

Abstract
Introduction
Cancer in the parathyroid gland is rare, but parathyroid cancer is occasionally seen in relation to genetic abnormalities. Due to a limited amount of evidence, the optimal handling of these cases is not clear. Furthermore, the presence of a malignant parathyroid tumor is rarely known at the time of the initial operation; therefore, re-operations are often necessary. The aim of this study was to present the case of a patient with a previously diagnosed jaw tumor and parathyroid carcinoma that presents as a recurrence of hyperparathyroid hypercalcemia.
Case Presentation
A 41-year-old patient who was already diagnosed with a parathyroid carcinoma and a jaw tumor caused by a CDC73 mutation, presented with biochemical evidence of increasing parathyroid hormone (PTH) and calcium levels after a previous total parathyroidectomy. The patient’s ionized calcium increased to 1.55 mmol/L and PTH increased to 16.0 pmol/L. A previous genetic analysis revealed a mutation in the CDC73 gene. There was no family history of hyperparathyroidism. We performed a sestamibi scintigraphy and an 11-C methionine (MET) positron emission tomography (PET) scan that showed a recurrence on the left side of the trachea. The patient underwent a third neck operation for the removal of a tumor on the left side of the trachea. The pathology report revealed that the tumor was a lymph node metastasis from the previous parathyroid carcinoma. The patient is currently enrolled in our follow-up regime. Hyperparathyroidism-jaw tumor (HPT-JT) syndrome is a rare autosomal dominant disorder characterized by a parathyroid adenoma or carcinoma, fibro-osseous lesions (ossifying fibroma) of the mandible and maxilla, and renal cysts and tumors. This autosomal dominant familial cancer syndrome has been reported with a variable and incomplete penetrance, and up to 10% of gene carriers do not show any clinical manifestations. Here we present a patient’s case and discuss the literature related to this condition.
Conclusions
The recurrence of hyperparathyroid hypercalcemia in HTP-JT syndrome after an initial total parathyroidectomy is a well-known condition necessitating careful management, an evaluation of any underlying genetic abnormality, and a family examination. A surgical treatment and surveillance of calcium and PTH measurements are necessary to prevent a recurrence.
Keywords: Parathyroid Cancer, Jaw Tumor, Hyperparathyroidism
1. Introduction
Hyperparathyroidism-jaw tumor syndrome (HPT-JT) is a rare autosomal dominant disorder characterized by a parathyroid adenoma or carcinoma, fibro-osseous lesions (ossifying fibroma) of the mandible and maxilla, and renal cysts and tumors.
In the common clinical setting, the presentation of hyperparathyroid hypercalcemia is almost always caused by primary hyperparathyroidism (PHPT). This disease is generally benign and asymptomatic or presents with only mild symptoms. After a clinical workup and an evaluation of PHPT complications such as osteoporosis and kidney stones, patients are recommended to either undergo an operation with a parathyroidectomy or undergo follow-up care according to international guidelines (1).
The development of pathologic parathyroid tissue is not completely understood. However, pathogenesis in parathyroid cells may be affected by various genetic and external factors (2). PHPT can occur in several familiar syndromes (e.g., multiple endocrine neoplasia [MEN]) (2). Furthermore, a few genetic abnormalities have been described as the causes of these different syndromes. One of the known mutations occurs in the CDC73 gene (2). The condition associated with this mutation is HPT-JT syndrome. It has been estimated that approximately 70% of patients affected by this mutation may develop PHPT, which in most patients is caused by a single parathyroid adenoma, although 10% - 15% of patients affected by the mutation may develop parathyroid cancer. Additionally, the mutation is associated with tumors at other sites including the jaw, kidneys, and uterus. If a parathyroid cancer is diagnosed, it is recommended than an en bloc resection including an ipsilateral hemithyroidectomy be performed (3). However, since parathyroid cancer is very rare, the surgeon rarely expects to find a cancer during the first operation, consequently, a second operation is often necessary to secure a proper local resection. Evidence is scarce regarding the optimal handling of these rare cases, and especially the handling of disseminated or recurrent parathyroid cancer. One way to monitor patients is with parathyroid hormone (PTH) and calcium measurements after a previous total parathyroidectomy (PTX). Any secondary increases in these biochemical measures are evidence suggesting a recurrence of the disease. The current case presentation concerns recurrent hyperparathyroid hypercalcemia after a previous total PTX due to HPT-JT syndrome.
2. Case Presentation
A 41-year-old man was admitted to the hospital after a multi-fracture traumatic episode with severe hyperparathyroid hypercalcemia. Ionized calcium (Ca2+) was 3.54 mmol/L (1.18 - 1.32 mmol/L) and plasma PTH was 111.6 pmol/L (1.6 - 6.9 pmol/L). The patient had a history of mandibular pain and loose teeth. X-rays of the mandible showed abnormal bone tissue as a result of a jaw tumor. A computerized tomography (CT) scan revealed bilateral kidney stones and dual energy X-ray absorptiometry (DXA) revealed osteoporosis with a T-score of -2.5 at the lumbar spine and -4.0 at the total hip. The patient also presented with moderate renal insufficiency (eGFR/creatinine ca 45 mL/minute).
Preoperatively, localization studies were performed with ultrasound of the neck, and sestamibi scintigraphy. On ultrasound, there was a tumor posterior to the left thyroid gland measuring 51 × 20 × 22 mm, without enlarged lymph nodes. The patient was operated on with a unilateral neck exploration in August 2009 with a resection of one very enlarged parathyroid gland (11.3 g). Postoperatively, the patient developed symptoms of hungry bones with low plasma calcium levels, a PTH elevation, and clinical symptoms of hypocalcemia. Medical treatment reversed these symptoms. The pathology report revealed a carcinoma in the removed parathyroid gland.
Genetic analyses revealed a mutation in CDC73 (c.358C > T) causing an amino acid change (p.R120X) with a stop of the transcription at exon 4. A family screening was performed showing that other family members were carriers of the same mutation. However, so far, none of the family members have shown any signs of hyperparathyroidism (HPT) (Figure 1).
Figure 1. Pedigree Chart of the Family.

Open circles (women) and squares (men) show unaffected individuals. The filled-in square indicates the index patient reported in this paper. Shaded circles and squares show carriers of the mutation not showing clinical signs of the disease.
A second operation was performed in December 2009 with a total PTX and a unilateral hemithyroidectomy. No further malignancy was found in the pathology report. As expected, the patient developed hypoparathyroidism and was treated with calcium, vitamin D3, and alfacalcidol.
During the follow up in March 2014, there was evidence of recurrent hyperparathyroid hypercalcemia. Ionized calcium increased to 1.55 mmol/L and PTH increased to 16.0 pmol/L. The patient also reported hypercalcemic symptoms with thirst and polyuria. Furthermore, a sestamibi scintigraphy and an 11-C MET PET scan concordantly revealed a probable recurrence on the left side of the trachea (Figure 2A - 2B). The patient subsequently underwent a third neck operation (April 2014) with the removal of a tumor on the left side of the trachea (Figure 3A - 3B). The pathology report revealed that this was a lymph node metastasis from the previous parathyroid carcinoma. The calcium values normalized after the surgery and returned to medical follow-up. Six months after the third operation, there was still measurable PTH levels and ionized calcium levels around the upper normal range (Figure 4). At present, we have no plans for further surgical intervention.
Figure 2. Probable Recurrence of the Trachea.
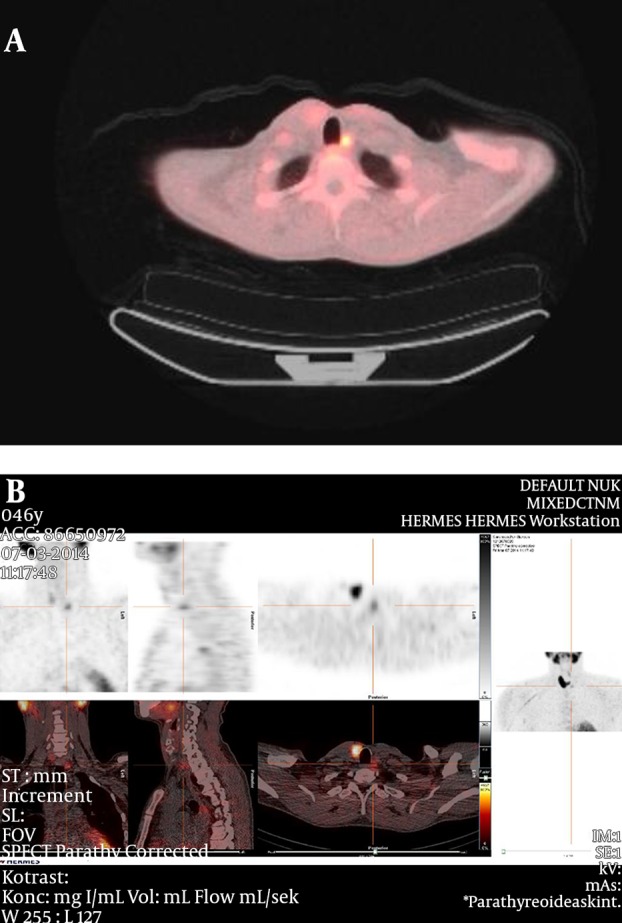
A, 11-C methionine (MET) positron emission tomography (PET) showing a recurrence on the left side of trachea; B, the sestamibi scintigraphy shows the localization of the recurrence.
Figure 3. Removal of a Tumor on the Left Side of the Trachea.
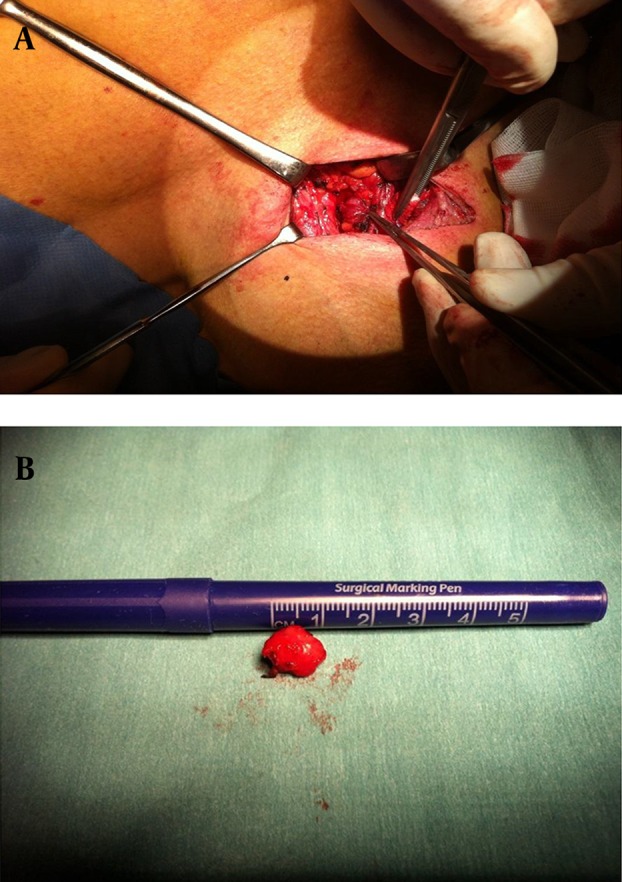
A, Intraoperative dissection of the PTH carcinoma; B, the specimen following removal.
Figure 4. Perioperative Calcium and PTH Levels.

A, Perioperative PTH levels; B, perioperative calcium levels. Points 4 and 5 are the intraoperative PTH measurements. Points 1, 2, and 3 are from 6 weeks, 2 weeks, and 1 week before the operation, respectively. Points 6, 7, and 8 are from 1, 2, and 30 days after the operation, respectively.
2.1. Pathology
The lower left parathyroid gland was removed in 2009. Macroscopically, the gland measured 45 × 25 × 8 mm and weighed 11.3 g (Figure 5A - 5C). An irregular surface and calcification was noted. The histological examination revealed a uniform hypercellular tumor intersected by broad fibrous bands and surrounded by a thick fibrous capsule. The tumor cells, which were chief cells, appeared well differentiated without any significant pleomorphism. Several foci showed capsular and vascular invasions. In a small area it was doubtful whether the resection had been sufficient. Immunohistochemistry revealed that tumor cells expressed PTH and the proliferation marker Ki-67 showed a proliferation of less than 1%. The parathyroid carcinoma diagnosis was based on these findings.
Figure 5. Lower Left Parathyroid Gland.

A, PTH carcinoma; B, PTH carcinoma: Vessel invasion; C, carcinoma with an invasion through the membrane; D, lymph node metastasis.
In April 2014 a lymph node to the left of the trachea was removed and a metastasis from the parathyroid carcinoma was confirmed (Figure 5D).
3. Discussion
HPT-JT syndrome is a rare autosomal dominant disorder characterized by parathyroid adenoma or carcinoma, fibro-osseous lesions (ossifying fibroma) of the mandible and maxilla, and renal cysts and tumors. This autosomal dominant familial cancer syndrome has been reported as a variable and incomplete penetrance, and up to 10% of gene carriers do not show any clinical manifestations (4).
The HPT-JT syndrome presents with various characteristics. Apart from parathyroid tumors, ossifying tumors of the jaw or mandible are frequent and must be distinguished from the osteoclastic brown tumors in PHPT. In about 15% of cases, patients may develop renal pathologies such as Wilms’ tumor, renal cell carcinoma, and polycystic disease (5). Uterine tumors are also common in women with HPT-JT (6).
Typically, only one of the four parathyroid glands is affected, but in some patients, tumors may be present in more than one gland synchronously. This is different from patients with MEN1, where all or most of the glands are involved simultaneously. Gland involvement is similar to the sporadic variety of PHPT (7). In most HPT-JT syndrome cases, multiple or single parathyroid adenomas are present, but a parathyroid carcinoma occurs in approximately 10 - 15% of these patients (4, 5, 7). The frequency of cancer seems to be much higher in HPT-JT syndrome compared to the absence of cancer in MEN1 and below 1% in sporadic HPT (7).
The clinical presentation of parathyroid cancers may differ from the presentation of sporadic PHPT. In some cases, the tumor is palpable, and during surgery, it often presents as a hard consistency and is usually larger than the typical parathyroid adenoma. Furthermore, the plasma levels of calcium and PTH are frequently much higher than in sporadic PHPT (7).
Histologically, the diagnosis of a parathyroid carcinoma is considered extremely difficult and is based upon several features. These features include the presence of a metastatic disease, a persistent locally aggressive and invasive disease, and the risk of recurrence in the area of previous surgery (7).
The genetic mutation in the CDC73 gene is responsible for parafibromin expression, a protein with tumor-suppressor activity (8). Gill et al. confirm that the complete loss of parafibromin by immunohistochemical staining is evident in both parathyroid carcinomas and HPT-JT–related tumors (adenomas and carcinomas), but not in other benign lesions (9). Several authors have concluded that the lack of parafibromin can be used as a marker to confirm parathyroid carcinoma and is associated with an increased risk of recurrence (10).
HPT in the HPT-JT syndrome seems to be more aggressive with severe hypercalcemia and the presence of hypercalcemic crises compared to MEN1 and sporadic HPT. Therefore, it is important to be aware of the symptoms related to the jaw and teeth in patients diagnosed with HPT and to perform kidney examinations. If positive, molecular testing and radiological studies amongst family members are indicated. If one or more relatives show positive for this mutation, they must be subjected to a regular surveillance of serum calcium and PTH levels that allows for an early diagnosis and treatment of hyperparathyroid hypercalcemia. Thus, this may aid in preventing the development of a parathyroid carcinoma. As an alternative to surgical treatment, medical treatment with calcimimetics may be considered. Parathyroid cancers are often highly differentiated tumors expressing the calcium-sensing receptor, and calcimimetics may reduce PTH secretion by acting on the calcium-sensing receptor. However, as this does not cure the disease, calcimimetics are often only used in patients awaiting surgery or in patients with an inoperable disease (11).
The CDC73 mutation found in our index patient has not previously been reported. Several family members were also found to be carriers of the mutation. None of them have, so far, developed signs of HPT. Accordingly, it has to be considered whether the mutation is actually of pathogenic importance. However, as the mutation is located in exon 4, causing an early termination of the transcript, it seems likely to be of pathogenic importance. Continuous follow-up examinations of family members in the upcoming years will help to resolve whether the penetrance of this mutation is lower than reported for other mutations in CDC73. As already advocated by Mehta et al., a life-long observation must be offered for these patients (12).
In cases of recurrence, we are advocates for an image-guided reoperation with a resection of the potential lymph node metastases. The sestamibi scintigraphy may be useful, but others have used PET-CT scans, magnetic resonance imaging (MRI), or ultrasound.
3.1. Conclusion
We have presented a case of a patient with HPT-JT syndrome and recurrent hyperparathyroid hypercalcemia after an initial total PTX. This condition is rare but well-known, and specialized management is needed that includes genetic examinations and a family history. In spite of a surgical cure, surveillance with calcium and PTH measurements are recommended to screen for disease recurrence.
Footnotes
Financial Disclosure:The authors have no financial interests related to the material in the manuscript and no other conflicts of interest.
References
- 1.Bilezikian JP, Khan AA, Potts Jr JT. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab. 2009;94(2):335–9. doi: 10.1210/jc.2008-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan K, Gomez Hernandez K, Mete O. Clinicopathological correlates of hyperparathyroidism. J Clin Pathol. 2015;68(10):771–87. doi: 10.1136/jclinpath-2015-203186. [DOI] [PubMed] [Google Scholar]
- 3.Sarquis MS, Silveira LG, Pimenta FJ, Dias EP, Teh BT, Friedman E, et al. Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008;143(5):630–40. doi: 10.1016/j.surg.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Abdulla AG, O'Leary EM, Isorena JP, Diaz MF, Yeh MW. Recurrent hyperparathyroidism and a novel nonsense mutation in a patient with hyperparathyriodism-jaw tumor syndrome. Endocr Pract. 2013;19(6):e134–7. doi: 10.4158/EP13187.CR. [DOI] [PubMed] [Google Scholar]
- 5.Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2003;253(6):634–42. doi: 10.1046/j.1365-2796.2003.01168.x. [DOI] [PubMed] [Google Scholar]
- 6.Masi G, Barzon L, Iacobone M, Viel G, Porzionato A, Macchi V, et al. Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr Relat Cancer. 2008;15(4):1115–26. doi: 10.1677/ERC-08-0066. [DOI] [PubMed] [Google Scholar]
- 7.Frank-Raue K, Haag C, Schulze E, Keuser R, Raue F, Dralle H, et al. CDC73-related hereditary hyperparathyroidism: five new mutations and the clinical spectrum. Eur J Endocrinol. 2011;165(3):477–83. doi: 10.1530/EJE-11-0003. [DOI] [PubMed] [Google Scholar]
- 8.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32(4):676–80. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 9.Gill AJ. Understanding the genetic basis of parathyroid carcinoma. Endocr Pathol. 2014;25(1):30–4. doi: 10.1007/s12022-013-9294-3. [DOI] [PubMed] [Google Scholar]
- 10.Kruijff S, Sidhu SB, Sywak MS, Gill AJ, Delbridge LW. Negative parafibromin staining predicts malignant behavior in atypical parathyroid adenomas. Ann Surg Oncol. 2014;21(2):426–33. doi: 10.1245/s10434-013-3288-8. [DOI] [PubMed] [Google Scholar]
- 11.Sharretts JM, Kebebew E, Simonds WF. Parathyroid cancer. Semin Oncol. 2010;37(6):580–90. doi: 10.1053/j.seminoncol.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta A, Patel D, Rosenberg A, Boufraqech M, Ellis RJ, Nilubol N, et al. Hyperparathyroidism-jaw tumor syndrome: Results of operative management. Surgery. 2014;156(6):1315–24. doi: 10.1016/j.surg.2014.08.004. discussion 1324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]