e.g. PD-1, CTLA-4, TIM3, LAG3, etc) that act to dampen their activation within tissues (Pardoll, 2012; Zou et al., 2016). Thus, combining immune checkpoint blockade with CAR T cells is a logical therapeutic combination that has demonstrated some promise in preclinical models. For example, in mouse models of sarcoma and breast cancer, Her2-specific CAR T cells have shown enhanced in vivo anti-tumor activity when combined with anti-PD-1 blockade (John et al., 2013). An alternative approach is to engineer CAR T cells with a “switch receptor” in which a truncated extracellular domain of PD-1 is fused with the intracellular signaling domain of an activation molecule, such as CD28 (Liu et al., 2016). With this strategy, engagement of PD-1 ligands by the switch receptor on T cells induces activation rather than inhibition, thereby leading to decreased T cell susceptibility to tumor-induced hypofunction and enhanced anti-tumor activity in vivo. However, additional features of the tumor microenvironment may also be involved in regulating CAR T cell hyporesponsiveness in tumors including a lack of sufficient immunostimulatory signals. To this end, CAR T cells can be engineered to express cytokines (e.g. IL-12) that may then act in an autocrine fashion to enhance T cell activity and persistence (Koneru et al., 2015a). These so-called “armored CARs” have demonstrated the potential for superior anti-tumor activity and persistence compared to conventional CAR T cells in xenograft models and await clinical investigation in patients (Koneru et al., 2015b). Thus, shifting the balance between immune suppressive and stimulatory signals within tumors may enhance CAR T cell efficacy.
T cell activity within tumor tissues can be regulated by arginase and indoleamine 2,3 dioxygenase (IDO), which catabolize arginine and tryptophan, respectively. Both tumor cells and myeloid cells within the tumor microenvironment can express arginase and IDO which act to deplete amino acids necessary for T cell proliferation and sustained activation (Bronte & Zanovello, 2005; Munn & Mellor, 2007). For example, neuroblastoma can suppress the activity of GD2-specific CAR T cells by overexpressing arginase II, which limits CAR T cell proliferation in the absence of sufficient quantities of arginine (Mussai et al., 2015). Similarly, IDO expressed within the tumor microenvironment can inhibit CAR T cell proliferation, cytotoxicity, and cytokine secretion that is reversed by inhibiting IDO activity (Ninomiya et al., 2015). Thus, altering metabolic components within the tumor microenvironment may be critical for maximizing the potential of CAR T cells in solid tumors.
Despite the well-recognized capacity of myeloid cells to be immunosuppressive, their inherent plasticity also makes them capable of mediating potent anti-tumor activity (Long & Beatty, 2013). In this regard, it is likely that CAR T cells may require myeloid cells for realizing their full anti-tumor potential in vivo. In a preclinical model of ovarian cancer, the efficacy of CAR T cells was determined to be dependent on macrophages that acquired tumoricidal properties as a result of IFN-γ produced by CAR T cells (Spear et al., 2012). Thus, while it is tempting to deplete myeloid cells because of their tendency to be immunosuppressive, it may be more prudent to combine CAR T cells with strategies that shift the behavior of tumor-infiltrating myeloid cells from pro- to anti-tumor. For example, CD40 agonists have shown potential for redirecting tumor-infiltrating myeloid cells with anti-tumor and anti-fibrotic activity (Beatty et al., 2011; Beatty et al., 2013; Long et al., 2016). Similarly, CD47 antagonists can disrupt inhibitory signals that prevent macrophages from attacking malignant cells and potentially stimulating endogenous T cell immunity (Willingham et al., 2012; Tseng et al., 2013; Liu et al., 2015). CSF1R inhibitors have also been reported to shift the biology of tumor-infiltrating myeloid cells from pro- to anti-tumor (Pyonteck et al., 2013). Small molecule inhibitors of BTK and PI3Kδ may also be effective at disrupting key immunoregulatory pathways established within tumors by myeloid cells and other leukocyte populations (Ali et al., 2014; Gunderson et al., 2016). Thus, CAR T cells may benefit from reinforcement provided by innate immunity, in particular myeloid cells, which if redirected from pro- to anti-tumor could enhance the efficacy of CAR T cells.
The anti-tumor activity of CAR T cells is dependent on expression of their target by malignant cells. Thus, downregulation or loss of target antigen expression on tumor cells may allow for immune escape. This possibility is supported by findings in a patient with glioblastoma undergoing operative resection after treatment with EGFRvIII-redirected CAR T cells (O’Rourke et al., 2015). Pathological findings of the surgically resected tumor tissue from this patient revealed T cell infiltration, but with evidence of EGFRvIII antigen loss within tumors suggestive of immune editing of malignant cells by tumor-infiltrating T cells. To address this mechanism of immune evasion, it may be necessary to combine CAR T cells that recognize multiple unique target antigens expressed by tumors. However, it remains unclear whether all malignant cells will need to express the CAR target since tumor cell death induced by CAR T cells may also invoke epitope spreading (i.e. development of an immune response against antigens distinct from the target antigen) and subsequently, reinvigorate endogenous tumor-specific TCR-dependent T cell immunity (Beatty, 2014b). For example, in preclinical models, EGFRvIII-specific CAR T cells have demonstrated the capacity to induce anti-tumor immunity that prevents rechallenge with EGFRvIII negative tumors (Sampson et al., 2014). Thus, CAR T cells could also act as a vaccine strategy.
5. Next steps in CAR T cell development for solid malignancies
Realizing the potential of CAR T cells in solid malignancies will require a multi-targeted approach that addresses issues of CAR design and safety as well as CAR T cell persistence, expansion, trafficking, and fate in vivo. Based on findings to date with CAR T cells in solid malignancies, it is likely that the success of CAR T cells will depend on rational combination treatment strategies that address each of the potential barriers described above. In the coming years, it is expected that significant effort will also continue toward (i) the development of CARs that recognize novel targets, (ii) the engineering of CAR T cells as delivery mechanisms to release factors that may alter the tumor microenvironment, and (iii) the design of more elegant CAR T cells that incorporate novel synthetic genes that direct the selectivity, potency, and effector activity of CAR T cells.
In solid tumors, CAR T cells also offer a novel tool to understand mechanisms of immune evasion in cancer. Dissecting these mechanisms will be benefited by approaches that monitor the fate of CAR T cells in vivo and within the tumor microenvironment. In addition, high throughput evaluation of combination strategies may be possible by incorporating parallel preclinical studies in relevant immunocompetent spontaneous mouse models of cancer. Together, the conduct of co-clinical trials in patients and mice has the potential to provide a seamless platform for advancing CAR T cells for solid tumors.
Beyond CAR T cell design and selection of rational combination strategies, it will also be critical to consider how to more effectively translate CAR T cells to solid malignancies. For example, immune profiling has revealed distinct tumor phenotypes that have been broadly classified as T cell-inflamed (or immunogenic) and non-inflamed (or immune privileged) based on robust and poor effector T cell infiltration, respectively (Gajewski, 2007; Beatty & Gladney, 2015; Joyce & Fearon, 2015). It is possible that the use of CAR T cells in T cell-inflamed tumors may allow for more effective CAR T cell recruitment to tumor tissue and enhanced efficacy. Certainly, defining unique characteristics of tumors –beyond just presence or absence of the CAR target protein – that may positively or negatively impact CAR T cell efficacy in solid tumors will be important for improving patient selection. Similarly, as the long-term safety profile of CAR T cells becomes better defined, CAR T cell therapies might be considered in the neoadjuvant setting for some malignancies where surgical resection of tumor tissue could provide a more informative “snapshot” of CAR T cells in the tumor microenvironment. Thus, there may be a role for refining patient selection and trial design beyond tumor expression of the CAR target for realizing the potential of CAR T cells in solid tumors.
6. Concluding remarks
Cellular immunotherapy using CAR T cells has demonstrated remarkable therapeutic potential for the treatment of patients with hematological malignancies. However, the translation of CAR T cells to solid malignancies has been met with several challenges. In this review, we discussed the current landscape of CAR T cell studies in patients with solid tumors. In addition, we defined many of the barriers that may impact the safety and efficacy of CAR T cells in solid tumors including the selection of the CAR target protein, expansion and persistence of adoptively transferred CAR T cells, trafficking of CAR T cells to tumor tissue, and the tumor microenvironment. Next steps in the development of CAR T cells for solid malignancies will involve a multidisciplinary effort focusing on fine-tuning of CAR design, combination therapies, and novel clinical study designs for monitoring the therapeutic potential and cellular fate of CAR T cells administered to patients. With this effort, CAR T cells have the potential to not only improve our understanding of the immunobiology of cancer but also to impact clinical outcomes for patients with a broad range of cancers.
Abbreviations
- CAIX
Carbonic anhydrase IX
- CAR
Chimeric antigen receptor
- EGFR
Epidermal growth factor receptor
- EGFRvIII
Epidermal growth factor receptor variant III
- HSV-tk
Herpes simplex thymidine kinase
- IDO
Indoleamine 2,3 dioxygenase
- IL
Interleukin
- MHC
Major histocompatibility
- synNotch
Synthetic Notch receptor
- TCR
T cell receptor
Footnotes
Financial support: This work was supported by a National Institutes of Health grant K08 CA138907, by a grant from Precision Medical Research Associates (Grant # PMRA_2014-0612.1), and by grant 2013107 from the Doris Duke Charitable Foundation,
Conflict of interest statement
G.L.B. has received research funding from Novartis, Incyte, and Biothera. The authors declare no other conflicts of interest.
References
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510(7505):407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24(8):717–727. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL. Engineered chimeric antigen receptor-expressing T cells for the treatment of pancreatic ductal adenocarcinoma. Oncoimmunology. 2014b;3:e28327. doi: 10.4161/onci.28327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014a;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, O’Hara MH, Nelson AM, McGarvey M, Torigian DA, Lacey SF, et al. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J Clin Oncol. 2015a;33(15_suppl):3007. [Google Scholar]
- Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology. 2015b;149(1):201–210. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107(6):2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonecchi R, Locati M, Mantovani A. Chemokines and cancer: a fatal attraction. Cancer Cell. 2011;19(4):434–435. doi: 10.1016/j.ccr.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- Bot A, Rossi JM, Jiang Y, Navale L, Shen Y-w, Sherman M, et al. Cyclophosphamide and fludarabine conditioning chemotherapy induces a key homeostatic cytokine profile in patients prior to CAR T cell therapy. 2015;126:4426. [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5(8):641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016;34(10):1112–1121. doi: 10.1200/JCO.2015.64.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127(9):1117–1127. doi: 10.1182/blood-2015-11-679134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, Rudolph C, Heberlein C, von Neuhoff N, Schrock E, Schambach A, et al. Tumor cells escape suicide gene therapy by genetic and epigenetic instability. Blood. 2004;104(12):3543–3549. doi: 10.1182/blood-2004-03-0852. [DOI] [PubMed] [Google Scholar]
- Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13(18 Pt 1):5256–5261. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263(1):68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6(3):270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinatorial targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73(10):2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109(43):17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180(10):7028–7038. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21(14):3149–3159. doi: 10.1158/1078-0432.CCR-14-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieback E, Charo J, Sommermeyer D, Blankenstein T, Uckert W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc Natl Acad Sci U S A. 2008;105(2):623–628. doi: 10.1073/pnas.0710198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015b;13:102. doi: 10.1186/s12967-015-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology. 2015a;4(3):e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- Lanitis E, Poussin M, Kattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209–1215. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology. 2013;2(12):e26860. doi: 10.4161/onci.26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6(4):400–413. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J, Wilkie S. CAR mechanics: driving T cells into the MUC of cancer. Cancer Res. 2009;69(11):4559–4562. doi: 10.1158/0008-5472.CAN-09-0564. [DOI] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8(1):74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med. 2014;6(228):228ra237. doi: 10.1126/scitranslmed.3007240. [DOI] [PubMed] [Google Scholar]
- Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. 2016;6(2):133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164(4):780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukasa A, Wykosky J, Ligon KL, Chin L, Cavenee WK, Furnari F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci U S A. 2010;107(6):2616–2621. doi: 10.1073/pnas.0914356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015;75(15):3043–3053. doi: 10.1158/0008-5472.CAN-14-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125(25):3905–3916. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke D, Desai A, Morrissette J, Martinez-Lage M, Nasrallah M, Brem S, et al. IMCT-15 pilot study of T cells redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. Neuro-Oncology. 2015;17:v110–v111. [Google Scholar]
- Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. 2011;18(5):1207–1213. doi: 10.1158/1078-0432.CCR-11-1591. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and anti-tumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2(2):216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, 2nd, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20(4):972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2(7):512–519. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2012;70(15):6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra323. doi: 10.1126/scitranslmed.aad5222. [DOI] [PubMed] [Google Scholar]
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol. 2012;188(12):6389–6398. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014 doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen HN, Cuevas C, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. 2015;28(5):638–652. doi: 10.1016/j.ccell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi JL, Haas AR, Beatty GL, Morgan MA, Stashwick CJ, O’Hara MH, et al. Abstract CT105: safety and feasibility of chimeric antigen receptor modified T cells directed against mesothelin (CART-meso) in patients with mesothelin expressing cancers. Cancer Res. 2015;75:CT105. [Google Scholar]
- Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler I, Newrzela S, Hartmann S, Schneider N, von Laer D, Koehl U, et al. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther. 2010;18(7):1330–1338. doi: 10.1038/mt.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016;6(1):80–95. doi: 10.1158/2159-8290.CD-15-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]