

Abstract
T cell based immunological memory has the potential to provide the host with life-long protection against pathogen re-exposure and thus offers tremendous promise for the design of vaccines targeting chronic infections or cancer. In order to exploit this potential in the design of new vaccines it is necessary to understand how and when memory T cells acquire their poised effector potential, and moreover, how they maintain these properties during homeostatic proliferation. To gain insight into the persistent nature of memory T cell functions, investigators have turned their attention to epigenetic mechanisms. Recent efforts have revealed that many of the properties acquired among memory T cells are coupled to stable changes in DNA methylation and histone modifications. Furthermore, it has recently been reported that the delineating features among memory T cells subsets are also linked to distinct epigenetic events providing exciting new hypotheses regarding their cellular ancestry. Here we review recent studies focused on epigenetic programs acquired during effector and memory T cell differentiation and discuss how these data may shed new light on the developmental path for generating long-lived CD8 T cell memory.
Introduction
Effector and Memory T Cell Differentiation
The diverse repertoire of naïve T cells in a host at any given time facilitates the surveillance of a wide array of antigens. Recent reports estimate there to be between 4,000 to 200,000 naïve CD8 T cells specific to a given antigen in human blood [1–4] and 15–1,000 naïve CD8 T cells specific to a given antigen in mice [5–7]. While the number of circulating naïve T cells recognizing a specific antigen may seem low, upon encountering their cognate antigen, naïve CD8 T cells undergo clonal expansion increasing their quantity roughly 105 fold [5]. During this highly proliferative expansion phase of the immune response, the cells acquire the ability to express effector molecules including interferon gamma (IFNγ), tumor necrosis factor- α (TNF-α), perforin (Prf), and granzymes as they undergo effector differentiation. Once the pathogen is cleared, roughly 90–95% of the antigen-specific effector cells die, the surviving cells develop into the pool of long-lived memory T cells (Figure 1.A).
Figure 1.
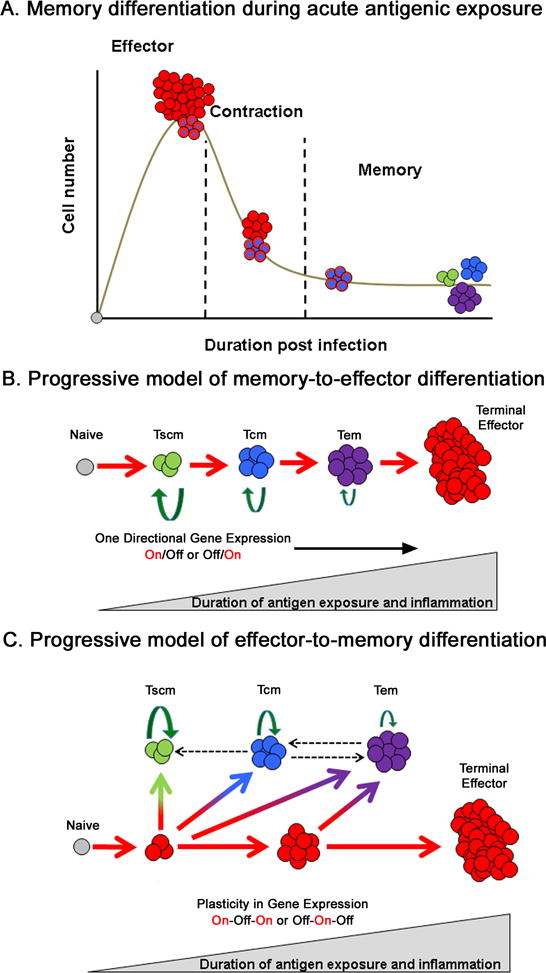
Models for generation of effector and memory CD8 T cells. (A) Cartoon representation of antigen-specific T cell clonal expansion and contraction in response to an acute infection. Upon cognate antigen presentation, naïve antigen-specific CD8 T cell proliferate and increase their numbers 1×105 fold as they differentiate into effector cells. Approximately 90–95% of these cells die during the contraction stage coinciding with a progressive change in phenotypic and functional properties of the surviving population of cells. CD8 T cells that survive the contraction stage of the immune response make up a heterogeneous pool of memory cells. The schematics (B) and (C) represent two distinct models of progressive memory T cell differentiation. According to the memory-to-effector differentiation model (B) the memory T cell subsets – stem-cell-like memory (Tscm), central-memory (Tcm) and effector-memory (Tem) arise during the early stages of activation of naïve T cells prior to effector stage of differentiation (Teff). The effector-to-memory differentiation model (C) suggests that early after activation of naïve T cells, memory precursor effector cells are generated which acquire effector functions but retain the potential to give rise to the memory subsets. In both models, strength and duration of exposure to antigen reduce the memory potential of the cells. Size of the
 arrow is reflective of the self-renewing capacity of the memory subsets.
arrow is reflective of the self-renewing capacity of the memory subsets.
At the memory stage of the immune response, life-long persistence of antigen-specific cells occurs without the requirement of antigen re-exposure. This observation raises many questions regarding when memory cells develop as well as how they remain poised to rapidly recall effector functions upon antigen re-exposure. Over the past two decades, major efforts have been made to identify the mechanisms that impart antigen-specific CD8 T cells with the ability to survive the contraction stage of the immune response and undergo life-long homeostasis. Based on the fact that memory CD8 T cell homeostasis is in part regulated by IL-15 and IL-7 signaling [8–12], Kaech et.al made the transformative discovery that a subset of effector cells express the IL7 receptor-α (IL-7Rα – CD127) during the early stages of CD8 T cell response, now referred to as memory precursor effector cells (Mpec), facilitating their survival through the contraction stage of the immune response [12]. From this finding, and that of other labs, it is now clear that the ability to survive the contraction stage of the immune response is a unique property attained by subsets of effector cells. Subsequent studies further demonstrated that in addition to expressing CD127 [12], Mpecs downregulate the expression of CD25 and KLRG1 and can be identified at a very early stage of the effector stage of differentiation [13, 14]. Compared to Teff, Mpecs are multipotent, and retain the potential to develop into memory T cells differing in their anatomical locations, effector functions, proliferative capacity, and longevity. The considerable diversity in the pool of memory T cells that can be derived from Mpec has propelled efforts to define the cellular and molecular mechanisms that dictate memory subset differentiation and acquisition of subset-specific functions.
Recent studies tracking the fate of single T cells have reported that a naïve CD8 T cell can differentiate into a heterogeneous mix of effector and of memory CD8 T cell populations [15–19], therefore, it is unlikely that the heterogeneity among effector and memory populations is pre-determined. Much more likely, heterogeneity among cell populations arises from events occurring during the primary immune response, thus raising the question whether a better understanding of these events would enable us to manipulate and enhance vaccine-generated T cell memory. To identify the events driving memory T cell development, it is necessary to first dissect the phenotypic and functional heterogeneity among the pool of memory T cells. Sallusto et.al made the landmark discovery in 1999 that the pool of memory T cells is composed of effector-memory (Tem) and central-memory (Tcm) subsets (Figure 1B and C) based on their anatomical localization and the expression of lymph node homing markers [20]. Specifically, Tcm express the chemokine receptor CCR7+ and the adhesion molecule CD62L+, facilitating their localization to lymphoid tissues, whereas Tem are CCR7− and CD62L− and distributed in non-lymphoid sites [20, 21]. In addition to the expression of lymph node homing markers, Tcm also express CD27, have higher homeostatic proliferative potential, and produce more IL-2. Tem on the other hand, do not express CD27, have reduced homeostatic proliferative potential, restricted IL-2 expression, and produce more effector cytokines upon stimulation [20, 22–24]. Recently, two additional memory CD8 T cell subsets have been identified: tissue resident memory T cells and stem-cell-like memory cells. Memory T cells that are non-circulating, reside at tissue sites of initial antigen encounter, and elicit a rapid protective response in situ upon antigen re-encounter have been termed tissue-resident memory T cells (Trm) [25–31]. Trm are maintained without replenishment from an influx of circulating memory T cells and share many functional properties with the Tem subset. These cells express CD103, CD69, and require TGFβ, TNFα, Il-15, and IL-33 for their generation [32–34]. Trm have intentionally been left out of figures 1.B and C as their relationship to the other memory subsets is not yet clear. Another newly identified subset of memory CD8 T cells, the stem-cell-like memory subset (Tscm), shares many phenotypic and functional properties with both naïve T and memory T cells (Figure 1.B and C). They have been termed stem-cell-like memory based on their ability to self-renew and their potential to give rise to other CD8 T cell memory subsets. Among the memory subsets, Tscm have the greatest proliferative response to homeostatic cytokines, therefore, are thought to have the greatest longevity of all the memory subsets. In addition, like other memory subsets, Tscm can rapidly respond to antigenic re-exposure, giving rise to Tem, Tcm, and effector T cells [22, 23, 35]. Transcriptional profiling studies suggest that among the memory subsets, the transcriptional profile of Tscm most closely resembles that of naïve T cells [23], and that of Tcm is between naïve and Tem [36]. While each of these individual subsets provides potent recall responses, the protective immunity is achieved by their collective efforts.
Although the functions that define each of these subsets are generally agreed upon, the developmental pathways and the origin of memory T cells remain an area of considerable discussion in the field. In an effort to better define the origin of long-lived memory T cells, the field has developed several models that may explain effector and memory T cell differentiation, each yielding a testable hypothesis. The widely accepted, progressive differentiation model argues that effector and memory cells develop along a common path linking the strength and duration of TCR signaling to the reduced plasticity in CD8 T cell developmental potential (Figure 1.B and 1.C) [14, 22, 37–39]. This model is further divided into two sub-models based on when memory T cells arise. The first sub-model proposes that naïve T cells develop directly into memory T cells then eventually into effector cells (Figure 1.B), following unidirectional programming of gene expression (off-on or on-off, Figure 3.A right panels). The opposing sub-model, describes the development of memory CD8 T cells from a population of effector cells, the Mpecs (Figure 1.C), and suggests bi-directionality in gene expression programs (off-on-off or on-off-on, Figure 3.A left panels). Such plasticity in gene expression programs would argue that memory subsets arise from cells that transit through an effector stage of differentiation without acquiring a terminal fate. The dichotomy in these models has led to considerable debate in the field, and it remains to be conclusively established whether or not effector and memory cells have distinct and fixed developmental lineages and functions.
Figure 3.
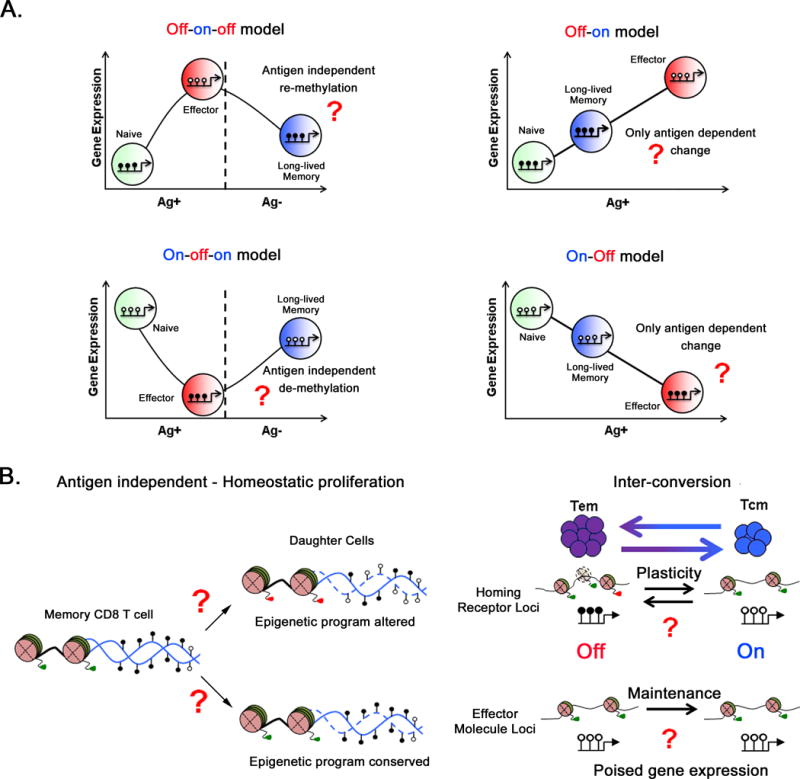
Maintenance versus plasticity of acquired transcriptional programs during memory T cell differentiation. (A) Unidirectional versus bidirectional Memory T cell differentiation models generate distinct hypotheses regarding the mechanism of transcriptional regulation. During unidirectional memory T cell differentiation, gene expression programs would arise during the antigen-dependent stage of the immune response. Such a model of on-off or off-on gene expression would predict only antigen-dependent epigenetic reprogramming events. Bidirectional memory T cell differentiation predicts effector gene expression programs are first acquired during the antigen-dependent stage of differentiation, but then further modified during the antigen-independent stage of the immune response. Such a model of off-on-off or on-off-on gene expression would predict antigen-independent epigenetic reprogramming events. (B) It remains to be determined whether the repressive epigenetic programs at homing receptor loci or the poised epigenetic programs at effector molecule loci acquired during memory CD8 T cell differentiation are maintained or erased in daughter cells during homeostatic proliferation or memory cell inter-conversion. These models also provide the opportunity to test whether stability and/or plasticity of the epigenetic programs facilitates the maintenance or pliability of transcriptional programs. Solid blue line indicates parental DNA and dashed blue line indicates the newly synthesized DNA strand. Repressive programs are shown as filled circles and red triangles, permissive programs are shown as open circles and green triangles.
Many key insights resulting in the development and evolution of the above described models for memory differentiation have been, in large part, borne out of genome-wide transcriptional profiling studies on T cells at the various stages of effector and memory differentiation [11, 14, 36, 40]. Based on these studies it has become clear that acquisition of subset-specific transcriptional programs is in part mediated by coordinated expression of transcription factors (recently reviewed in [41–45]), resulting in gene expression programs that can be selectively maintained from parent cell to daughter cell during the prolifertive burst of an effector response as well as memory cell homeostasis. While many of the acquired gene expression programs in memory CD8 T cells are long-lived, several of the program may undergo further adaptation, allowing memory CD8 T cells to exhibit phenotypic and functional plasticity [46–50]. Because of the well-accepted role epigenetic mechanisms play in regulating cell fates, several investigators have turned their attention to epigenetic mechanisms in an effort to better understand gene expression plasticity during memory T cell generation, and more broadly, the lineage relationship between effector and memory T cells.
Epigenetic Regulation of Transcription
The genome of eukaryotic cells is packaged into tightly folded structures known as nucleosomes (figure 2.A) and their relative density near transcriptional control elements orchestrates cell-type specific gene expression programs by controlling the accessibility of transcription factors to the DNA [51–53] (figure 2.B). As cells undergo differentiation, chromatin accessibility is modified by ATP-dependent nucleosomal remodeling (assembly, disassembly, sliding), changing the chromatin landscape which controls the genes targeted by transcriptional machinery [54–56]. Such alteration in nucleosomal density is regulated by covalent modifications to DNA and histones (figure 2.B), which can be faithfully propagated from parent to daughter cell during cell division. As such, maintaining acquired cell type specific gene expression programs is intimately linked to the stability of epigenetic modifications and is critical for maintaining cellular fates. Here we briefly describe a few of the better understood epigenetic mechanisms and discuss them in the context of effector and memory T cell differentiation.
Figure 2.
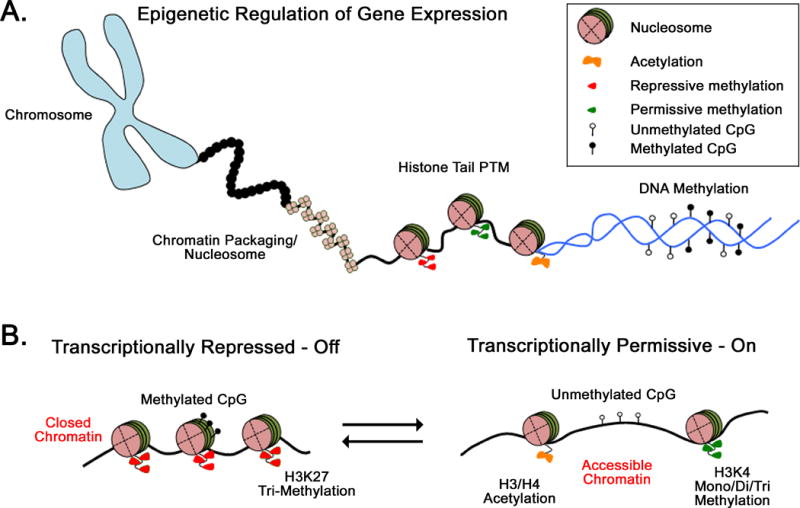
Chromatin organization, accessibility and gene expression. (A) A cartoon overview of various levels of chromatin organization. To accommodate the ~2m long human genomic DNA into the nucleus of a cell, the DNA exists as highly condensed structures called chromosomes. Dense packaging of the DNA into the chromosome in mediated in part by nucleosomes, composed of a histone octamer with DNA wrapped around it. Modifications to histone tails (acetylation-orange bowtie, repressive-red triangle or permissive-green triangle methylation) alters nucleosomal density near the promoter of a gene and regulates the accessibility of the chromatin. Finally, cytosine methylation at CpGs within the promoter is thought to provide the cell-division transmissible component of epigenetic transcriptional regulation. (B) Transcriptionally repressed versus active genetic locus. A transcriptionally repressed gene is characterized by the presence of nucleosome, enrichment of repressive histone modifications (lack of acetylation, H3K27 methylation-red), and DNA methylation in the promoter sequence. These modifications maintain a compact chromatin state and restrict gene expression. A transcriptionally permissive gene is characterized by absence of nucleosomes near the transcriptional start site, enrichment of permissive histone modifications (acetylation and H3K4 methylation), and CpG demethylation within the promoter.
Histone Modifications
Nucleosomal architecture is driven by the “wrapping” of DNA around histones. Cues for nucleosome assembly arise from the post-translational modifications (PTM) of histones at the N-terminal tail, and can serve to alter the affinity of the nucleosome for DNA. These modifications are major epigenetic mechanisms affecting DNA accessibility in the nucleosome [57] and include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, etc. It is now well accepted that a combination of these modifications can mark active, repressed, or poised genes in the chromosome. For example, H3K4 methylation is coupled to transcriptional activation [58, 59], whereas H3K27 trimethylation is associated with repressive chromatin (figure 2.B) [60]. A system of methyltransferases and demethylases work to modify histones and maintain permissive/repressive conformations of the chromatin. In addition to methylation of the histone tail, acetylation of the lysine residues in histone also serves as an epigenetic regulatory mechanism. Histone acetylation, mediated by histone acetyltransferases (HATs), decreases the affinity of the nucleosome for DNA, leading to an open chromatin state (Figure 2B). The balance between accessible and inaccessible chromatin states can, in part, be directed by HATs and the antagonizing activity of histone deacetylases (HDACs), which ultimately regulates the ability of transcriptional machinery to access promoter and enhancer regions of the genome [61–63].
DNA Methylation
DNA methylation is another major epigenetic mechanism that provides instructions for maintaining chromatin accessibility during cell division. In mammals, DNA methylation predominantly occurs at cytosine residues within the context of CpG dinucleotides. When present at promoters, CpG methylation is often associated with transcriptional silencing of a gene (Figure 2B) [64]. The palindromic nature of the CpG dinucleotide can serve as a template for the propagation of methylation patterns from the parental DNA strand to the newly synthesized daughter strand, thus providing a mechanism for stable maintenance of epigenetic states. Faithful duplication of methylation patterns from parent to daughter cell is performed by the maintenance methyltransferase DNMT1, whereas establishment of new DNA methylation programs is performed by a group of de novo DNA methyltransferase enzymes (DNMT3) that can target unmethylated CpGs [64].
Cytosine methylation can be further modified by covalent addition of a hydroxyl group resulting in 5-hydroxymethyl cytosine (5hmC) [65]. While less is known about the role of 5hmC in transcriptional regulation, it has been found at transcription start sites and enhancer regions of actively transcribed genes [65, 66]. Conversion of 5mC to 5hmC is catalyzed by members of the ten eleven translocation (TET) family of enzymes. Subsequent modification of 5hmC into 5-formyl (5fC) or 5-carboxy cytosine (5caC) has been reported to promote base excision repair, providing a potential mechanism for active DNA demethylation (without cell division) [66]. Additionally, cell division dependent passive DNA demethylation has also been shown to occur as a result of reduced affinity of DNMT1 for hemi-5hmC [65]. Such potential mechanisms for DNA demethylation may be especially relevant for effector and memory T cell differentiation given the significant level of proliferation that antigen-specific T cells undergo during the effector stage of the immune response. This mechanism should also be kept in mind as we consider the antigen-independent self-renewal that memory T cells undergo during homeostatic proliferation. The observation of active and passive DNA demethylation challenges the previous dogma that, once acquired, DNA methylation programs are persistent [65].
While recent studies have provided new mechanistic insight into how epigenetic programs can be modified, in general, the cell-division transmissible nature of epigenetic programming facilitates the stable maintenance of lineage specific traits. A very recent study has shed new light on the relative longevity of these two major epigenetic mechanisms (histone PTMs vs. DNA methylation) during cellular proliferation. In this study, Bintu et al. used an in vitro model of targeted chromatin modifier recruitment coupled to time-lapse microscopy to provide compelling evidence in line with previous reports, suggesting that HDACs impart short-term epigenetic memory and DNA methyltransferases provide long-lived epigenetic memory [67]. In their study, the authors show that histone de-acetylation mediated by HDAC4 was stable only for a few days, with rapid reacquisition of acetylation observed in the absence of HDAC at the locus [67, 68], while DNMT3-mediated DNA methylation established long-term memory persisting for at least 30 days (presumably this epigenetic program could have been maintained longer). Such maintenance occurs even in the absence of DNMT3 recruitment to the genetic locus. There is also accumulating evidence suggesting that cross-talk between epigenetic modifications, for example the targeted recruitment of DNMTs by histone methyltransferases, which enhance the levels of DNA methylation at these sites, and extend the boundary of the epigenetically silenced locus [69, 70]. These studies underscore the role that various epigenetic mechanisms play in lineage-specific gene expression and cell fate commitment. In the next section we provide a brief overview of how epigenetic mechanisms are thought to contribute to early developmental processes. For a more in-depth review of epigenetics in early development we suggest the following articles [71–74].
Role of Epigenetics in Development
Much of what we have learned about the role of epigenetics in cell fate determination has come from studies of embryonic development. During early embryonic development, the pluripotent potential of zygote-derived cells allows the cells to acquire appropriate gene expression programs, permitting further differentiation into various cell lineages. Prior to the amazing lineage commitment process that a zygotic cell undertakes, its genome remains pliable, lacking a specific lineage program, and maintains epigenetic programs that facilitate expression of only totipotency-associated genes. Upon differentiation, totipotency-associated genes are stably silenced, and lineage-defining genes become expressed. As the cell undergoes lineage specification, the associated permissive and repressive gene expression programs become maintained in every subsequent generation of daughter cells.
Symmetric cell divisions of a zygote ultimately lead to the formation of an inner cell mass (ICM) that later develops into the embryo, yolk sac, allantois, and amnion. The ICM represents the first differentiated lineage during zygotic development. In vitro propagation of ICM cells leads to the derivation of embryonic stem cells (ESCs). Human and mouse embryonic stem cells (hESCs and mESCs) retain their pluripotency, and have been used to investigate the relationship between epigenetic modification and changes in gene expression programs during cell fate commitment [75–78]. Utilizing these models, it has been shown that pluripotency-associated transcription factors, such as Oct4, regulate the expression of H3K9 demethylases, which in turn maintain an open chromatin structure of undifferentiated ESCs [79, 80]. Accumulating evidence suggests that SirT1 mediates deacetylation of histones H4, H3, and H1 [81], and establishes a heterochromatin state, silencing lineage-specific genes in stem cells. Silencing of lineage-defining genes in ESCs is also maintained in part by bivalent chromatin modifications enriched for both permissive and repressive histone methylation. These bivalent histone modifications allow for the repression of developmentally important transcription factors, while simultaneously poising them for re-expression in later developmental stages [82]. Of particular relevance, such bivalent structures have recently been identified during CD8 T cell differentiation [83].
In addition to the histone PTMs discussed above, DNA methylation also contributes to the coordinated silencing of pluripotency-associated genes during ESC differentiation. During the first week of embryonic development, the primordial germ cell genome undergoes significant levels of DNA demethylation. This large-scale DNA demethylation event occurs in two distinct phases. The paternal DNA in the sperm undergoes an active demethylation occurring without cell division), whereas maternal DNA methylation programs undergo passive demethylation subsequent rounds of cell division [84–87]. Such resetting of the methylome allows for the expression of a diverse set of genes that leads to the formation of pluripotent cells. The methylation program is re-acquired following implantation of the embryo and is likely mediated by DNMT3B [88, 89]. Notably, this classic example of epigenetic reprogramming may provide clues to the mechanism for changes in the phenotype and function of effector and memory T cells, in particular changes that occur after antigen clearance.
Specific deletion of DNMTs in ESCs has been performed to further elucidate the biological consequence of DNA methylation reprogramming in cell-fate decisions and commitment. The cumulative results from the various deletion studies support a model whereby DNMT3 enzymes provide a DNA methylation program that can be inherited in daughter cells [90, 91], and while ESCs appear to maintain their transcriptional programs in the absence of DNA methylation, they are significantly impaired in their ability to differentiate into other cell types [91]. Additionally, maintenance of DNA methylation appears to be essential for embryonic development [92, 93] likely owing to the role it plays in maintaining the repressive programs of pluripotency associated genes (including Oct4, Nanog) [94]. Collectively, these studies suggest that DNA methylation is a critical epigenetic mechanism for maintaining cell fate decisions.
Not surprisingly, epigenome reprogramming has also been observed during cellular differentiation at later stages of development. The significance of these reprogramming events has been highlighted in hematopoietic stem cells (HSC), where conditional knock out of the de novo DNA methyltransferase DNMT3A results in increased self-renewal and development of myeloid and lymphoid associate malignancies [95], possibly due to an inability of the cells to acquire silencing program at HSC associated regulatory elements in adult cells. Interestingly though, deletion of the maintenance DNA methyltransferase DNMT1 leads to a dramatic defect in the self-renewal of HSC and skews their development towards myeloid cells, hinting that HSC may by default carry myeloid-specific DNA methylation programs [73, 96, 97]. Further evidence of the role DNA methylation programming plays in HSC lineage commitment has come from studies in TET2 deletion models, where absence of DNA demethylation leads to aberrant increase in proliferation and myeloid differentiation of HSC [98–101]. Together, these studies provide compelling evidence that epigenetic reprogramming events control lineage differentiation by regulating the multipotentency of stem cells and the acquisition of lineage-specific functions in adult cells.
Epigenetic Reprograming during T cell Lineage Development
Similar to its contribution in somatic cell fate commitment during embryonic development, several studies provide evidence that epigenetic programming plays a key role in regulating the later stages of lymphoid cell differentiation. Conditional knock out of DNMT1 has been shown to lead to a decreased proliferative capacity and survival of naïve T lymphocytes [102]. In addition to affecting naïve T cell homeostasis, DNA methylation mediated by DNMTs also maintains the lineage-specific functions of T cells during an immune response. One of the studies to test the hypothesis that epigenetic modifications were associated with acquired changes in CD4 T cell function was performed by Bird et al. The authors demonstrated that under Th1 polarizing conditions, Ifnγ locus becomes epigenetically open, whereas the Il-4 locus remained closed [103]. It was subsequently reported that T-bet mediated removal of HDAC at the IFNγ locus allows for the expression of IFNγ and Th1 differentiation [104]. More recently, Vahedi et al. carried out a genome-wide study of the epigenetic regulation of active lineage-specific enhancer elements and identified STAT proteins as key modulators of chromatin accessibility regulating Th1 and Th2 subset differentiation [105]. Deletion of DNMT1 in CD4 T cells has also been shown to result in the enhanced expression of effector cytokines (IFNγ, IL-2, IL-3, IL-4) by naive CD4 T cells upon activation, compared to WT CD4 T cells. Additionally, deletion of DNMT1 from CD8 T cells leads to an upregulation of these Th2 effector cytokines by CD8 T cells, which normally do not express them [106]. Notably, the aberrant cytokine expression by CD8 T cells in this study was not due to upregulation of transcription factor known to promote the cytokine expression, but rather was attributed to increased accessibility of the genomic loci and DNA demethylation.
In addition to controlling proliferation and function of T cells, study of DNA methylation programming during T cell differentiation has provided mechanistic insight into its role in mediating T cell lineage stability. Demethylation of promoter regions enhances the stability of lineage-specific transcription factor–promoter interaction, and mediates T cell lineage stability as in the case of Th2 and regulatory T cells [107, 108]. Recent studies have demonstrated that acquisition of DNMT3A mediated DNA methylation program at the CD4 locus in DN thymocytes and its maintenance by DNMT1 is necessary for “securing the lineage border” between CD4 and CD8 T cell subsets by stably silencing CD4 expression in cytotoxic T cells [109–111]. In addition, data from these studies suggests that DNA demethylation of the proximal enhancer, possibly mediated by TET enzymes, contributes to the re-expression of the gene in helper T cells [111]. In a separate study, Josefowicz et al. identified that DNMT1 mediated methylation Foxp3 is required for the stable suppression of its expression in CD8 T cells [112]. Collectively, these studies, and many others, have provided compelling evidence that DNA methylation and histone modifications regulate T cell lineage specification. Recently, these concepts have been extended to questions surrounding the adaptive nature of CD8 T cell gene expression programs during acute versus chronic antigen stimulation, and the maintenance of poised effector programs in long-lived memory T cells.
Epigenetics in CD8 T Cell Memory Differentiation
Maintenance of the poised effector state during homeostatic proliferation and an ability to persist in both lymphoid and non-lymphoid tissues is a hallmark of T cell memory, but are these long-lived acquired traits facilitated by epigenetic reprogramming? Initial insight into the role of epigenetic regulation in memory CD8 T cell differentiation and function came from studies focused on examining transcriptional regulation of mouse Ifnγ, GzmB, and Prf during the effector and memory stages of an immune response [113–115]. Using models of in vitro T cell activation [114] and in vivo acute viral infection [113, 115], these studies collectively demonstrated that increased abundance of transcriptional machinery near these genes in antigen-specific effector and memory CD8 T cells was coupled to acquisition and maintenance of an open chromatin state [113–115]. From these data, it was revealed that the “poising” of chromatin in memory CD8 T cells is coupled to the rapid re-expression of effector molecules upon antigen re-exposure. Several studies have been undertaken to identify the epigenetic mechanisms modulating chromatin accessibility. Using in vitro and in vivo experimental systems, Northrop et.al demonstrated that the rapid expression of IFNγ from stimulated murine memory CD8 T cells is facilitated by acquisition of histone hyperacetylation marks at the Ifnγ locus [116, 117]. Similarly, Araki et.al demonstrated that increased expression of the transcription factor EOMES, and its target genes PRF and GZMB in resting and activated human memory CD8 T cells relative to naïve cells is associated with enrichment of histone 3 acetylation at their proximal promoter and exons 1 [118]. From these studies, it was concluded that histone acetylation marks are associated with accessible chromatin at these loci, providing epigenetic memory for heightened recall of effector responses in memory CD8 T cells [116–118]. To further examine the breadth of epigenetic control of poised transcription at effector-related loci in memory cells, Araki et al. used ChipSeq to perform a genome-wide assessment of H3K4me3 and H3K27me3 histone modifications in human naïve and polyclonal memory CD8 T cells [83]. The authors found that permissive histone modifications (H3K4me3) were associated with actively transcribed genes while repressive histone modifications (H3K27me3) were associated with repressed genes. In addition, some non-expressed genes were associated with bivalent histone modifications, having both H3K4me3 and H3K27me3 marks. Interestingly, they also identified permissive histone marks associated with genes that are not expressed [83]. This was the first study to perform a whole-genomic assessment of the epigenetic programming in T cells and paved the way for future studies investigating the histone PTMs that accompany CD8 T cell subset differentiation and the acquisition of effector function during effector and memory cell generation.
Recently, the Turner and Restifo labs each performed a comprehensive genome-wide analysis of histone modifications acquired in various effector and memory CD8 T cell subsets[119, 120]. Using the murine model of acute influenza infection, Russ et al. utilized ChIP-Seq to map histone modifications in antigen-specific naïve, effector, and bulk memory CD8 T cells ex vivo [119]. The authors observed bivalent histone marks (both H3K4me3 and H3K27me3) deposited at transcription factors loci such as T-bet, EOMES, and genes related to cell proliferation and metabolism in naïve CD8 T cells [119]. These bivalent loci rapidly transitioned to a permissive state via the loss of H3K27me3, which was associated with the transcriptional activation of these genes prior to the initiation of cell division. However, it still remains to be determined why some loci have bivalent histone marks while others carry repressive or permissive marks. Unlike many of the canonical effector cytokines, naïve CD8 T cells rapidly upregulate the expression of TNFα upon stimulation, and accordingly, the promoter of TNFα in resting naïve cells is enriched with permissive histone modifications. Likewise, the temporal delay in expression of granzymes and IFNγ in activated CD8 T cells is associated with progressive enrichment of permissive histone modifications [119, 121]. Surprisingly, this permissive state was diminished at the loci of granzymes in resting memory CD8 T cells [119, 121]. From these data, it was concluded that H3K27me3 acts as an epigenetic brake at the loci of lineage-defining transcription factors and effector molecules. While highly informative, these analyses were performed on a pool of memory CD8 T cells raising the possibility that the difference in histone modification in effector and memory CD8 T cells is not wholly generalizable to the distinct subsets of memory cells.
To further explore the possibility that phenotypic and functional differences among the various memory CD8 T cell subsets are coupled to distinct epigenetic programs, Crompton et al. recently performed a genome-wide assessment of H3K4me3 and H3K27me3 modifications for in vitro generated mouse Tcm, Tem, and Tscm CD8 T cells [120]. The authors found that Tcm and Tem were highly enriched for permissive (H3K4me3) histone modifications at effector molecule loci including IFNγ, GZMB, and PRF [120]. Notably though, Tscm possess permissive histone modifications at levels similar to naïve CD8 T cells at many of the examined effector molecule loci [120]. Based on these data, as well as results from their prior studies, the authors proposed that progressive acquisition of permissive histone modifications during naïve to memory differentiation provides support for a unidirectional model memory-to-effector differentiation that results in a linear path for development of Tscm to Tcm to Tem and lastly terminally differentiated effector CD8 T cells (Figure 1.B) [120]. Despite using different strategies to generate effector and memory cells, both the Russ et al. and Crompton et al. studies reached the conclusion that effector molecule loci acquire permissive marks during a effector and memory differentiation, and that histone PTMs modulate both differentiation and effector functions of CD8 T cell memory subsets. While indeed permissive histone modifications at effector molecule loci correlate with their expression in effector cells, several of these modifications did not correlate with the poised state of resting memory CD8 T cells [83, 116–118] suggesting that additional mechanisms are utilized to maintain effector programs in long-lived memory CD8 T cells.
To further explore the molecular mechanisms that mediate stability of the acquired transcriptional programming in effector and memory CD8 T cell subsets, several investigators have turned their attention to DNA methylation programming [122, 123]. Like the above described histone modification studies, initial examination of DNA methylation in effector and memory CD8 T cells was focused on the dynamic change in transcriptional regulation of IFNγ expression. Using bisulfite DNA sequencing, Kersh et.al reported that the Ifnγ gene undergoes dynamic changes in the methylation state in antigen-specific CD8 T cells transitioning through the effector and memory stage of an immune response [124]. While the Ifnγ locus is heavily methylated in naïve CD8 T cells, it was unmethylated in effector cells. Surprisingly, memory CD8 T cells were methylated at this locus but could rapidly lose these methylation marks within several hours following re-stimulation [124]. These data provide evidence that methylation of effector molecule loci is quite pliable enabling rapid re-expression of these molecules in memory CD8 T cells upon re-stimulation. Locus-specific DNA methylation studies provide great insight into the molecular mechanisms responsible for the maintenance of transcriptional programs of targeted genes. However, genome-wide DNA methylation analysis is required for an unbiased identification of the DNA methylation program in CD8 memory subsets and to dissect the role of dynamic DNA methylation programming in the differentiation of effector and memory CD 8 T cells.
In a recent study, Scharer et al. used a genome-wide approach to gain insight in the changing DNA methylation profile accompanying naïve to effector CD8 T cell differentiation [125]. The authors report that during acute lymphocytic choriomeningitis virus (LCMV) infection of mice, many promoters and putative distal enhancer regions of effector genes, including Ifnγ and GzmB, become demethylated during effector CD8 T cell differentiation [125]. Additionally, the study also identified regions in transcription factor binding motifs that were differentially methylated in naïve versus effector cells. Methylation of putative binding sites for the transcription factors TCF4, FOXA1, BCL6 etc. was speculated to silence naïve-specific transcriptional programs in developing effector CD8 T cells. At the same time, demethylation of binding sites for the transcription factors c-JUN, NFATc1, IRF etc. was associated with the generation of effector CD8 T cells [125]. These data provide evidence that changes in epigenetic programming may modulate the ability of transcriptional machinery to bind potential regulatory elements linked to CD8 T cell function and fate decision [125]. Interestingly, a subset of promoters contained both methylated and demethylated CpGs in effector cells, raising the possibility that these regions may contain both negative and positive regulatory elements that lead to differential gene expression depending on the lineage. Similar to histone modification analyses, this study also identified functional classes of genes that could be coordinately regulated by DNA methylation [125]. However, this study did not distinguish between the different memory CD8 T cell subsets. Therefore, future studies are required to determine the contribution of DNA methylation in memory CD8 T cell subset function and differentiation. Additionally, given that DNA methylation can reinforce the stability of acquired transcriptional programs [122, 123], future studies are needed to address the long-term stability of the acquired methylation programs in memory T cells. Taken together, histone modification and DNA methylation profiling studies provide evidence that epigenetic programs can be adapted in response to TCR signaling [118, 124], and demonstrate that the rapid recall response manifested by memory CD8 T cells is coupled to epigenetic programs that create a poised transcriptional state of the effector molecule loci.
Epigenetic Plasticity and Transcriptional Reprogramming in CD8 T Cells
Plasticity of naïve CD8 T cells enables them to differentiate into a diverse pool of effector CD8 T cells. However, duration and strength of the antigenic stimuli as well as the level of inflammation reduces the developmental potential of antigen-specific CD8 T cells (models discussed in figures 1B and C). Studies have shown that this reduced developmental potential is accompanied by significant and stable changes to the transcriptional program of these antigen-experienced cells. One is therefore inclined to ask: are these acquired, subset-specific transcriptional programs fixed? In other words, is the effector differentiation pathway a one-way street? Or instead, are these transcriptional programs pliable, with the possibility of further (antigen-dependent or independent) modifications? Gene expression patterns in effector and memory cells suggest that certain lineage defining genes (e.g. Lck in CD8 T cells) remain upregulated in effector and memory CD8 T cells, following the activation of a naïve cell. Genes with similar linear expression profiles would fall under the “off-on” or “on-off” category (figure 3.A, right panels), and support the fixed nature of the transcriptional program. However, several other genes are differentially expressed during the CD8 T cell effector-memory response and fall under the “off-on-off” (e.g. PDCD-1, IFNγ) or the “on-off-on” (e.g. IL-7Ra, Bcl-2) category (figure 3.A, left panels), indicating plasticity in the acquired transcriptional program [126]. In order to uncover the true nature of these acquired transcriptional programs, it is necessary to investigate mechanisms that regulate transcriptional plasticity in naïve and memory T cell populations.
There are many clear examples of epigenetic programs acquired and maintained in effector and memory CD8 T cell. However, is there any evidence that these programs regulate transcriptional plasticity, or the lack thereof, in effector and memory cells? Absence of any epigenetic re-programming during the antigen independent stage of the immune response would support a unidirectional model of memory differentiation where gene expression and memory cell fate gets “locked in” during antigen driven phase of the immune response (Figure 1.B and 3.A, right panels). However, epigenetic re-programming during the antigen independent stage of the immune response would be consistent with a bi-directional model of memory differentiation and gene expression (Figure 1.C) and could provide a mechanistic explanation of the plasticity in gene expression pattern of the differentially expressed genes (off-on-off or on-off-on) (Figure 3.A, left panels). Indeed, studies have revealed that transcriptional plasticity observed in the case of effector genes is in part regulated by epigenetic modification. Kersh et.al demonstrated that epigenetic reprogramming observed for the off-on-off expression of the effector molecule IFNγ correlates with the pliable nature of Ifnγ locus methylation in naïve, effector, and memory CD8 T (Figure 3.A) [124]. While methylated in naïve CD8 T cells, the locus becomes de-methylated during the effector stage of the immune response and is methylated in resting memory CD8 T cells. Following re-stimulation, the Ifnγ locus can rapidly undergo demethylation to allow for the rapid transition to an epigenetically permissive state [124]. Together with permissive histone marks, the demethylation of the locus may allow for the rapid re-expression of IFNγ upon re-stimulation [113, 115–120, 124].
While indeed memory CD8 T cells generated during acute antigen exposure retain developmental plasticity, such plasticity is lost with increasing levels of TCR signaling and duration of exposure as occurs during chronic viral infection. Many chronic viral infections and cancers initially elicit a robust CD8 T cell response, but sustained stimulation to the antigen-specific T cells drives them to a nonfunctional state of differentiation often referred to as T cell exhaustion. Development of T cell exhaustion specifically reduces the proliferative capacity of the cells as well as restricts their ability to recall effector cytokine expression. Acquisition of an exhaustion differentiation program in T cells occurs progressively during the prolonged exposure to antigen and is coincident with upregulation of inhibitory receptors [127, 128]. Recent evidence suggests that exhausted CD8 T cells maintain aspects of their non-functional state even in the absence of any antigenic stimulation [129, 130]. Initial assessment of the molecular mechanisms regulating the functional impairment of exhausted T cells has come from comparing the transcriptional profiles between functional memory T cells and exhausted CD8 T cells [127]. This study demonstrated that exhausted CD8 T cells have a gene expression profile distinct from naïve, effector, and functional memory CD8 T cells. Thus, the reported stability in the exhausted state of differentiation is likely coupled to stable changes in gene regulation.
Recent work by Youngblood et.al. has clearly shown that the phenomenon of epigenetic plasticity regulating the gene expression is limited not only to effector molecule loci, but is also observed at other genes, including the immune-regulatory PDCD1 gene, which encodes for the programmed cell death protein-1 (PD-1) [131, 132]. It is established that PD-1 inhibitory receptor is upregulated by naïve cells shortly after activation and is down-regulated following the clearance of antigen. However, during chronic infection, antigen-specific CD8 T cells persistently express PD-1, which contributes to their progressive functional impairment i.e. exhaustion. Using models of acute and chronic viral infection in mice, it was shown that the PD-1 locus undergoes transient demethylation during the effector phase of a T cell response to an acute infection. Following viral control, the PD-1 locus progressively reacquired DNA methylation during the effector-to-memory stage of the immune response (Figure 3.A). These observed changes in the methylation status of the PD-1 locus correlated with the downregulated expression of PD-1 on antigen-specific CD8 T cells [131, 132]. However, during chronic infection the PD-1 locus remained demethylated in exhausted CD8 T cells even after the virus was cleared and PD-1 expression was reduced [131, 132]. Additionally, the authors reported that maintenance of the demethylated state of the PD-1 locus was coupled to the transcriptionally poised state of the gene, which facilitated the rapid re-expression of PD-1 by LCMV–specific CD8 T cells from chronically infected mice compared with memory CD8 T cells from acutely infected mice [131]. Although changes in IFNγ and PD-1 expression are critical for effector function and T-cell exhaustion, many other genes related to effector and memory function also undergo dynamic changes in transcriptional regulation. Therefore, it will be important for future studies to address the plasticity of DNA methylation programming in the various T-cell subsets generated under conditions of acute and chronic antigen exposure.
As a follow up to the prior analysis of PD-1 methylation status in exhausted CD8 T cell in mice, Youngblood et.al examined the methylation status of the PD-1 locus in HIV-specific CD8 T cells. Surprisingly, the authors observed that in HIV elite controllers and infected individuals who had controlled viral load following highly active antiretroviral treatment, the PD-1 locus remained demethylated decades after no antigen was detectable [132]. Interestingly though, in contrast to HIV elite controllers, where prolonged exposure to antigen at some point may have programmed the PD-1 locus to remain demethylated, recent prime–boost vaccination studies have demonstrated that secondary and tertiary memory CD8 T cells generated in response to sequential acute antigen exposure are fully functional and do not express PD-1 despite repeated antigen exposure [133, 134]. This suggests that repeated antigen exposure by itself, is not sufficient to fully erode memory CD8 T plasticity [133, 134]. It will be of great interest for future studies to determine if regulators of epigenetic plasticity contribute to the reduced functionality of exhausted CD8 T cells and maintained functionality of memory CD8 T cells generated from prime-boost vaccination.
Implications of Memory Generation from Epigenetic Reprogramming
While there is still much to be learned about the mechanisms governing memory T cell plasticity, several observations about their acquired properties may help guide our immediate and long-term questions. As mentioned earlier, antigen-independent homeostatic proliferation is a unique feature of memory CD8 T cells and many acquired properties are maintained during this self-renewal. While many of the poised programs appear to persist over long periods of time, one can speculate that such homeostatic proliferation also offers the cells an opportunity to erase established repressive programs through passive mechanisms (demethylation by blocking maintenance methylation) (Figure 3.B). Evidence of such mechanisms regulating the reprogramming of developed T cells may be very important for enhancing the rejuvenation of exhausted T cells and may also provide support for a bi-directional model of memory T cell differentiation. Several studies addressing the stability of memory T cell subset fates have found evidence for inter-conversion between memory subsets. A classic example of this inter-conversion is the upregulation of CD62L on Tem cells as they develop into Tcm cells [46–50]. It remains to be determined whether the stability of the transcriptional programs in homeostatically proliferating memory CD8 T cells or their adaptability during inter-conversion (Figure 3.B) is indeed associated with the stability or plasticity of epigenetic programs. It has been suggest that maintenance of CD8 T cell exhaustion; even in the absence of antigen is coupled to the loss of epigenetic plasticity [131, 132], which poses a potential challenge for sustaining long-lived immunity after use of immunotherapies aimed at rejuvenating exhausted CD8 T cells. If developmental plasticity of functional memory T cells is indeed mediated by epigenetic modifications, can that knowledge be applied to reprogram exhausted T cells? Further more, could these epigenetically reprogrammed CD8 T cells acquire new or alter their functions? Exploration of the mechanisms controlling dynamic epigenetic programming will broaden our understanding of the molecular basis of memory CD8 T cell differentiation, and aid in rational vaccine design and advancement of immunotherapy strategies.
Acknowledgments
We thank Dr. Scott Hale (University of Utah) for his critical review of this manuscript. This work is supported by the National Institutes of Health (NIH) grant 1R01AI114442-01A1 (to B.A.Y), and funds from American Lebanese Syrian Associated Charities (ALSAC) (to B.A.Y).
References
- 1.Alanio C, Lemaitre F, Law HK, Hasan M, Albert ML. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood. 2010;115:3718–3725. doi: 10.1182/blood-2009-10-251124. [DOI] [PubMed] [Google Scholar]
- 2.Coulie PG, Karanikas V, Lurquin C, Colau D, Connerotte T, Hanagiri T, Van Pel A, Lucas S, Godelaine D, Lonchay C, Marchand M, Van Baren N, Boon T. Cytolytic T-cell responses of cancer patients vaccinated with a MAGE antigen. Immunol Rev. 2002;188:33–42. doi: 10.1034/j.1600-065x.2002.18804.x. [DOI] [PubMed] [Google Scholar]
- 3.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 4.Letsch A, Keilholz U, Schadendorf D, Nagorsen D, Schmittel A, Thiel E, Scheibenbogen C. High frequencies of circulating melanoma-reactive CD8+ T cells in patients with advanced melanoma. Int J Cancer. 2000;87:659–664. [PubMed] [Google Scholar]
- 5.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28:859–869. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotturi MF, Scott I, Wolfe T, Peters B, Sidney J, Cheroutre H, von Herrath MG, Buchmeier MJ, Grey H, Sette A. Naive precursor frequencies and MHC binding rather than the degree of epitope diversity shape CD8+ T cell immunodominance. J Immunol. 2008;181:2124–2133. doi: 10.4049/jimmunol.181.3.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, Mathis D, Butz EA. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. 2002;195:1515–1522. doi: 10.1084/jem.20020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubinstein MP, Lind NA, Purton JF, Filippou P, Best JA, McGhee PA, Surh CD, Goldrath AW. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood. 2008;112:3704–3712. doi: 10.1182/blood-2008-06-160945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 12.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 13.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. The Journal of experimental medicine. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plumlee CR, Sheridan BS, Cicek BB, Lefrancois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity. 2013;39:347–356. doi: 10.1016/j.immuni.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, Zehn D, Bevan MJ, Schepers K, Schumacher TN. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. 2010;207:1235–1246. doi: 10.1084/jem.20091175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, de Boer RJ, Schumacher TN. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340:635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 18.Schepers K, Swart E, van Heijst JW, Gerlach C, Castrucci M, Sie D, Heimerikx M, Velds A, Kerkhoven RM, Arens R, Schumacher TN. Dissecting T cell lineage relationships by cellular barcoding. J Exp Med. 2008;205:2309–2318. doi: 10.1084/jem.20072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, Drexler I, Hofer T, Riddell SR, Busch DH. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014;41:116–126. doi: 10.1016/j.immuni.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 21.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 22.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–4266. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 25.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107:17872–17879. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, Farber DL. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014;7:501–510. doi: 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–467. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. 2006;176:2079–2083. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 29.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, Carter JB, Fisher DC, Kupper TS. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4:117ra117. doi: 10.1126/scitranslmed.3003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 33.Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39:687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–1305. doi: 10.1038/nm1326. [DOI] [PubMed] [Google Scholar]
- 36.Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175:5895–5903. doi: 10.4049/jimmunol.175.9.5895. [DOI] [PubMed] [Google Scholar]
- 37.Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- 38.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. 2013;25:556–563. doi: 10.1016/j.coi.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong WF, Kohu K, Chiba T, Sato T, Satake M. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology. 2011;132:157–164. doi: 10.1111/j.1365-2567.2010.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naito T, Tanaka H, Naoe Y, Taniuchi I. Transcriptional control of T-cell development. Int Immunol. 2011;23:661–668. doi: 10.1093/intimm/dxr078. [DOI] [PubMed] [Google Scholar]
- 44.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huster KM, Koffler M, Stemberger C, Schiemann M, Wagner H, Busch DH. Unidirectional development of CD8+ central memory T cells into protective Listeria-specific effector memory T cells. Eur J Immunol. 2006;36:1453–1464. doi: 10.1002/eji.200635874. [DOI] [PubMed] [Google Scholar]
- 47.Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marzo AL, Yagita H, Lefrancois L. Cutting edge: migration to nonlymphoid tissues results in functional conversion of central to effector memory CD8 T cells. J Immunol. 2007;179:36–40. doi: 10.4049/jimmunol.179.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179:6704–6714. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 50.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 51.Bai L, Morozov AV. Gene regulation by nucleosome positioning. Trends Genet. 2010;26:476–483. doi: 10.1016/j.tig.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bell O, Tiwari VK, Thoma NH, Schubeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 54.Cairns BR. Chromatin remodeling complexes: strength in diversity, precision through specialization. Curr Opin Genet Dev. 2005;15:185–190. doi: 10.1016/j.gde.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Littman DR, Crabtree GR. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature. 2002;418:195–199. doi: 10.1038/nature00876. [DOI] [PubMed] [Google Scholar]
- 56.Chi T. A BAF-centred view of the immune system. Nat Rev Immunol. 2004;4:965–977. doi: 10.1038/nri1501. [DOI] [PubMed] [Google Scholar]
- 57.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 58.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 59.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 63.Verdone L, Agricola E, Caserta M, Di Mauro E. Histone acetylation in gene regulation. Brief Funct Genomic Proteomic. 2006;5:209–221. doi: 10.1093/bfgp/ell028. [DOI] [PubMed] [Google Scholar]
- 64.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 65.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, Elowitz MB. Dynamics of epigenetic regulation at the single-cell level. Science. 2016;351:720–724. doi: 10.1126/science.aab2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katan-Khaykovich Y, Struhl K. Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002;16:743–752. doi: 10.1101/gad.967302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288:34287–34294. doi: 10.1074/jbc.R113.512517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 71.Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015;15:7–17. doi: 10.1038/nri3777. [DOI] [PubMed] [Google Scholar]
- 72.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 74.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 75.Chen YF, Tseng CY, Wang HW, Kuo HC, Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology. 2012;55:1193–1203. doi: 10.1002/hep.24790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei H, Tan G, Manasi, Qiu S, Kong G, Yong P, Koh C, Ooi TH, Lim SY, Wong P, Gan SU, Shim W. One-step derivation of cardiomyocytes and mesenchymal stem cells from human pluripotent stem cells. Stem Cell Res. 2012;9:87–100. doi: 10.1016/j.scr.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 78.Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, Zhang X, Coyne M, Fostel JL, Holmes L, Meldrim J, Guttman M, Epstein C, Park H, Kohlbacher O, Rinn J, Gnirke A, Lander ES, Bernstein BE, Meissner A. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 81.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 82.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 83.Araki Y, Wang Z, Zang C, Wood WH, 3rd, Schones D, Cui K, Roh TY, Lhotsky B, Wersto RP, Peng W, Becker KG, Zhao K, Weng NP. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet. 2005;14(Spec No 1):R47–58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- 85.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 86.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241:172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 87.O’Neill C. The epigenetics of embryo development. Animal Frontiers. 2015;5 [Google Scholar]
- 88.Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Li G, Ci W, Li W, Zhou Q, Aluru N, Tang F, He C, Huang X, Liu J. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157:979–991. doi: 10.1016/j.cell.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hirasawa R, Sasaki H. Dynamic transition of Dnmt3b expression in mouse pre- and early post-implantation embryos. Gene Expr Patterns. 2009;9:27–30. doi: 10.1016/j.gep.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 90.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 93.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 94.Nakanishi MO, Hayakawa K, Nakabayashi K, Hata K, Shiota K, Tanaka S. Trophoblast-specific DNA methylation occurs after the segregation of the trophectoderm and inner cell mass in the mouse periimplantation embryo. Epigenetics. 2012;7:173–182. doi: 10.4161/epi.7.2.18962. [DOI] [PubMed] [Google Scholar]
- 95.Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, Goodell MA. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125:629–638. doi: 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, Andrade-Navarro MA, Jacobsen SE, Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 98.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, Melnick A, Godley LA, Aifantis I, Levine RL. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, Godley L, Opolon P, Tilly H, Solary E, Duffourd Y, Dessen P, Merle-Beral H, Nguyen-Khac F, Fontenay M, Vainchenker W, Bastard C, Mercher T, Bernard OA. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 100.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 103.Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA, Sider JR, Gajewski TF, Wang CR, Reiner SL. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 104.Chang S, Collins PL, Aune TM. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol. 2008;181:8372–8381. doi: 10.4049/jimmunol.181.12.8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O’Shea JJ. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol. 2004;173:4402–4406. doi: 10.4049/jimmunol.173.7.4402. [DOI] [PubMed] [Google Scholar]
- 107.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murayama A, Sakura K, Nakama M, Yasuzawa-Tanaka K, Fujita E, Tateishi Y, Wang Y, Ushijima T, Baba T, Shibuya K, Shibuya A, Kawabe Y, Yanagisawa J. A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. EMBO J. 2006;25:1081–1092. doi: 10.1038/sj.emboj.7601012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chappell C, Beard C, Altman J, Jaenisch R, Jacob J. DNA methylation by DNA methyltransferase 1 is critical for effector CD8 T cell expansion. J Immunol. 2006;176:4562–4572. doi: 10.4049/jimmunol.176.8.4562. [DOI] [PubMed] [Google Scholar]
- 110.Feng Y, Rudensky AY. DNA methylation secures CD4(+) and CD8(+) T cell lineage borders. Nat Immunol. 2015;16:681–683. doi: 10.1038/ni.3207. [DOI] [PubMed] [Google Scholar]
- 111.Sellars M, Huh JR, Day K, Issuree PD, Galan C, Gobeil S, Absher D, Green MR, Littman DR. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat Immunol. 2015;16:746–754. doi: 10.1038/ni.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol. 2009;182:6648–6652. doi: 10.4049/jimmunol.0803320. [DOI] [PubMed] [Google Scholar]
- 113.Denton AE, Russ BE, Doherty PC, Rao S, Turner SJ. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc Natl Acad Sci U S A. 2011;108:15306–15311. doi: 10.1073/pnas.1112520108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Juelich T, Sutcliffe EL, Denton A, He Y, Doherty PC, Parish CR, Turner SJ, Tremethick DJ, Rao S. Interplay between chromatin remodeling and epigenetic changes during lineage-specific commitment to granzyme B expression. J Immunol. 2009;183:7063–7072. doi: 10.4049/jimmunol.0901522. [DOI] [PubMed] [Google Scholar]
- 115.Zediak VP, Johnnidis JB, Wherry EJ, Berger SL. Cutting edge: persistently open chromatin at effector gene loci in resting memory CD8+ T cells independent of transcriptional status. J Immunol. 2011;186:2705–2709. doi: 10.4049/jimmunol.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006;177:1062–1069. doi: 10.4049/jimmunol.177.2.1062. [DOI] [PubMed] [Google Scholar]
- 117.Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181:865–868. doi: 10.4049/jimmunol.181.2.865. [DOI] [PubMed] [Google Scholar]
- 118.Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, Stock AT, Croom HA, Cullen JG, Nguyen ML, Rowe S, Olson MR, Finkelstein DB, Kelso A, Thomas PG, Speed TP, Rao S, Turner SJ. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity. 2014;41:853–865. doi: 10.1016/j.immuni.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, Patel SJ, Sukumar M, Palmer DC, Peng W, Wang E, Marincola FM, Klebanoff CA, Zhao K, Tsang JS, Gattinoni L, Restifo NP. Lineage relationship of CD8 T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol. 2015 doi: 10.1038/cmi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nguyen ML, Hatton L, Li J, Olshansky M, Kelso A, Russ BE, Turner SJ. Dynamic regulation of permissive histone modifications and GATA3 binding underpin acquisition of granzyme A expression by virus-specific CD8(+) T cells. Eur J Immunol. 2016;46:307–318. doi: 10.1002/eji.201545875. [DOI] [PubMed] [Google Scholar]
- 122.Youngblood B, Hale JS, Ahmed R. Memory CD8 T cell transcriptional plasticity. F1000Prime Rep. 2015;7:38. doi: 10.12703/P7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunol Rev. 2014;261:157–168. doi: 10.1111/imr.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, Ahmed R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol. 2006;176:4083–4093. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- 125.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419–3429. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology. 2013;139:277–284. doi: 10.1111/imm.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 128.Youngblood B, Wherry EJ, Ahmed R. Acquired transcriptional programming in functional and exhausted virus-specific CD8 T cells. Curr Opin HIV AIDS. 2012;7:50–57. doi: 10.1097/COH.0b013e32834ddcf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86:8161–8170. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, Zehn D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14:603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- 131.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, Ahmed R. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400–412. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, Bordi R, Procopio FA, Miura T, Allen TM, Sidney J, Sette A, Walker BD, Ahmed R, Boss JM, Sekaly RP, Kaufmann DE. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013;191:540–544. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]