

Abstract
A novel metabolite identification strategy is presented for the combined NMR/MS analysis of complex metabolite mixtures. The approach first identifies metabolite candidates from 1D or 2D NMR spectra by NMR database query, which is followed by the determination of the masses (m/z) of their possible ions, adducts, fragments, and characteristic isotope distributions. The expected m/z ratios are then compared with the MS1 spectrum for the direct assignment of those signals of the mass spectrum that contain information about the same metabolites as the NMR spectra. In this way, the mass spectrum can be assigned with very high confidence and it provides at the same time validation of the NMR-derived metabolites. The method was first demonstrated on a model mixture and it was then applied to human urine collected from a pool of healthy individuals. A number of metabolites could be detected that had not been reported previously further extending the list of known urine metabolites. The new analysis approach, which is termed NMR/MS Translator, is fully automated and takes only few seconds on a computer workstation. The NMR/MS Translator synergistically uses the power of NMR and MS enhancing the accuracy and efficiency of the identification of those metabolites compiled in databases.
Keywords: metabolomics/metabonomics, complex mixture analysis, NMR and MS hybrid approach, human urine metabolome
Graphical Abstract
1. INTRODUCTION
Profiling of metabolites in the context of metabolomics and metabonomics is a promising approach with many powerful applications in basic biological, biomedical, environmental, agricultural, and nutritional research.1,2,3,4 Mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy are the two leading analytical methods in metabolomics.5 This is because of their high-resolution power allowing detection of hundreds of signals from individual metabolites without the need of extensive sample purification or hyphenation.6 Despite of having this common property, metabolomics studies that integrate NMR- and MS-based analysis are still rare. Some approaches that make synergistic use of MS and NMR use specially designed hardware, such as LC-MS-NMR,7 chemical methods,8 or statistical methods.9,10,11 However, for the majority of applications that combine the two methods, they are applied essentially independently from each other. Only at the end, the metabolites identified by each method are compared with each other with the goal to increase the total number of detected metabolites in complex mixtures, since some metabolites show up only in one of either technique.12,13
NMR spectroscopy is a powerful tool for the independent identification and elucidation of the structure of metabolites.14 Still, the knowledge of the chemical formula of a molecule based on accurate mass determination by mass spectrometry before starting the structure elucidation process by NMR can significantly enhance both the accuracy and efficiency of this process. Such combinations of NMR and MS have been employed in the context of organic chemical synthesis to elucidate the structure of purified, i.e. isolated, reaction products and by natural product researchers to determine the structures of isolated compounds.15 So far, such combined MS/NMR methods could not be commonly applied to structure elucidation of unknown metabolites, because the purification of unknown metabolites from their complex matrix can be a challenge. Recently, we introduced the SUMMIT MS/NMR approach, which specifically addresses this challenge by finding the structures of unknown metabolites in complex mixtures without requiring a pre-purification step.16 The approach uses the accurate mass information provided by MS to generate all structures consistent with the chemical formulas. The prediction of the NMR spectra of all structures and comparison with the experimental NMR spectra permits the accurate identification of the structures present in the sample of interest.
Querying of metabolomics databases represents the most efficient approach to the analysis of complex metabolite mixtures. Over the last decade both NMR and MS metabolomics databases increased the total number of metabolites available in their libraries along with the accuracy of their querying platforms leading to greatly improved querying results.17,18,19,20,21 A general goal is to expand metabolite libraries with newly discovered metabolites so that database querying can identify an evergrowing number of metabolites observed in real-world applications.
Unfortunately, there has been a lack of strategies to rapidly identify known metabolites detected both in NMR and MS spectra. Traditionally, known metabolites are identified in the NMR spectrum by using NMR metabolomics databases, whereas known metabolites in the MS spectrum are identified via MS metabolomics databases. Metabolites, along with their names, identified by both methods are then compared with each other. This approach is labor-intensive and does not fully capitalize on the power of these two analytical approaches even when the two data sets stem from the same sample.
Here, we introduce a metabolite identification approach, which relies on a combined NMR/MS strategy for the analysis of catalogued, i.e. known, metabolites. It integrates the power of NMR and MS for the accurate identification of metabolites in a new and more effective way. We demonstrate the new approach, which we term “NMR/MS Translator”, for a model mixture and then apply it to human urine as an example of a highly complex metabolite mixture.
2. EXPERIMENTAL SECTION
2.1. Sample preparation
A model mixture was prepared by dissolving the following 26 metabolites: lysine, isoleucine, histidine, alanine, glutamine, ornithine, carnitine, shikimate, arginine, glutamate, fructose, ribose, galactose, glucose, lactose, sucrose, inosine, threonine, valine, leucine, proline, cysteine, methionine, adenosine, serine, and citrulline. For NMR analysis, the final concentration of each metabolite in the model mixture was 1 mM in 600 μL D2O with 20 mM phosphate buffer. Pooled normal human urine was obtained from Innovative Research, Inc. 1 mL lyophilized urine was dissolved in 600 uL D2O with 20 mM phosphate buffer and 0.1 mM DSS. Both samples were transferred to a 5-mm tube for NMR experiments.
For MS1 analysis, 1 mM model mixture was dissolved in 200 μL H2O of which 10 μL was diluted 10-fold by 50%/50% (v/v) ACN/H2O solution with 0.1% formic acid. For urine, the sample was diluted 100-fold by 50%/50% (v/v) ACN/H2O solution with 0.1% formic acid. The resulting solutions were both centrifuged at 13,000 rpm and 4 °C for 5 min and the supernatants were used for direct infusion MS1.
2.2 NMR experiments and processing
2D 13C-1H HSQC22 spectra of the 26-compound model mixture was collected with N1=512 and N2=800 complex points using a cryogenically cooled probe at 600 MHz proton frequency at 298 K. The spectral width along the indirect and the direct dimensions were 24143.564 Hz and 6009.615 Hz, respectively. The number of scans per t1 increment was 16. The transmitter frequency offsets were 80 ppm in the 13C dimension and 4.7 in the 1H dimension. The total measurement time was 5 hours.
2D 13C-1H HSQC and 2D 13C-1H HSQC-TOCSY23 spectra of human urine were collected with N1=512 and N2=1024 complex points. The spectral width along the indirect and the direct dimensions were 32193.432 Hz and 8012.820 Hz, respectively. The numbers of scans per t1 increment were 16 for 13C-1H HSQC and 32 for 13C-1H HSQC-TOCSY. The transmitter frequency offset were 80 ppm in the 13C dimension and 4.7 in the 1H dimension. The total measurement time was 10 hours for 13C-1H HSQC and 20 hours for and 13C-1H HSQC-TOCSY. 2D 1H-1H TOCSY24 spectrum of the same sample was collected with N1=256 and N2=1024 complex points. The spectral width along both dimensions was 8012.820 Hz. The number of scans per t1 increment was 16. The transmitter frequency offset was 4.7 ppm in both of the 1H dimensions. The total measurement time was 4 hours. The TOCSY mixing time was set to 90 ms for both 1H-1H TOCSY and 13C-1H HSQC-TOCSY. All spectra were collected using a cryogenically cooled probe at 800 MHz proton frequency at 298 K. All data were zero-filled, Fourier transformed, and phase and baseline corrected using NMRPipe.25
2.3. Mass spectrometry experiments and processing
Direct infusion studies were conducted in positive ion mode and negative ion mode detection on a Bruker maXis 4G ESI Q-TOF instrument (electrospray ionization quadrupole time-of-flight mass spectrometer). The instrument was calibrated with Agilent Low-Concentration Tuning Mix (Part No. G1969-85000) before sample analysis achieving a mass accuracy of ± 5 ppm. The samples were directly infused to the ESI source at 2 μL/min. The settings for the Q-TOF mass spectrometer were as follows: capillary voltage, 4500 V (for positive ion mode) and −4000V (for negative ion mode); end plate offset, −500 V; drying gas flow (N2), 4.0 L/min; drying gas temperature, 200 °C; and nebulizer gas (N2), 0.5 bar.
3.3. Reconstruction of MS1 spectrum from 13C-1H HSQC
The 2D 13C-1H HSQC spectra of the model mixture and human urine were queried using the COLMAR 13C-1H HSQC database26 with default query parameters (0.03 ppm 1H and 0.3 ppm 13C chemical shift cutoff values). Then for each of the identified metabolites, monoisotopic m/z ratios of its commonly observed ions, its fragments, and adduct features were calculated and used to assign lines in the experimental MS1 spectrum within an m/z error < 30 ppm and an intensity threshold corresponding to > 1000 ion counts. The following ion features were considered in this study: [M+H]+, [M+NH4]+, [M+Na]+, [M+H-2H2O]+, [M+H-H2O]+, [M+K]+, [M+ACN+H]+, [M+ACN+Na]+, [M+2Na-H]+, [M+2H]2+, [M+3H]3+, [M+H+Na]2+, [M+2H+Na]3+, [M+2Na]2+, [M+2Na+H]3+, [M+Li]+, [M+CH3OH+H]+, [M-H]−, [M-H2O-H]−, [M+Na-2H]−, [M+Cl]−, [M+K-2H]−, [M+FA-H]−, [M-2H]2−, [M-3H]3−, [M+CH3COO]−, [M+F]−.
Since the first isotopic peak of each metabolite is dominated by partially 13C-labeled molecules, these peaks were also assigned. However, this time the intensity threshold value was dynamically adapted based on the experimental intensity of its monoisotopic peak (ion-countmono) and the number of carbons in the metabolite (NC). Since natural abundance of 13C is 1.1%, the theoretical intensity of the isotope peak (ion countiso) is approximately NC*0.011*ion-countmono. Isotopic m/z ratios were assigned in the experimental MS1 spectra within an m/z error < 30 ppm and intensity threshold value range between 0.25*ion-countiso and 4*ion-countiso. The m/z error interval of 30 ppm adopted in this study has been widely accepted in the literature as a reasonable maximum mass error tolerance for Q-TOF instruments.20
3. RESULTS
3.1. Workflow of the NMR/MS Translator
The general strategy of NMR/MS Translator relies (i) on the accurate identification of metabolites by NMR metabolomics databases followed (ii) by automated assignments of all commonly observed ions, adducts, fragments, and isotope features of the identified metabolites in the experimental MS1 spectrum of the same sample. The 13C-1H HSQC version of the approach is schematically shown in Figure 1. A sample is first divided into two parts. The first part is used to acquire a 13C-1H HSQC NMR spectrum,22 while the rest of the material is used to acquire a MS1 spectrum (without fragmentation, i.e. no MS/MS spectra) in positive and negative ion mode each. Once the 13C-1H chemical shifts of all detected cross-peaks are peak-picked, the cross-peak list is queried against the COLMAR 13C-1H HSQC web server,26 which returns a list of identified metabolites. For every identified metabolite, the m/z ratios of its most likely observable ions ([M+H]+, [M-H]− …), adducts ([M+Na]+, [M+K]+, [M+Cl]− …), fragments ([M+H-H2O]+,[M-H2O-H]−) and 13C isotope features are calculated and assigned to the corresponding signals in the experimental MS1 spectra within the mass error and ion count threshold value. In this way, metabolites are identified from NMR spectra using NMR metabolomics databases and assigned to their most probable m/z ratios in the MS1 spectra. After all NMR and MS data have been collected, NMR/MS Translator takes only few seconds on a computer workstation.
Figure 1.
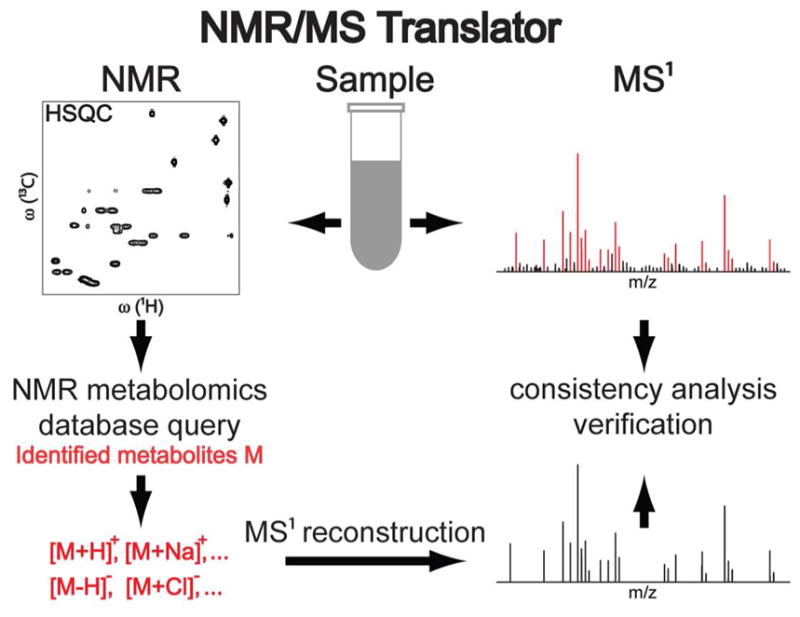
Schematic representation of the proposed NMR/MS Translator for the rapid identification of metabolites observed both in 2D 13C-1H HSQC NMR and MS1 spectra of the same complex metabolite mixture. Metabolites are identified by querying 13C-1H chemical shifts of the metabolites in COLMAR 13C-1H HSQC metabolomics database. For each identified metabolite, the MS1 m/z ratios of their most likely observable ions (including adducts, fragments, and 13C isotope features) are automatically calculated and simultaneously assigned to their signals in the experimental MS1 spectrum within a reasonable mass error and ion count threshold value.
3.2. Application to Model Mixture
The NMR/MS Translator is first demonstrated on the 26-compound model mixture consisting of metabolites commonly encountered in nature. 2D 13C-1H HSQC spectrum of the model mixture (Figure S1) was collected on a 600 MHz solution-state NMR spectrometer. 13C-1H HSQC cross-peaks of the mixture were manually picked and compiled in a cross-peak list. Direct infusion MS1 spectra (in positive and negative mode) of the model mixture were collected by using an ESI Q-TOF mass spectrometer. All detected m/z ratios were uploaded and interfaced with the COLMAR 13C-1H HSQC database. Upon querying of the 13C-1H HSQC peak list against the COLMAR 13C-1H HSQC database, the web server successfully identified all 26 metabolites, calculated their m/z ratios and assigned them to experimental MS1 spectra. For each of the 26 metabolites at least one consistent m/z ratio was observed in the experimental MS1 spectra. The detailed list of the different MS1 metabolite features assigned by the NMR/MS Translator is contained in Table S1. In the MS1 spectra, the majority of metabolites appeared as multiple different ion, adduct, or fragment features, which would make the manual performance of this procedure time-consuming. Figure 2 shows the agreement of experimental MS1 spectra with its NMR-based reconstruction. The NMR-based reconstruction of mass spectra by the NMR/MS Translator is for assignment purposes only and, hence, the intensities in the NMR-reconstructed MS1 are taken from the experimental MS1 spectrum. Overall, the agreement is excellent with almost all major peaks in the MS1 spectra observed either in positive or negative ion mode explained satisfactorily by the NMR-identified compounds.
Figure 2.

Application of the NMR/MS Translator to MS1 spectra (recorded in positive and negative ion mode) of a 26-compound model mixture. All of the 26 metabolites are positively identified by querying the 2D 13C-1H HSQC spectrum of the mixture against the COLMAR 13C-1H HSQC database. Automated reconstruction of the mass spectrum demonstrated that each of the 26 metabolites has at least one consistent m/z ratio in the experimental MS1 spectra. The NMR reconstruction is purely for assignment purposes and, hence, the ion count in the NMR-based reconstructed MS1 is taken from the experimental MS1 spectra.
3.3. Application to Healthy Human Urine
Next the NMR/MS Translator was applied to human urine collected from a pool healthy individuals. 2D 13C-1H HSQC (Figure S2) and direct infusion MS1 spectra (in positive and negative modes) of the model mixture were collected by using a 800 MHz solution-state NMR spectrometer and an ESI Q-TOF mass spectrometer, respectively. 13C-1H HSQC cross-peaks of human urine were manually picked and used to compile a HSQC cross-peak list. Upon querying of the HSQC peak list against the COLMAR 13C-1H HSQC database, the web server identified 87 urine metabolites. An additional 11 metabolites could be identified by manual analysis of unassigned cross-peaks of the 2D HSQC. These metabolites are mostly pH sensitive metabolites, whose resonances were significantly shifted despite of the buffer used. Metabolite identifications were independently confirmed using 13C-1H HSQC-TOCSY23 and 1H-1H TOCSY24 experiments using the 1H(13C)-TOCCATA database.27 The detailed list of the 98 identified urine metabolites by 13C-1H HSQC is given in Table S2. Calculated m/z ratios of the identified urine metabolites are automatically assigned to their most consistent experimental MS1 spectra. It is found that 88 of the 98 metabolites display at least one line in the experimental MS1 spectra that is consistent with the NMR-based predicted mass (m/z ratio). Hence, the NMR and MS information display a considerable amount of overlap.
The detailed list of the different features of metabolites assigned by the NMR/MS Translator is given in Table S3. Figure 3 shows the agreement of the experimental MS1 spectra with their NMR-based reconstructed counter-parts. Many of the major peaks are well-explained by the NMR-derived compounds. Still, there are some peaks that could not be explained in the experimental MS1 spectra by the NMR information (Figure 3). Similarly there are many peaks in the 13C-1H HSQC spectrum that remained unidentified. Both observations can be attributed, at least in part, to the incompleteness of the NMR metabolomics databases used. The total number 13C-1H HSQC peaks detected is 1012, but only 437 of them belong to the 98 identified metabolites. Assuming a similar number of HSQC cross-peaks per compound, another ~130 metabolites is estimated to be present that show up in the 13C-1H HSQC spectrum, but whose identity is not known.
Figure 3.

Application of the NMR/MS Translator to MS1 spectra (recorded in positive and negative ion mode) of human urine. 98 metabolites are positively identified by querying a 2D 13C-1H HSQC spectrum against the COLMAR 13C-1H HSQC database. Automated reconstruction of the mass spectrum demonstrated that 88 of these metabolites have at least one consistent m/z ratio in the experimental MS1 spectra.
Many of the 98 metabolites observed in the human urine 13C-1H HSQC spectrum were observed in previous urine NMR-metabolomics studies.28 29 30 31 Interestingly, here we identified 8 additional metabolites that were missed in a recent comprehensive study of human urine based on 1D 1H NMR,32 presumably because of extensive peak overlaps in the 1D 1H NMR spectrum. Some of these 8 metabolites were also previously detected and identified in other urine studies. The missed metabolites are quinic acid, 4-acetamidobutyric acid, isethionic acid, 3-hydroxyisobutyric acid, galactitol, fructose, ribose, and allose. Except for isethionic acid, all of these metabolites are consistent with at least one line in the experimental MS1 spectra (Table S3).
4. DISCUSSION
The choice of the instrumental technique(s) selected for a particular metabolomics study depends on multiple factors, including the accessibility to instrumentation, the specific questions at hand, and the training and background of the scientists involved. The NMR/MS Translator can significantly benefit the research of diverse groups of scientists, namely those who traditionally focus on (A) NMR-based metabolomics, (B) MS-based metabolomics, or (C) combined NMR/MS metabolomics approaches.
For group A, 13C-1H HSQC is among the most commonly used 2D NMR experiment applied for metabolomics studies. However, automated metabolite identification based on 13C-1H HSQC alone can be prone to false positive identifications, particularly when the spectrum is highly congested as is the case in many real-world metabolomics samples, such as urine. While false positives can often be identified and eliminated by using a 2D 1H-1H TOCSY and/or a 2D 13C-1H HSQC-TOCSY spectrum and comparing connectivity information of molecules, this may not always provide clear-cut identifications. For instance, for human urine, identification of creatine solely based on 13C-1H HSQC alone was challenging. Although experimentally observed cross-peaks were most similar to creatine cross-peaks in the COLMAR 13C-1H HSQC database, these experimental peaks were also similar to creatine-phosphate. Unfortunately, differentiation between creatine and creatine-phosphate metabolites by comparing their TOCSY pattern is not possible (because both molecules do not have any TOCSY cross-peaks). However, when we analyzed the MS1 spectra of human urine by the NMR/MS Translator, we observed a unique m/z ratio only for creatine, which strongly suggests that creatine, and not creatine-phosphate, is present. Hence, when connectivity information from 2D NMR spectra is unavailable or uninformative, the use of accurate mass information increases the accuracy of metabolite identification in NMR-based metabolomics.
For group B, the NMR/MS Translator increases the number of metabolites that can be identified by MS-based metabolomics. High-resolution mass spectrometry (MS1) provides chemical formula information, which corresponds to a potentially very large number of stable isobaric metabolites. To address this degeneracy, additional information is required, such as the one obtained from MS/MS fragmentation patterns. The fragment masses are used as fingerprints for the identification of the specific structures by comparing them with fragmentation patterns of known compounds stored in databases, such as METLIN20 and HMDB.19 Although excellent progress has been made in the compilation of MS metabolomics databases, MS/MS spectra of many of the metabolites in their commonly observed adduct and fragment forms are presently not available in those databases. A majority of metabolites in these databases have MS/MS data for the precursor ion [M+H]+ in positive ion mode, and for the precursor ion [M-H]− in negative ion mode. On the other hand, in real metabolomics samples such as human urine, metabolites also appear in many other different adduct and fragment forms, and in many cases they do not appear in [M+H]+ and [M-H]− forms at all (Table S3). For instance, in this study, 88 human urine metabolites appeared in total in 327 different MS features (Table S3), but only 51 of these features have MS/MS spectra in the METLIN database. These 51 MS/MS spectra belong to 48 different metabolites. Therefore, even if the MS/MS approach works optimally, it can identify only 48 metabolites in human urine, while the NMR/MS Translator identified 88 metabolites. Taken together, the NMR/MS Translator approach applied to human urine is capable of identifying a larger number of metabolites than what is currently possible by the MS/MS approach.
For group C, the NMR/MS Translator also makes the co-analysis of NMR and MS spectra faster and more consistent. In the past, combined NMR/MS metabolomics studies lacked comprehensive strategies for the rapid identification of metabolites that are simultaneously detected in NMR and MS spectra of the same complex metabolite mixture. The NMR/MS Translator addresses this challenge by providing a self-consistent and accurate identification of metabolites by NMR metabolomics databases followed by automated assignments of all ions, adducts, fragments, and observable isotope features of the identified metabolites in the experimental MS1 spectrum of the same sample. Traditional NMR/MS strategies independently pursue the identification of known metabolites in the NMR spectrum by using NMR metabolomics databases and identification of metabolites in the mass spectrum by MS metabolomics databases with the help of MS/MS libraries. This approach is not only labor-intensive, but also might lead to contradictory results. For example, a recent human urine study,32 which followed the traditional procedure, combined LC-MS/MS and direct flow injection (DFI)-MS/MS with 1D 1H NMR to test the consistency of these analytical techniques. It was found that there were only 28 metabolites that could be commonly identified by NMR and DFI/LC-MS/MS. This low number is likely due to the limited size of MS/MS databases and the challenges of obtaining high-quality MS/MS spectra in real-world complex metabolite mixtures as discussed recently.20 Briefly, when a precursor ion is in adduct form (e.g. [M+Na]+) or the intensity of the precursor ion is low, then obtaining a high-quality MS/MS spectrum is hampered.20 Furthermore, if there is another ion in the vicinity of the precursor ion of interest (within 1–2 m/z), then the signal(s) of the neighboring ion can contaminate the MS/MS spectrum.20 In human urine, metabolites appeared in many different adduct and fragment features (Table S3), which could complicate identification of urine metabolites by MS/MS. Another advantage of the NMR/MS Translator is that it enables a much faster analysis of redundant MS information of the different ion, adducts, fragment, and isotope features belonging to the same molecular formula over manual analysis.
In the present study, we used standard direct-infusion ESI MS for a proof of concept of the NMR/MS Translator, but more sophisticated MS approaches such as LC-MS can be applied to further optimize the number of metabolites detected by mass spectrometry. For instance metabolites that could not be detected in direct infusion spectrum because of severe ion suppression can be detected with LC-MS. Application of the NMR/MS Translator to LC-MS is straightforward representing a natural extension of the method.
Since the MS input data does not rely on MS/MS, the urine metabolites identified from MS alone is, according to the Metabolomics Standard Initiative,33 at “Level 2–3” (putatively annotated compounds or classes). The combination with 2D NMR via the NMR/MS Translator promotes these hits to “Level 1” (identified compounds). The NMR/MS Translator revealed that there remain many unexplained signals in both the NMR and MS spectra of human urine. The NMR/MS Translator rapidly determines the signals of the known metabolites and thereby pinpoints the signals belonging to unknown metabolites. The latter can be further investigated by the recent SUMMIT MS/NMR approach geared specifically toward the characterization of new metabolites that are not contained in databases. The combination of these two strategies should provide an efficient framework that takes full advantage of database information minimizing the “rediscovery” rate of already catalogued metabolites.
5. CONCLUSION
An integrated NMR/MS approach, termed NMR/MS Translator, was presented that allows the accurate and automated identification of catalogued metabolites detected in NMR and MS1 spectra of the same complex metabolite mixture. We anticipate that the NMR/MS Translator will find a useful role in addition or even as an alternative to existing metabolite identification protocols in MS and NMR-based analysis of complex mixtures as encountered in metabolomics and metabonomics.
Supplementary Material
Acknowledgments
We thank Dr. Arpad Somogyi and Dr. Cao Yu (Campus Chemical Instrument Center Mass Spectrometry Facility at The Ohio State University) for their expert help in the acquisition of mass spectra and their helpful discussion. We thank Dr. Lei Bruschweiler-Li for expert help in the preparation of the samples. This work was supported by the National Institutes of Health (grant R01 GM 066041 and SECIM (Southeast Center for Integrated Metabolomics) grant U24 DK097209-01A1).
Footnotes
This material is available free of charge via the Internet at http://pubs.acs.org. Figure S1 - 2D 13C-1H HSQC spectrum of 26-compound model mixture, Figure S2 - 2D 13C-1H HSQC spectrum of human urine, Table S1 - List of identified metabolites in the model mixture by the NMR/MS Translator, Table S2 - List of identified metabolites in human urine by 2D 13C-1H HSQC, Table S3 - List of metabolites in human urine determined by the NMR/MS Translator.
References
- 1.Holmes E, Wilson ID, Nicholson JK. Metabolic Phenotyping in Health and Disease. Cell. 2008;134:714–717. doi: 10.1016/j.cell.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 2.Bundy JG, Davey MP, Viant MR. Environmental metabolomics: a critical review and future perspectives. Metabolomics. 2009;5:3–21. [Google Scholar]
- 3.Obata T, Fernie AR. The use of metabolomics to dissect plant responses to abiotic stresses. Cell Mol Life Sci. 2012;69:3225–3243. doi: 10.1007/s00018-012-1091-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie G, Li X, Li H, Jia W. Toward Personalized Nutrition: Comprehensive Phytoprofiling and Metabotyping. J Proteome Res. 2013;12:1547–1559. doi: 10.1021/pr301222b. [DOI] [PubMed] [Google Scholar]
- 5.Lenz EM, Wilson ID. Analytical Strategies in Metabonomics. J Proteome Res. 2007;6:443–458. doi: 10.1021/pr0605217. [DOI] [PubMed] [Google Scholar]
- 6.Bingol K, Brüschweiler R. Multidimensional Approaches to NMR-Based Metabolomics. Anal Chem. 2014;86:47–57. doi: 10.1021/ac403520j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corcoran O, Spraul M. LC-NMR-MS in drug discovery. Drug Discov Today. 2003;8:624–631. doi: 10.1016/s1359-6446(03)02749-1. [DOI] [PubMed] [Google Scholar]
- 8.Tayyari F, Gowda GAN, Gu H, Raftery D. 15N-Cholamine A Smart Isotope Tag for Combining NMR- and MS-Based Metabolite Profiling. Anal Chem. 2013;85:8715–8721. doi: 10.1021/ac401712a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crockford DJ, Holmes E, Lindon JC, Plumb RS, Zirah S, Bruce SJ, Rainville P, Stumpf CL, Nicholson JK. Statistical Heterospectroscopy, an Approach to the Integrated Analysis of NMR and UPLC-MS Data Sets: Application in Metabonomic Toxicology Studies. Anal Chem. 2006;78:363–371. doi: 10.1021/ac051444m. [DOI] [PubMed] [Google Scholar]
- 10.Pan Z, Gu H, Talaty N, Chen H, Shanaiah N, Hainline BE, Cooks RG, Raftery D. Principal component analysis of urine metabolites detected by NMR and DESI-MS in patients with inborn errors of metabolism. Anal Bioanal Chem. 2007;387:539–549. doi: 10.1007/s00216-006-0546-7. [DOI] [PubMed] [Google Scholar]
- 11.Marshall DD, Lei S, Worley B, Huang Y, Garcia-Garcia A, Franco R, Dodds ED, Powers R. Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics. 2014:1–12. doi: 10.1007/s11306-014-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia J, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS. The Human Serum Metabolome. PLoS One. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng KK, Akasaki Y, Lecommandeur E, Lindsay RT, Murfitt S, Walsh K, Griffin JL. Metabolomic Analysis of Akt1-Mediated Muscle Hypertrophy in Models of Diet-Induced Obesity and Age-Related Fat Accumulation. J Proteome Res. 2015;14:342–352. doi: 10.1021/pr500756u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bingol K, Zhang F, Bruschweiler-Li L, Brüschweiler R. Carbon Backbone Topology of the Metabolome of a Cell. J Am Chem Soc. 2012;134:9006–9011. doi: 10.1021/ja3033058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen SH, Jensen AG, Cornett C, Bjornsdottir I, Taylor S, Wright B, Wilson ID. High-Performance Liquid Chromatography On-Line Coupled to High-Field NMR and Mass Spectrometry for Structure Elucidation of Constituents of Hypericum perforatum L. Anal Chem. 1999;71:5235–5241. [Google Scholar]
- 16.Bingol K, Bruschweiler-Li L, Yu C, Somogyi A, Zhang F, Brüschweiler R. Metabolomics Beyond Spectroscopic Databases: A Combined MS/NMR Strategy for the Rapid Identification of New Metabolites in Complex Mixtures. Anal Chem. 2015 doi: 10.1021/ac504633z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Q, Lewis IA, Hegeman AD, Anderson ME, Li J, Schulte CF, Westler WM, Eghbalnia HR, Sussman MR, Markley JL. Metabolite identification via the Madison Metabolomics Consortium Database. Nat Biotechnol. 2008;26:162–164. doi: 10.1038/nbt0208-162. [DOI] [PubMed] [Google Scholar]
- 18.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Wenger RK, Yao H, Markley JL. BioMagResBank. Nucl Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong Y, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucl Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu ZJ, Schultz AW, Wang J, Johnson CH, Yannone SM, Patti GJ, Siuzdak G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat Protocols. 2013;8:451–460. doi: 10.1038/nprot.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bingol K, Zhang F, Bruschweiler-Li L, Brüschweiler R. TOCCATA: A Customized Carbon Total Correlation Spectroscopy NMR Metabolomics Database. Anal Chem. 2012;84:9395–9401. doi: 10.1021/ac302197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodenhausen G, Ruben DJ. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem Phys Lett. 1980;69:185–189. [Google Scholar]
- 23.Lerner L, Bax A. Sensitivity-enhanced two-dimensional heteronuclear relayed coherence transfer NMR spectroscopy. J Magn Reson. 1986;69:375–380. [Google Scholar]
- 24.Braunschweiler L, Ernst RR. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J Magn Reson. 1983;53:521–528. [Google Scholar]
- 25.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 26.Bingol K, Li DW, Bruschweiler-Li L, Cabrera OA, Megraw T, Zhang F, Brüschweiler R. Unified and Isomer-Specific NMR Metabolomics Database for the Accurate Analysis of 13C-1H HSQC Spectra. ACS Chem Biol. 2015;10:452–459. doi: 10.1021/cb5006382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bingol K, Bruschweiler-Li L, Li DW, Brüschweiler R. Customized Metabolomics Database for the Analysis of NMR 1H-1H TOCSY and 13C-1H HSQC-TOCSY Spectra of Complex Mixtures. Anal Chem. 2014;86:5494–5501. doi: 10.1021/ac500979g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bales JR, Higham DP, Howe I, Nicholson JK, Sadler PJ. Use of high-resolution proton nuclear magnetic resonance spectroscopy for rapid multi-component analysis of urine. Clin Chem. 1984;30:426–32. [PubMed] [Google Scholar]
- 29.Holmes E, Loo RL, Stamler J, Bictash M, Yap IKS, Chan Q, Ebbels T, De Iorio M, Brown IJ, Veselkov KA, Daviglus ML, Kesteloot H, Ueshima H, Zhao L, Nicholson JK, Elliott P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature. 2008;453:396–400. doi: 10.1038/nature06882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronwald W, Klein MS, Zeltner R, Schulze BD, Reinhold SW, Deutschmann M, Immervoll AK, Boger CA, Banas B, Eckardt KU, Oefner PJ. Detection of autosomal dominant polycystic kidney disease by NMR spectroscopic fingerprinting of urine. Kidney Int. 2011;79:1244–1253. doi: 10.1038/ki.2011.30. [DOI] [PubMed] [Google Scholar]
- 31.Rai RK, Sinha N. Fast and Accurate Quantitative Metabolic Profiling of Body Fluids by Nonlinear Sampling of 1H-13C Two-Dimensional Nuclear Magnetic Resonance Spectroscopy. Anal Chem. 2012;84:10005–10011. doi: 10.1021/ac302457s. [DOI] [PubMed] [Google Scholar]
- 32.Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P, Dame ZT, Poelzer J, Huynh J, Yallou FS, Psychogios N, Dong E, Bogumil R, Roehring C, Wishart DS. The Human Urine Metabolome. PLoS One. 2013;8:e73076. doi: 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriot P, Nicholls AW, Reily MD, Thaden JJ, Viant MR. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.