

Little is known about biomarker trajectories over time in dementia with Lewy bodies (DLB). Sarro et al. report that higher baseline β-amyloid deposition predicts faster grey matter atrophy in an Alzheimer’s disease-like pattern and faster clinical decline. Patients with DLB and high β-amyloid could benefit from emerging anti-amyloid agents.
Keywords: DLB, structural MRI, beta-amyloid, amyloid imaging, brain atrophy

Little is known about biomarker trajectories over time in dementia with Lewy bodies (DLB). Sarro et al. report that higher baseline β-amyloid deposition predicts faster grey matter atrophy in an Alzheimer’s disease-like pattern and faster clinical decline. Patients with DLB and high β-amyloid could benefit from emerging anti-amyloid agents.
Abstract
Alzheimer’s disease pathology frequently coexists with Lewy body disease at autopsy in patients with probable dementia with Lewy bodies. More than half of patients with probable dementia with Lewy bodies have high amyloid-β deposition as measured with 11 C-Pittsburgh compound B binding on positron emission tomography. Biomarkers of amyloid-β deposition precede neurodegeneration on magnetic resonance imaging during the progression of Alzheimer’s disease, but little is known about how amyloid-β deposition relates to longitudinal progression of atrophy in patients with probable dementia with Lewy bodies. We investigated the associations between baseline 11 C-Pittsburgh compound B binding on positron emission tomography and the longitudinal rates of grey matter atrophy in a cohort of clinically diagnosed patients with dementia with Lewy bodies ( n = 20), who were consecutively recruited to the Mayo Clinic Alzheimer’s Disease Research Centre. All patients underwent 11 C-Pittsburgh compound B positron emission tomography and magnetic resonance imaging examinations at baseline. Follow-up magnetic resonance imaging was performed after a mean (standard deviation) interval of 2.5 (1.1) years. Regional grey matter loss was determined on three-dimensional T 1 -weighted magnetic resonance imaging with the tensor-based morphometry-symmetric normalization technique. Linear regression was performed between baseline 11 C-Pittsburgh compound B standard unit value ratio and longitudinal change in regional grey matter volumes from an in-house modified atlas. We identified significant associations between greater baseline 11 C-Pittsburgh compound B standard unit value ratio and greater grey matter loss over time in the posterior cingulate gyrus, lateral and medial temporal lobe, and occipital lobe as well as caudate and putamen nuclei, after adjusting for age ( P < 0.05). Greater baseline 11 C-Pittsburgh compound B standard unit value ratio was also associated with greater ventricular expansion rates ( P < 0.01) and greater worsening over time in Clinical Dementia Rating Scale, sum of boxes ( P = 0.02). In conclusion, in patients with probable dementia with Lewy bodies, higher amyloid-β deposition at baseline is predictive of faster neurodegeneration in the cortex and also in the striatum. This distribution is suggestive of possible interactions among amyloid-β, tau and α-synuclein aggregates, which needs further investigation. Furthermore, higher amyloid-β deposition at baseline predicts a faster clinical decline over time in patients with probable dementia with Lewy bodies.
Introduction
Dementia with Lewy bodies (DLB) represents the second most common neurodegenerative form of dementia in the elderly, and is prevalent in up to 30% of all dementia cases in the community ( Zaccai et al. , 2005 ). The aggregation of aberrant α-synuclein in Lewy bodies and in Lewy neurites is the pathological hallmark of DLB, but it often coexists with the presence of Alzheimer’s disease pathology at autopsy in the form of both tau neurofibrillary tangles and amyloid-β neuritic plaques ( McKeith et al. , 2005 ; Schneider et al. , 2007 ). Little is known about how the evolution of Alzheimer’s disease-pathology relates to α-synuclein formation ( Wolozin and Behl, 2000 ; Harding and Halliday, 2001 ) and the temporal sequencing of Alzheimer’s disease-related biomarkers in patients with DLB, many of whom will have pathologic and biomarker characteristics of Alzheimer’s disease.
In the Lewy bodies disease spectrum of disorders, autopsy studies highlighted that co-occurrence of amyloid-β plaques and phosphorylated tau together with α-synuclein deposition are associated with greater cognitive decline ( Kraybill et al. , 2005 ; Nelson et al. , 2009 ; Ferman et al. , 2015 ; Howlett et al. , 2015 ), and shorter survival ( Kotzbauer et al. , 2012 ; Graff-Radford et al. , 2015 ) .
Amyloid PET labelling tracer 11 C-Pittsburgh compound B (PiB) is an in vivo biomarker of amyloid-β deposition in DLB ( Fodero-Tavoletti et al. , 2007 ; Burack et al. , 2010 ; Driscoll et al. , 2012 ; Kantarci et al. , 2012 c ). Meta-analysis from multiple cohorts indicates that 68% of patients with probable DLB have abnormal PiB retention ( Petrou et al. , 2015 ). PiB retention is on average higher in patients with DLB than Parkinson’s disease and Parkinson’s disease dementia ( Gomperts et al. , 2008 , 2012 ), but lower than patients with Alzheimer’s disease ( Kantarci et al. , 2012 b ; Catafau and Bullich, 2015 ). Furthermore, PiB retention has been shown to be associated with older age, higher prevalence of APOE ε4 carriers ( Maetzler et al. , 2009 ), impaired cognitive performance ( Gomperts et al. , 2008 , 2012 ; Maetzler et al. , 2009 ) and lower PET glucose metabolism ( Claassen et al. , 2011 ) in patients with DLB. Villemagne et al. (2011) suggested that greater amyloid-β deposition on PET is associated with a faster development of diagnostic clinical features after the onset of cognitive impairment in patients with DLB.
Atrophy in the limbic, temporal and parietal association cortices on structural MRI is a marker of neurodegeneration associated with tau neurofibrillary tangle pathology of Alzheimer’s disease and DLB ( Jack et al. , 2004 ; Burton et al. , 2009 ; Murray et al. , 2013 ). In several studies, patients with probable DLB showed an Alzheimer’s disease-like pattern of atrophy ( Whitwell et al. , 2007 b ; Sabattoli et al. , 2008 ; Zhong et al. , 2014 ) while autopsy confirmation later showed that the presence of tau neurofibrillary tangles and their Braak stage ( Braak and Braak, 1991 ) of Alzheimer’s disease primarily explained the cross-sectional ( Burton et al. , 2009 ; Kantarci et al. , 2012 a ; Murray et al. , 2013 ) and longitudinal ( Nedelska et al. , 2015 ) atrophy observed in the medial temporal structures.
Amyloid imaging with PiB retention and its relation to atrophy on MRI has been widely studied in Alzheimer’s disease dementia ( Archer et al. , 2006 ; Jack et al. , 2009 ; Chetelat et al. , 2010 ), amnestic mild cognitive impairment and preclinical Alzheimer’s disease ( Andrews et al. , 2013 ; Knopman et al. , 2013 ; Petersen et al. , 2013 , 2016 ; Whitwell et al. , 2013 ; Jack et al. , 2014 ; Araque Caballero et al. , 2015 ). Patients with probable DLB and Parkinson’s disease dementia with high PiB retention have an Alzheimer’s disease-like pattern of atrophy ( Shimada et al. , 2013 ) but longitudinal imaging studies investigating whether amyloid-β deposition influences rate of atrophy in patients with probable DLB are lacking.
The aim of this study is therefore to determine whether higher amyloid-β deposition at baseline measured by PiB-PET is associated with increased rates of atrophy in the limbic, neocortical regions and subcortical grey matter nuclei on MRI in patients with probable DLB. To pursue these goals, we analysed the longitudinal rates of atrophy on MRI in cortical and subcortical grey matter, with a customized atlas subdivision, which includes regions of interest involved in the pathophysiology of DLB and/or Alzheimer’s disease such as basal ganglia, posterior cingulate gyrus and medial temporal lobe, temporal, occipital, parietal and frontal neocortex ( Minoshima et al. , 2001 ; Whitwell et al. , 2007 b ; Burton et al. , 2009 ; Teune et al. , 2010 ; Zhong et al. , 2014 ; McCleery et al. , 2015 ; Nedelska et al. , 2015 ; Mak et al. , 2016 ). Furthermore, we investigated whether amyloid-β deposition at baseline is associated with a faster cognitive and functional decline in patients with probable DLB.
Materials and methods
Subjects
Between November 2007 and August 2013, consecutive patients with a clinical diagnosis of probable DLB ( n = 22) according to the Third Consortium Criteria for DLB ( McKeith et al. , 2005 ) were recruited to the Mayo Clinic Alzheimer’s disease Research Centre. Each patient underwent a baseline structural MRI and PiB-PET scans, and was followed longitudinally until a follow-up structural MRI scan was performed between 1 to 5 years from the baseline. Patients who had poor MRI scan quality for analysis at one of the time points were excluded ( n = 2).
The diagnosis of probable DLB was made after a comprehensive neurological and neuropsychological evaluation, and confirmed in the setting of a consensus meeting composed of behavioural neurologists, neuropsychologists, and nurses who evaluated the patient. Standardized measures of cognitive and functional performance at baseline such as Mini-Mental State Examination (MMSE) and Clinical Dementia Rating Scale, Sum of Boxes (CDR-SOB) were collected. Furthermore, the clinical characteristics of DLB at baseline were collected as follows: presence of parkinsonism was recorded, and the degree of extrapyramidal motor impairment was assessed through the Unified Parkinson’s Disease Rating Scale part III (UPDRS-III); visual hallucinations were characterized by being fully formed, not restricted to a single episode and not related to another medical issue, treatment or advanced dementia; the fluctuations were considered to be present if they scored 3 to 4 on the Mayo Fluctuations Questionnaire ( Ferman et al. , 2004 ); and probable rapid eye movement (REM) sleep behaviour disorder was defined as previously published ( Boeve et al. , 2011 ). Data concerning acetylcholinesterase inhibitor (AChEI), atypical neuroleptics and dopaminergic treatments during the study period were collected by a neurology resident (L.S.) from each patient’s chart, and further reviewed by a behavioural neurologist (J.G.R.). The daily dose of dopaminergic agents was expressed as levodopa equivalent dose ( Tomlinson et al. , 2010 ).
Patients were excluded from this study if they had: a history of traumatic brain injury, hydrocephalus, or intracranial mass; neurological and psychiatric diseases other than probable DLB, history of chemotherapy, head radiation therapy or substance abuse. Patients were not excluded if they had a history of cerebrovascular disease, and none of them developed major cerebrovascular lesions during the study period.
The Mayo Clinic Institutional Review Board approved this study. All subjects provided written informed consent before participating in any research activity.
MRI acquisition
MRI examinations were performed at 3 T (GE Healthcare). A 3D high resolution magnetization prepared rapid gradient echo (MPRAGE) acquisition with repetition time/echo time/inversion time of 7/3/900 ms; flip angle of 8°; ∼1 mm cubed resolution was performed for anatomical segmentation and labelling.
11 C-Pittsburgh compound B-PET acquisitions
A PET/CT scanner (DRX; GE Healthcare) operating in 3D mode was used to acquire PET images. A CT image was obtained for attenuation correction. Subjects were injected with PiB (mean, 596 MBq; range, 292–729 MBq). Following a 40-min PiB uptake period, a 20-min PiB scan consisting of four 5-min dynamic frames was obtained.
Structural MRI analysis
Regional grey matter loss across the entire brain was estimated with an automated, in-house developed implementation of tensor-based morphometry using symmetric diffeomorphic normalization (TBM-SyN) ( Cash et al. , 2015 ). This method uses the symmetric diffeomorphic registration between serial MRIs ( Avants et al. , 2008 ) to compute mappings between each subject’s pair of serial preprocessed T 1 -weighted MRI scans. The annualized log determinants of the Jacobians of these deformations were averaged within each cortical region of interest across both hemispheres, and assessed within the group in study ( Cash et al. , 2015 ). Magnitude of atrophy rates from an Automated Anatomical Labelling atlas ( Tzourio-Mazoyer et al. , 2002 ), customized and modified in-house to fit our template ( Vemuri et al. , 2008 ) were applied to the annualized log Jacobian images. An additional region of interest to label the ventricles was created.
11 C-Pittsburgh compound B-PET image analysis
PiB-PET methods of analysis are consistent to what has been described in a previous study in probable DLB ( Kantarci et al. , 2012 b ). Cerebellar uptake was used as an internal reference region of interest for PiB-PET uptake normalization. The global PiB retention standard uptake value ratio (SUVR) was obtained from the bilateral parietal (including posterior cingulate and precuneus), temporal, prefrontal, orbitofrontal, and anterior cingulate regions as previously described ( Jack et al. , 2008 ; Lowe et al. , 2009 ).
Statistical analysis
All subjects were required to have data from two imaging visits that passed the quality control—one baseline scan plus one follow-up scan. We averaged regional atrophy rates from the in-house modified Automated Anatomical Labelling atlas to derive atrophy rates in 10 grey matter regions of interest plus the ventricles. Derived grey matter regions of interest included the following: posterior cingulate gyrus, medial temporal, temporal (excluding medial temporal), occipital, parietal (excluding posterior cingulate gyrus) and frontal lobes, as well as putamen, caudate, pallidum and thalamus nuclei. We used separate linear regression models of global PiB SUVR at baseline on annualized log Jacobian, adjusting for age at baseline, to assess the relationship between global PiB SUVR and the rate of change in grey matter in each region of interest. Annualized log Jacobian ×100 was used as an estimate of the annual per cent change in grey matter volume. The same was repeated to assess the relationship between global PiB SUVR and the rate of change in ventricular volume. To assess associations between baseline global PiB SUVR and annual per cent change in cognitive and functional measures such as MMSE and CDR-SOB, we first obtained annual per cent change by performing a linear regression on log of cognition for each subject using age as on the time scale. We used the transformation 100 × [exp (slope) − 1], which allows us to interpret the slope as an approximate annual percentage change. The next step was to calculate the Spearman rank correlations and associated P -values between annual percentage change in cognition and baseline global PiB SUVR. We fit a linear regression between annual percentage change in cognition and baseline global PiB SUVR to estimate the annualized change in cognition for a 1-unit decrease in PiB. To assess the relationship between baseline cognition and baseline PiB, as well as between change in cognition and change in grey matter volume, we again used Spearman’s rank correlations and associated P -values. Analyses were performed using statistical software R v3.1.1.
Results
Subjects characteristics
Table 1 summarizes the characteristics of the 20 patients with probable DLB included in this study. Our sample had a mean baseline age of 70 years with a low frequency of females (15%), and a high frequency of visual hallucinations (80%), fluctuations (85%), parkinsonism (95%) and probable REM sleep behaviour disorder (95%). At baseline, mean MMSE score was 22 in our sample, while mean CDR-SOB was 4.0. APOE ε4 allele carrier frequency of was 40%. Average global PiB SUVR at baseline was 1.53.
Table 1.
Demographic and clinical characteristics of patients with probable DLB at baseline
| Mean (SD) or count (%) | |
|---|---|
| DLB included, n | 20 |
| Females, n (%) | 3 (15) |
| Age at baseline MRI, years | 70 (7.5) |
| Time between MRI scans, years | 2.5 (1.14) |
| Education, years | 15 (3.3) |
| APOE ε4 carriers, n (%) | 8 (40) |
| CDR-SOB score | 4.0 (1.6) |
| MMSE | 22 (5) |
| UPDRS-III | 9 (5) |
| Visual hallucinations, n (%) | 16 (80) |
| Fluctuations, n (%) | 17 (85) |
| Parkinsonism, n (%) | 19 (95) |
| Probable RBD, n (%) | 19 (95) |
| PiB ratio at baseline | 1.53 (0.35) |
| Treatment with dopaminergic agent only, n (%) | 1 (5) |
| Treatment with AChEI, n (%) | 19 (95) |
| AChEI only, n (%) | 10 (50) |
| AChEI + dopaminergic agent, n (%) | 9 (45) |
| Treatment with atypical neuroleptics, n (%) | 6 (30) |
SD = standard deviation.
The average time interval between the baseline and follow-up MRI scans was 2.5 years. During this period, all of the participants were exposed to AChEI treatment. In particular, 90% of the patients with DLB ( n = 18) were continuously treated with AChEI for the whole study duration, while one subject (5%) who was only on dopaminergic therapy at baseline, was introduced to AChEI therapy during the inter-scan interval and remained on this therapeutic regimen without further discontinuations. Another subject (5%), who was on AChEI therapy at baseline, suspended treatment before the follow-up scan and started a dopaminergic therapy. Neither of the two abovementioned subjects behaved as outlier in the clinical or imaging data. Furthermore, 75% of patients with DLB ( n = 15) were on dopaminergic therapy during the study period. Five of them (25% of the sample) were not on dopaminergic drugs at baseline and initiated this therapy during the interscan interval. None of the patients suspended the dopaminergic therapy during the interscan interval. In our sample, the levodopa equivalent dose range at baseline was 180–800 mg, while the levodopa equivalent dose range at follow up was 180–1000 mg. Atypical neuroleptic drugs were administered to six (30%) patients during the study period ( Table 1 ).
Baseline 11 C-Pittsburgh compound B-PET retention and rate of grey matter atrophy
After adjusting for age, an association between higher global PiB SUVR at baseline and greater decline in grey matter volume expressed as annualized log Jacobian percentage was found in the following cortical regions of interest: the posterior cingulate gyrus ( P = 0.02), medial temporal lobe ( P = 0.01) and temporal lobe (this region excludes the medial temporal region; P < 0.05), and occipital lobe ( P = 0.02) but not the frontal ( P = 0.17) nor parietal lobes (this region excludes the posterior cingulate gyrus; P = 0.78). A similar association of higher global PiB SUVR at baseline and higher grey matter atrophy rates was present also in subcortical grey matter regions such as the caudate ( P < 0.01) and putamen ( P < 0.01) nuclei but not in the globus pallidus ( P = 0.92) or thalamus ( P = 0.20) ( Table 2 ). A graphical representation of these associations is shown in Fig. 1 . Removing the outliers for each analysis had an influence on the estimated effect by less than 1 standard error. Furthermore, an association between higher PiB retention at baseline and greater annual per cent increase in ventricular volume was observed ( P < 0.01; Fig. 2 ). Figure 3 shows four patients with DLB who exemplify TBM-SyN findings of patients with high and low PiB retentions at different ages.
Table 2.
Relationship between regional annualized log Jacobian and global PiB SUVR, controlling for age
| PiB SUVR estimate (SE) a | PiB SUVR P -value | |
|---|---|---|
| Cortical grey matter regions | ||
| Posterior cingulate gyrus | −0.0190 (0.008) | 0.023 |
| Medial temporal lobe | −0.0218 (0.008) | 0.012 |
| Occipital lobe | −0.0128 (0.005) | 0.016 |
| Temporal lobe | −0.0151 (0.007) | 0.047 |
| Frontal lobe | −0.0063 (0.004) | 0.166 |
| Parietal lobe | −0.0024 (0.009) | 0.783 |
| Subcortical grey matter regions | ||
| Caudate | −0.0260 (0.008) | 0.007 |
| Putamen | −0.0189 (0.006) | 0.009 |
| Globus pallidus | 0.0019 (0.018) | 0.917 |
| Thalamus | −0.0109 (0.008) | 0.196 |
| Ventricles | 0.0487 (0.016) | 0.007 |
SE = standard error.
a Estimate of the change in annualized log Jacobian for a 1-unit increase in PiB SUVR. We multiply by 100 to get approximate per cent change.
Annualized log Jacobian estimates × 100 can be interpreted as annualized percentage change in grey matter volume. Values in bold indicate P < 0.05.
Figure 1.

Associations between baseline PiB retention and grey matter atrophy rates. Linear regression model between baseline global PiB SUVR ( x -axes) and regional grey matter annualized log Jacobian ( y -axes) in patients with DLB, after adjusting for age. Only in the regions of interest with significant association ( P < 0.05) are displayed. Regression line is shown for a 70-year-old.
Figure 2.
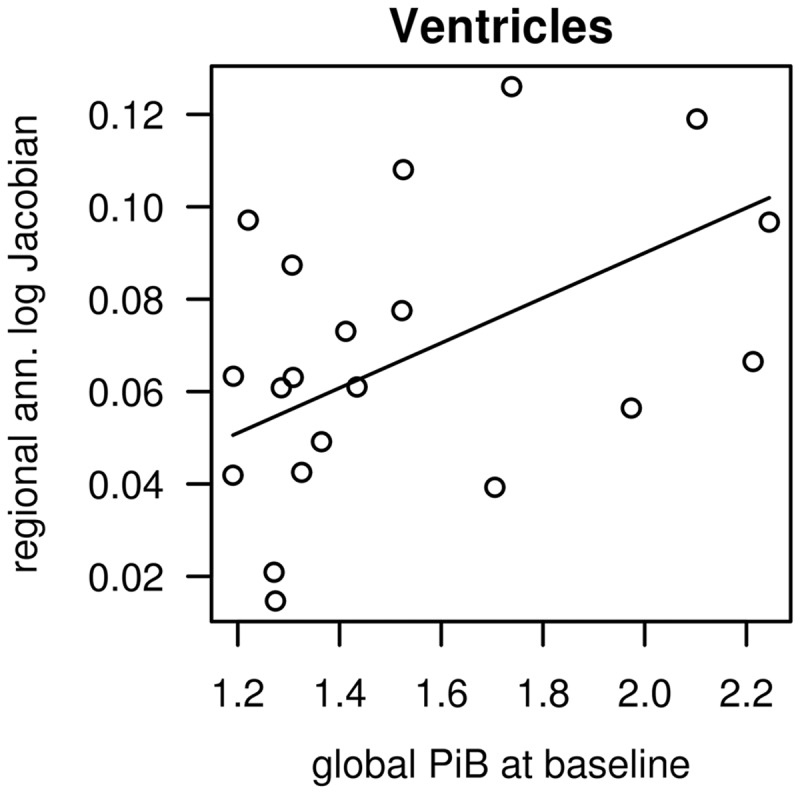
The association between baseline PiB retention and ventricular volume rate of change. Linear regression model between baseline global PiB SUVR ( x -axis) and annualized log Jacobian in ventricular volume ( y -axis) in patients with DLB, after adjusting for age, are shown. Regression line is shown for a 70-year-old.
Figure 3.

TBM-SyN maps of four subjects included in the analysis. ( A ) Subject is a 79-year-old female, with high baseline global PiB retention (PiB SUVR = 2.2). During an inter-scan interval of 2 years, the annual percentage change (APC) in grey matter showed diffuse volume shrinkage in the cortex and basal ganglia. In particular, in the medial temporal lobe the APC grey matter loss was 5%, while the volume ventricle APC expanded by 6%. This can be seen in the figure where, in the medial temporal lobe, the hippocampi are coloured in blue (volume shrinking), while the ventricles are coloured in red (volume expansion). ( B ) Subject is a 62-year-old female with a high baseline global PiB retention (PiB SUVR = 2.1), and an inter-scan interval of 2 years. Grey matter volume loss was found diffusely in the cortical and subcortical structures, with 2% APC grey matter shrinkage (coloured in blue) in medial temporal lobe and a 12% APC of expansion (coloured in red) in the ventricles. ( C ) Subject is a 70-year-old male, with low baseline global PiB retention (PiB SUVR = 1.3), and an inter-scan interval of 3 years. Minimal grey matter volume shrinkage was found throughout the brain. In particular, no loss of volume was found in medial temporal lobe (APC = 0%; no/minimal blue colouring), while ventricles volume did show a volume expansion (APC = 6%; coloured in red). ( D ) Subject is a 53-year-old male, with low baseline global PiB retention (PiB SUVR = 1.2), and an inter-scan interval of 3 years. Minimal grey matter volume shrinkage was found throughout the brain. In particular, no loss of grey matter volume was found in medial temporal lobe (APC = 0%; no/minimal blue colouring), while ventricles volume expanded (APC = 9%; coloured in red).
Baseline 11 C-Pittsburgh compound B-PET retention and global cognitive and functional measures
Higher PiB retention at baseline was not associated with MMSE (rho = −0.4, P = 0.08) or CDR-SOB (rho = −0.09, P = 0.71) scores at baseline. However, a correlation was found between higher global PiB retention at baseline and the annual per cent increase (i.e. worsening) in CDR-SOB scores (rho = 0.51; P = 0.02). No correlation was found between global PiB retention at baseline and MMSE (rho = −0.26; P = 0.29; data were missing at the second time point in one subject) annual per cent of change.
Brain atrophy rates and change in global cognitive and functional measures
Greater annual per cent of ventricles volume increase was associated with greater annual per cent increase (i.e. worsening) in CDR-SOB (rho = 0.54; P = 0.01). On the other hand, no correlation was found between the brain atrophy rates and annual changes in MMSE.
Discussion
In this study, higher PiB retention at baseline was associated with greater grey matter loss over time in regions such as the posterior cingulate gyrus, temporal lobe, occipital lobe and striatum, and with greater ventricular expansion over time. Furthermore, higher global PiB retention at baseline correlated with greater decline in CDR-SOB in patients with probable DLB.
Evidence from in vivo studies tracking biomarker trajectories during the progression of Alzheimer’s disease supports the hypothesis that amyloid-β deposition on PET precedes atrophy on MRI ( Jack et al. , 2013 , 2016 ; Knopman et al. , 2013 ; Villemagne et al. , 2013 ). Assuming that Alzheimer’s disease-related biomarkers would follow a similar trajectory in DLB, this study was designed to investigate the relationship between baseline PiB retention and atrophy progression on MRI in patients with a clinical diagnosis of probable DLB. Longitudinal MRI studies in patients with a spectrum of Alzheimer’s disease and Lewy body disease pathologies at autopsy indicate that ante-mortem atrophy rates in the limbic and association neocortices ( Nedelska et al. , 2015 ), as well as ventricular expansion rates ( Whitwell et al. , 2007 a ; Nedelska et al. , 2015 ) are primarily driven by Alzheimer’s disease pathology. Furthermore, longitudinal studies indicate that global atrophy ( Mak et al. , 2015 a ) and cortical thinning rates ( Mak et al. , 2015 b ) in patients with clinically diagnosed probable DLB are lower than patients with Alzheimer’s disease dementia and not different from controls. These observations, along with our findings, suggest that patients with probable DLB, who have additional amyloid-β deposition, develop higher rates of ventricular expansion and higher rates of brain atrophy, with an Alzheimer’s disease-like pattern.
Congruent with this interpretation, a cross-sectional analysis demonstrated that in patients within the Lewy body disease spectrum (probable DLB and Parkinson’s disease dementia) with high PiB retention ( n = 6), the pattern of atrophy overlapped with that observed in patients with Alzheimer’s disease dementia ( n = 13) by more than 95%, whereas patients with low PiB retention ( n = 9) showed no cortical atrophy compared to cognitively normal and Alzheimer’s disease subjects ( Shimada et al. , 2013 ). Despite the small sample size and a cross-sectional design, the findings are consistent with the current study that there is an association between higher PiB retention at baseline and increased rates of cortical atrophy. Several longitudinal studies in non-demented older adults also support our finding that higher PiB retention at baseline is associated with greater atrophy rates ( Chetelat et al. , 2012 ; Andrews et al. , 2013 ; Dore et al. , 2013 ; Araque Caballero et al. , 2015 ; Petersen et al. , 2016 ).
In addition to the higher rates of atrophy in the cortex, we also identified an association between higher PiB retention at baseline, and higher rates of atrophy in the striatum, which typically has reduced dopaminergic activity in DLB. In vivo striatum dopaminergic denervation with loss of presynaptic striatal dopamine transporter (DAT), as measured by single-photon emission tomography (SPECT) and PET, is a consistent finding among patients with probable DLB, compared to clinically normal controls and patients with Alzheimer’s disease dementia ( Walker et al. , 2002 , 2004 ; O'Brien et al. , 2004 ; McKeith et al. , 2007 ). While the dopaminergic degeneration has been widely documented to affect the striatum of patients with DLB ( Walker et al. , 2004 ; Klein et al. , 2010 ), a cholinergic network alteration involving the striatum has been postulated in the Lewy body disease spectrum of disorders ( Langlais et al. , 1993 ) but its mechanism is still poorly understood ( Bohnen and Albin, 2011 ). In this setting, our finding of an association between higher PiB retention and higher atrophy rates in the caudate and putamen nuclei suggest an accelerated subcortical neuronal injury involving the dopaminergic and possibly the cholinergic system in patients with higher amyloid-β load. Higher burden of both α-synuclein ( Duda et al. , 2002 a ; Jellinger and Attems, 2006 ) and amyloid-β deposition ( Jellinger and Attems, 2006 ; Edison et al. , 2008 ; Kalaitzakis et al. , 2008 ) has been observed in the striatum of patients with DLB, suggesting a possible interaction between α-synuclein and amyloid-β aggregation. Furthermore, tau and α-synuclein deposits may be aggregating topographically in close proximity in synucleinopathies ( Duda et al. , 2002 b ; Ishizawa et al. , 2003 ; Kotzbauer et al. , 2004 ), supporting direct cross-seeding between the two types of pathologies ( Guo et al. , 2013 ), which could influence striatal degeneration in DLB patients.
Another finding in this study was the correlation between higher PiB retention at baseline and greater rates of functional disease progression on CDR-SOB, which also correlated with the ventricular expansion rates. Cognitive rating of disease progression using MMSE, in contrast, was not related to PiB retention at baseline. The simplest explanation may be attributed to reduced statistical power given the heterogeneity of DLB and the relatively small sample size. Cognitive test performance may be more sensitive to other factors that we could not account for, such as the influence of other pathologies (e.g. α-synuclein, tau) and other non-neurological factors (e.g. mood, frailty, pain, sleepiness). Although it may be argued that AChEI might mitigate the relationship between performance and grey matter volume by improving cognitive scores, this seems unlikely given that all patients had exposure to AChEI. Variability in the relationship between amyloid-β and disease severity is evidence in the literature with some studies showing higher amyloid-β deposition in those with lower CDR ( Foster et al. , 2010 ) and MMSE scores ( Compta et al. , 2011 ), while others show no such association ( Compta et al. , 2011 ; Shimada et al. , 2013 ). The relationship between disease progression and PiB may be suggestive of a more complex relationship between amyloid-β, tau and α-synuclein that is not easily disentangled by examining only amyloid-β. Furthermore, since PiB imaging does not distinguish between amyloid-β deposition of diffuse more than neuritic plaques ( Kantarci et al. , 2012 c ), it is also not known whether this distinction may make a difference in the effects of amyloid protein aggregation in DLB, and may contribute to some of this variability. Overall our findings are in agreement with the hypothesis that patients with probable DLB with concomitant underlying Alzheimer’s disease pathology could show a more aggressive clinical phenotype of the disease with increased atrophy rates and functional decline ( Nelson et al. , 2009 ; Clinton et al. , 2010 ; Gomperts et al. , 2012 ; De Beer et al. , 2015 ; Ferman et al. , 2015 ; Graff-Radford et al. , 2015 ; Howlett et al. , 2015 ).
There are limitations to take into account, including potential bias related to the small sample size and ascertainment of subjects from a referral clinic-based cohort. Future studies that include ante-mortem imaging and post-mortem pathological analysis, as well as bigger sample sizes are needed to better understand PiB retention as a surrogate marker of amyloid-β deposition in DLB ( Kantarci et al. , 2012 c ).
Current results, although preliminary, provide evidence for a faster rate of grey matter loss in DLB patients with high amyloid-β deposition. More work is needed to better establish whether amyloid-β deposition is also related to a high level of tau and α-synuclein aggregation, suggesting a synergistic or additive relationship, or if amyloid-β deposition provides a unique contribution to neuronal loss and grey matter atrophy. Also, our findings are a step forward towards understanding the variability of disease progression in DLB, and in vivo biomarkers for α-synuclein may provide further insight into the pathogenesis of this complex disease. Furthermore, in vivo demonstration of a relationship between higher amyloid pathology in patients with probable DLB and progression of neuronal damage and clinical decline provides a rationale for targeting the subset of DLB patients with significant amyloid burden, together with patients with Alzheimer’s disease dementia, with emerging anti-amyloid agents.
Funding
This study was funded by the NIH [R01 AG040042, R01 AG11378, P50 AG16574, P50 AG44170, U01 AG06786, C06 RR018898], Foundation Dr. Corinne Schulerand, the Mangurian Foundation, The Elsie and Marvin Dekelboum Family Foundation, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program. The funding sources had no role in study design, collection, analysis, interpretation, or decision to submit this paper. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Conflict of interest
L. Sarro, E.S. Lundt, T.G. Lesnick, S.A. Przybelski and J. Graff-Radford report no disclosures. M.L. Senjem reports stock/options in Gilead Sciences, Inovio Pharmaceuticals Inc., outside the submitted work. Dr Boeve has served as an investigator for clinical trials sponsored by GE Healthcare and FORUM Pharmaceuticals. He receives royalties from the publication of a book entitled Behavioural Neurology Of Dementia (Cambridge Medicine, 2009). He serves on the Scientific Advisory Board of the Tau Consortium. He has consulted for Isis Pharmaceuticals. He receives research support from the NIH (U01 AG045390, U54 NS092089, P50 AG016574, UO1 AG006786, RO1 AG041797), and the Mangurian Foundation. Dr Lowe is a consultant for Bayer Schering Pharma, Piramal Imaging Inc, and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, NCI), the Elsie and Marvin Dekelboum Family Foundation, the Liston Family Foundation, and the MN Partnership for Biotechnology and Medical Genomics. Dr Ferman is funded by the NIH (Mayo Clinic Alzheimer's Disease Research Center/Project 1-P50-AG16574/P1 [Co-I]). Dr Knopman serves as Deputy Editor for Neurology ® ; serves on a Data Safety Monitoring Board for Lundbeck Pharmaceuticals and for the Dominantly Inherited Alzheimer’s Disease Treatment Unit. He is participating in clinical trials sponsored by Lilly Pharmaceuticals and Tau Rx Pharmaceuticals. He receives research support from the NIH. Dr Comi has received honoraria from Teva, Sanofi, Genzyme, Merck Serono, Biogen, Bayer, Serono Symposia International Foundation, Excemed, Roche, Almirall, Chugai, Receptos, Forward Pharma, outside the submitted work. Dr Filippi is Editor in Chief of the Journal of Neurology; serves on scientific advisory boards for Teva Pharmaceutical Industries; has received compensation for consulting services and/or speaking activities from Biogen Idec, Excemed, Novartis, and Teva Pharmaceutical Industries; and has received research support from Biogen Idec, Teva Pharmaceutical Industries, Novartis, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, Cure PSP, Alzheimer's Drug Discovery Foundation (ADDF), the Jacques and Gloria Gossweiler Foundation (Switzerland), and ARiSLA (Fondazione Italiana di Ricerca per la SLA). Dr Petersen chaired a Data Monitoring Committee of Pfizer, Inc. and Janssen Alzheimer Immunotherapy and serves as a consultant for Hoffman La Roche, Inc., Merck, Inc., Genentech, Inc., Biogen, Inc., Eli Lilly and Co. and receives research support from the NIH (P50 AG016574 [PI] and U01 AG006786 [PI], R01 AG011378 [Co-I], and U01 AG024904 [Co-I]). Dr Jack reports consulting services for Eli Lilly Co, funding from the NIH (R01 AG011378, U01 AG024904, RO1 AG041851, R01 AG037551, R01 AG043392, and U01 AG006786) and research support from the Alexander Family Professorship of Alzheimer’s Disease Research. Dr Kantarci serves on the data safety monitoring board for Pfizer Inc. and Janssen Alzheimer’s Immunotherapy Takeda Global Research & Development Center, Inc.; and she is funded by the NIH.
Glossary
Abbreviations
- AChEI =
acetylcholinesterase inhibitor
- CDR-SOB =
Clinical Dementia Rating scale, Sum of Boxes
- DLB =
dementia with Lewy bodies
- MMSE =
Mini-Mental State Examination
- PiB =
11 C-Pittsburgh compound B
- SUVR =
standardized uptake value ratio
- TBM-SyN =
tensor-based morphometry using symmetric diffeomorphic image normalization
- UPDRS-III =
Unified Parkinson’s Disease Rating Scale part III
References
- Andrews KA , Modat M , Macdonald KE , Yeatman T , Cardoso MJ , Leung KK , et al. . Atrophy rates in asymptomatic amyloidosis: implications for Alzheimer prevention trials . PLoS One 2013. ; 8 : e58816 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque Caballero MA , Brendel M , Delker A , Ren J , Rominger A , Bartenstein P , et al. . Mapping 3-year changes in gray matter and metabolism in Abeta-positive nondemented subjects . Neurobiol Aging 2015. ; 36 : 2913 – 24 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer HA , Edison P , Brooks DJ , Barnes J , Frost C , Yeatman T , et al. . Amyloid load and cerebral atrophy in Alzheimer's disease: an 11C-PIB positron emission tomography study . Ann Neurol 2006. ; 60 : 145 – 7 . [DOI] [PubMed] [Google Scholar]
- Avants BB , Epstein CL , Grossman M , Gee JC . Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain . Med Image Anal 2008. ; 12 : 26 – 41 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF , Molano JR , Ferman TJ , Smith GE , Lin SC , Bieniek K , et al. . Validation of the Mayo Sleep Questionnaire to screen for REM sleep behavior disorder in an aging and dementia cohort . Sleep Med 2011. ; 12 : 445 – 53 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI , Albin RL . The cholinergic system and Parkinson disease . Behav Brain Res 2011. ; 221 : 564 – 73 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H , Braak E . Neuropathological stageing of Alzheimer-related changes . Acta Neuropathol 1991. ; 82 : 239 – 59 . [DOI] [PubMed] [Google Scholar]
- Burack MA , Hartlein J , Flores HP , Taylor-Reinwald L , Perlmutter JS , Cairns NJ . In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia . Neurology 2010. ; 74 : 77 – 84 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton EJ , Barber R , Mukaetova-Ladinska EB , Robson J , Perry RH , Jaros E , et al. . Medial temporal lobe atrophy on MRI differentiates Alzheimer's disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis . Brain 2009. ; 132 ( Pt 1 ): 195 – 203 . [DOI] [PubMed] [Google Scholar]
- Cash DM , Frost C , Iheme LO , Unay D , Kandemir M , Fripp J , et al. . Assessing atrophy measurement techniques in dementia: Results from the MIRIAD atrophy challenge . Neuroimage 2015. ; 123 : 149 – 64 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catafau AM , Bullich S . Amyloid PET imaging: applications beyond Alzheimer's disease . Clin Transl Imaging 2015. ; 3 : 39 – 55 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G , Villemagne VL , Bourgeat P , Pike KE , Jones G , Ames D , et al. . Relationship between atrophy and beta-amyloid deposition in Alzheimer disease . Ann Neurol 2010. ; 67 : 317 – 24 . [DOI] [PubMed] [Google Scholar]
- Chetelat G , Villemagne VL , Villain N , Jones G , Ellis KA , Ames D , et al. . Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition . Neurology 2012. ; 78 : 477 – 84 . [DOI] [PubMed] [Google Scholar]
- Claassen DO , Lowe VJ , Peller PJ , Petersen RC , Josephs KA . Amyloid and glucose imaging in dementia with Lewy bodies and multiple systems atrophy . Parkinsonism Relat Disord 2011. ; 17 : 160 – 5 . [DOI] [PubMed] [Google Scholar]
- Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM . Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline . J Neurosci 2010. ; 30 : 7281 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compta Y , Parkkinen L , O'Sullivan SS , Vandrovcova J , Holton JL , Collins C , et al. . Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain 2011. ; 134 ( Pt 5 ): 1493 – 505 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beer M , Teunissen C , Van der Flier W , Sikkes S , Lemstra A . Dementia with Lewy bodies and Alzheimer’s disease: an unfortunate couple . Am J Neurodegener Dis 2015. ; 4 : 1 – 178 . 26389015 [Google Scholar]
- Dore V , Villemagne VL , Bourgeat P , Fripp J , Acosta O , Chetelat G , et al. . Cross-sectional and longitudinal analysis of the relationship between Abeta deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease . JAMA Neurol 2013. ; 70 : 903 – 11 . [DOI] [PubMed] [Google Scholar]
- Driscoll I , Troncoso JC , Rudow G , Sojkova J , Pletnikova O , Zhou Y , et al. . Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques . Acta Neuropathol 2012. ; 124 : 823 – 31 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE , Giasson BI , Mabon ME , Lee VM , Trojanowski JQ . Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases . Ann Neurol 2002a. ; 52 : 205 – 10 . [DOI] [PubMed] [Google Scholar]
- Duda JE , Giasson BI , Mabon ME , Miller DC , Golbe LI , Lee VM , et al. . Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred . Acta Neuropathol 2002b. ; 104 : 7 – 11 . [DOI] [PubMed] [Google Scholar]
- Edison P , Rowe CC , Rinne JO , Ng S , Ahmed I , Kemppainen N , et al. . Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography . J Neurol Neurosurg Psychiatry 2008. ; 79 : 1331 – 8 . [DOI] [PubMed] [Google Scholar]
- Ferman TJ , Aoki N , Murray ME , Boeve BF , Graff-Radford N , Van Gerpen J , et al. . disease trajectory and cognitive profiles of three pathologic subtypes of DLB . Am J Neurodegener Dis 2015. ; 4 ( Suppl 1 ): 1 – 178 . 26389015 [Google Scholar]
- Ferman TJ , Smith GE , Boeve BF , Ivnik RJ , Petersen RC , Knopman D , et al. . DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging . Neurology 2004. ; 62 : 181 – 7 . [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT , Smith DP , McLean CA , Adlard PA , Barnham KJ , Foster LE , et al. . In vitro characterization of Pittsburgh compound-B binding to Lewy bodies . J Neurosci 2007. ; 27 : 10365 – 71 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster ER , Campbell MC , Burack MA , Hartlein J , Flores HP , Cairns NJ , et al. . Amyloid imaging of Lewy body-associated disorders . Mov Disord 2010. ; 25 : 2516 – 23 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN , Locascio JJ , Marquie M , Santarlasci AL , Rentz DM , Maye J , et al. . Brain amyloid and cognition in Lewy body diseases . Mov Disord 2012. ; 27 : 965 – 73 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN , Rentz DM , Moran E , Becker JA , Locascio JJ , Klunk WE , et al. . Imaging amyloid deposition in Lewy body diseases . Neurology 2008. ; 71 : 903 – 10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford J , Lesnick T , Boeve BF , Przybelski S , Ferman TJ , Jones D , et al. . Predicting Survival in Dementia with Lewy Bodies with Hippocampal Volumetry . Am J Neurodegener Dis 2015. ; 4 ( Suppl 1 ): 1 – 178 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , et al. . Distinct alpha-synuclein strains differentially promote tau inclusions in neurons . Cell 2013. ; 154 : 103 – 17 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ , Halliday GM . Cortical Lewy body pathology in the diagnosis of dementia . Acta Neuropathol 2001. ; 102 : 355 – 63 . [DOI] [PubMed] [Google Scholar]
- Howlett DR , Whitfield D , Johnson M , Attems J , O'Brien JT , Aarsland D , et al. . Regional multiple pathology scores are associated with cognitive decline in lewy body dementias . Brain Pathol 2015. ; 25 : 401 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa T , Mattila P , Davies P , Wang D , Dickson DW . Colocalization of tau and alpha-synuclein epitopes in Lewy bodies . J Neuropathol Exp Neurol 2003. ; 62 : 389 – 97 . [DOI] [PubMed] [Google Scholar]
- Jack CR Jr , Lowe VJ , Senjem ML , Weigand SD , Kemp BJ , Shiung MM , et al. . 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment . Brain 2008. ; 131 ( Pt 3 ): 665 – 80 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr , Lowe VJ , Weigand SD , Wiste HJ , Senjem ML , Knopman DS , et al. . Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease . Brain 2009. ; 132 ( Pt 5 ): 1355 – 65 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr , Shiung MM , Gunter JL , O'Brien PC , Weigand SD , Knopman DS , et al. . Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD . Neurology 2004. ; 62 : 591 – 600 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr , Therneau TM , Wiste HJ , Weigand SD , Knopman DS , Lowe VJ , et al. . Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study . Lancet Neurol 2016. ; 15 : 56 – 64 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr , Wiste HJ , Knopman DS , Vemuri P , Mielke MM , Weigand SD , et al. . Rates of beta-amyloid accumulation are independent of hippocampal neurodegeneration . Neurology 2014. ; 82 : 1605 – 12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , et al. . Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers . Lancet Neurol 2013. ; 12 : 207 – 16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA , Attems J . Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 2006. ; 112 : 253 – 60 . [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME , Graeber MB , Gentleman SM , Pearce RK . Striatal beta-amyloid deposition in Parkinson disease with dementia . J Neuropathol Exp Neurol 2008. ; 67 : 155 – 61 . [DOI] [PubMed] [Google Scholar]
- Kantarci K , Ferman TJ , Boeve BF , Weigand SD , Przybelski S , Vemuri P , et al. . Focal atrophy on MRI and neuropathologic classification of dementia with Lewy bodies . Neurology 2012a. ; 79 : 553 – 60 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K , Lowe VJ , Boeve BF , Weigand SD , Senjem ML , Przybelski SA , et al. . Multimodality imaging characteristics of dementia with Lewy bodies . Neurobiol Aging 2012b. ; 33 : 2091 – 105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K , Yang C , Schneider JA , Senjem ML , Reyes DA , Lowe VJ , et al. . Antemortem amyloid imaging and beta-amyloid pathology in a case with dementia with Lewy bodies . Neurobiol Aging 2012c. ; 33 : 878 – 85 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JC , Eggers C , Kalbe E , Weisenbach S , Hohmann C , Vollmar S , et al. . Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo . Neurology 2010. ; 74 : 885 – 92 . [DOI] [PubMed] [Google Scholar]
- Knopman DS , Jack CR Jr , Wiste HJ , Weigand SD , Vemuri P , Lowe VJ , et al. . Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis . JAMA Neurol 2013. ; 70 : 1030 – 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer PT , Cairns NJ , Campbell MC , Willis AW , Racette BA , Tabbal SD , et al. . Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia . Arch Neurol 2012. ; 69 : 1326 – 31 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer PT , Giasson BI , Kravitz AV , Golbe LI , Mark MH , Trojanowski JQ , et al. . Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation . Exp Neurol 2004. ; 187 : 279 – 88 . [DOI] [PubMed] [Google Scholar]
- Kraybill ML , Larson EB , Tsuang DW , Teri L , McCormick WC , Bowen JD , et al. . Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both . Neurology 2005. ; 64 : 2069 – 73 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ , Thal L , Hansen L , Galasko D , Alford M , Masliah E . Neurotransmitters in basal ganglia and cortex of Alzheimer's disease with and without Lewy bodies . Neurology 1993. ; 43 : 1927 – 34 . [DOI] [PubMed] [Google Scholar]
- Lowe VJ , Kemp BJ , Jack CR Jr , Senjem M , Weigand S , Shiung M , et al. . Comparison of 18F-FDG and PiB PET in cognitive impairment . J Nucl Med 2009. ; 50 : 878 – 86 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maetzler W , Liepelt I , Reimold M , Reischl G , Solbach C , Becker C , et al. . Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics . Neurobiol Dis 2009. ; 34 : 107 – 12 . [DOI] [PubMed] [Google Scholar]
- Mak E , Li S , Williams GB , Watson R , Firbank M , Blamire A , et al. . Differential Atrophy of hippocampal subfields: a comparative study of dementia with lewy bodies and Alzheimer disease . Am J Geriatr Psychiatry 2016. : 24 : 136 – 43 . [DOI] [PubMed] [Google Scholar]
- Mak E , Su L , Williams GB , Watson R , Firbank M , Blamire AM , et al. . Longitudinal assessment of global and regional atrophy rates in Alzheimer's disease and dementia with Lewy bodies . Neuroimage Clin 2015a. ; 7 : 456 – 62 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak E , Su L , Williams GB , Watson R , Firbank MJ , Blamire AM , et al. . Progressive cortical thinning and subcortical atrophy in dementia with Lewy bodies and Alzheimer's disease . Neurobiol Aging 2015b. ; 36 : 1743 – 50 . [DOI] [PubMed] [Google Scholar]
- McCleery J , Morgan S , Bradley KM , Noel-Storr AH , Ansorge O , Hyde C . Dopamine transporter imaging for the diagnosis of dementia with Lewy bodies . Cochrane Database Syst Rev 2015. ; 1 : CD010633 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith I , O'Brien J , Walker Z , Tatsch K , Booij J , Darcourt J , et al. . Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study . Lancet Neurol 2007. ; 6 : 305 – 13 . [DOI] [PubMed] [Google Scholar]
- McKeith IG , Dickson DW , Lowe J , Emre M , O'Brien JT , Feldman H , et al. . Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium . Neurology 2005. ; 65 : 1863 – 72 . [DOI] [PubMed] [Google Scholar]
- Minoshima S , Foster NL , Sima AA , Frey KA , Albin RL , Kuhl DE . Alzheimer's disease versus dementia with Lewy bodies: cerebral metabolic distinction with autopsy confirmation . Ann Neurol 2001. ; 50 : 358 – 65 . [DOI] [PubMed] [Google Scholar]
- Murray ME , Ferman TJ , Boeve BF , Przybelski SA , Lesnick TG , Liesinger AM , et al. . MRI and pathology of REM sleep behavior disorder in dementia with Lewy bodies . Neurology 2013. ; 81 : 1681 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelska Z , Ferman TJ , Boeve BF , Przybelski SA , Lesnick TG , Murray ME , et al. . Pattern of brain atrophy rates in autopsy-confirmed dementia with Lewy bodies . Neurobiol Aging 2015. ; 36 : 452 – 61 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT , Kryscio RJ , Jicha GA , Abner EL , Schmitt FA , Xu LO , et al. . Relative preservation of MMSE scores in autopsy-proven dementia with Lewy bodies . Neurology 2009. ; 73 : 1127 – 33 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JT , Colloby S , Fenwick J , Williams ED , Firbank M , Burn D , et al. . Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with Lewy bodies . Arch Neurol 2004. ; 61 : 919 – 25 . [DOI] [PubMed] [Google Scholar]
- Petersen RC , Aisen P , Boeve BF , Geda YE , Ivnik RJ , Knopman DS , et al. . Mild cognitive impairment due to Alzheimer disease in the community . Ann Neurol 2013. ; 74 : 199 – 208 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC , Wiste HJ , Weigand SD , Rocca WA , Roberts RO , Mielke MM , et al. . Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community . JAMA Neurol 2016. ; 73 : 85 – 92 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrou M , Dwamena BA , Foerster BR , MacEachern MP , Bohnen NI , Muller ML , et al. . Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review . Mov Disord 2015. ; 30 : 928 – 35 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabattoli F , Boccardi M , Galluzzi S , Treves A , Thompson PM , Frisoni GB . Hippocampal shape differences in dementia with Lewy bodies . Neuroimage 2008. ; 41 : 699 – 705 . [DOI] [PubMed] [Google Scholar]
- Schneider JA , Arvanitakis Z , Bang W , Bennett DA . Mixed brain pathologies account for most dementia cases in community-dwelling older persons . Neurology 2007. ; 69 : 2197 – 204 . [DOI] [PubMed] [Google Scholar]
- Shimada H , Shinotoh H , Hirano S , Miyoshi M , Sato K , Tanaka N , et al. . beta-Amyloid in Lewy body disease is related to Alzheimer's disease-like atrophy . Mov Disord 2013. ; 28 : 169 – 75 . [DOI] [PubMed] [Google Scholar]
- Teune LK , Bartels AL , de Jong BM , Willemsen AT , Eshuis SA , de Vries JJ , et al. . Typical cerebral metabolic patterns in neurodegenerative brain diseases . Mov Disord 2010. ; 25 : 2395 – 404 . [DOI] [PubMed] [Google Scholar]
- Tomlinson CL , Stowe R , Patel S , Rick C , Gray R , Clarke CE . Systematic review of levodopa dose equivalency reporting in Parkinson's disease . Mov Disord 2010. ; 25 : 2649 – 53 . [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N , Landeau B , Papathanassiou D , Crivello F , Etard O , Delcroix N , et al. . Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain . Neuroimage 2002. ; 15 : 273 – 89 . [DOI] [PubMed] [Google Scholar]
- Vemuri P , Whitwell JL , Kantarci K , Josephs KA , Parisi JE , Shiung MS , et al. . Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem braak neurofibrillary tangle stage . Neuroimage 2008. ; 42 : 559 – 67 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , et al. . Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study . Lancet Neurol 2013. ; 12 : 357 – 67 . [DOI] [PubMed] [Google Scholar]
- Villemagne VL , Ong K , Mulligan RS , Holl G , Pejoska S , Jones G , et al. . Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias . J Nucl Med 2011. ; 52 : 1210 – 17 . [DOI] [PubMed] [Google Scholar]
- Walker Z , Costa DC , Walker RW , Lee L , Livingston G , Jaros E , et al. . Striatal dopamine transporter in dementia with Lewy bodies and Parkinson disease: a comparison . Neurology 2004. ; 62 : 1568 – 72 . [DOI] [PubMed] [Google Scholar]
- Walker Z , Costa DC , Walker RW , Shaw K , Gacinovic S , Stevens T , et al. . Differentiation of dementia with Lewy bodies from Alzheimer's disease using a dopaminergic presynaptic ligand . J Neurol Neurosurg Psychiatry 2002. ; 73 : 134 – 40 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL , Jack CR Jr , Parisi JE , Knopman DS , Boeve BF , Petersen RC , et al. . Rates of cerebral atrophy differ in different degenerative pathologies . Brain 2007a. ; 130 ( Pt 4 ): 1148 – 58 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL , Tosakulwong N , Weigand SD , Senjem ML , Lowe VJ , Gunter JL , et al. . Does amyloid deposition produce a specific atrophic signature in cognitively normal subjects? Neuroimage Clin 2013. ; 2 : 249 – 57 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL , Weigand SD , Shiung MM , Boeve BF , Ferman TJ , Smith GE , et al. . Focal atrophy in dementia with Lewy bodies on MRI: a distinct pattern from Alzheimer's disease . Brain 2007b. ; 130 ( Pt 3 ): 708 – 19 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B , Behl C . Mechanisms of neurodegenerative disorders: part 1: protein aggregates . Arch Neurol 2000. ; 57 : 793 – 6 . [DOI] [PubMed] [Google Scholar]
- Zaccai J , McCracken C , Brayne C . A systematic review of prevalence and incidence studies of dementia with Lewy bodies . Age Ageing 2005. ; 34 : 561 – 6 . [DOI] [PubMed] [Google Scholar]
- Zhong J , Pan P , Dai Z , Shi H . Voxelwise meta-analysis of gray matter abnormalities in dementia with Lewy bodies . Eur J Radiol 2014. ; 83 : 1870 – 4 . [DOI] [PubMed] [Google Scholar]