

Significance
We designed this study to elevate fetal hemoglobin for the treatment of β-thalassemia and sickle cell disease (SCD). It has long been known that some individuals who are compound heterozygotes of β-thalassemia or SCD with deletional hereditary persistence of fetal hemoglobin (HPFH) have minimal hematological abnormalities and mild clinical manifestation compared with the homozygous patients. We used CRISPR-Cas9 to modify normal bone marrow hematopoietic stem and progenitor cells (HSPCs) to the deletional HPFH genotype. The erythroid cells derived from such modified HSPCs showed significantly higher γ-globin expression compared with the nondeletion-modified cells. This study provides proof of concept for developing a potential new approach to autologous transplantation therapy for the treatment of homozygous β-thalassemia and SCD.
Keywords: engineered nucleases, deletion, colony assay, fetal hemoglobin, erythroid differentiation
Abstract
Hereditary persistence of fetal hemoglobin (HPFH) is a condition in some individuals who have a high level of fetal hemoglobin throughout life. Individuals with compound heterozygous β-thalassemia or sickle cell disease (SCD) and HPFH have milder clinical manifestations. Using RNA-guided clustered regularly interspaced short palindromic repeats-associated Cas9 (CRISPR-Cas9) genome-editing technology, we deleted, in normal hematopoietic stem and progenitor cells (HSPCs), 13 kb of the β-globin locus to mimic the naturally occurring Sicilian HPFH mutation. The efficiency of targeting deletion reached 31% in cells with the delivery of both upstream and downstream breakpoint guide RNA (gRNA)-guided Staphylococcus aureus Cas9 nuclease (SaCas9). The erythroid colonies differentiated from HSPCs with HPFH deletion showed significantly higher γ-globin gene expression compared with the colonies without deletion. By T7 endonuclease 1 assay, we did not detect any off-target effects in the colonies with deletion. We propose that this strategy of using nonhomologous end joining (NHEJ) to modify the genome may provide an efficient approach toward the development of a safe autologous transplantation for patients with homozygous β-thalassemia and SCD.
Sickle cell disease (SCD) and β-thalassemia are common genetic diseases caused by the coinheritance of two mutant β-globin alleles (in homozygous or compound heterozygous combination) (1). Both are chronic diseases with considerable morbidity and mortality. Curative allogeneic bone marrow or stem cell transplantation is only available for a small subset of patients who have suitable donors (2). Autologous transplant of stem cells transduced with a corrective β-globin gene has been successful in a few patients (3, 4), and larger clinical trials are in progress (5). However, the long-term safety of the known random viral integrations into multiple genomic sites needs to be determined (6, 7). An alternate approach to modify the hematopoietic stem cells that does not use integrating viral vectors needs to be explored.
The syndrome of hereditary persistence of fetal hemoglobin (HPFH) comprises a large number of genetic mutations primarily of the β-globin gene cluster, resulting in elevated fetal hemoglobin (HbF) levels persisting into adulthood (8). These disorders can be due to point mutations in the upstream promoter region of a γ-globin gene or, often, to different deletions affecting the β-globin gene clusters (9). They have also been broadly classified into the heterocellular or the pancellular form according to the distribution of HbF in some or all red blood cells, respectively (10). Some of the deletion group that exhibit pancellular distribution of hemoglobin seem to have no serious ill effect (11). Several individuals homozygous for deletional HPFH express only HbF and are healthy (12, 13). Individuals doubly heterozygous for this type of HPFH and β-thalassemia or SCD have minimal or mild clinical consequences in contrast to the serious illness in patients homozygous for these diseases (14, 15). Recently developed sequence-specific endonucleases afford the possibility of engineering these beneficial HPFH deletions in homozygous β-thalassemia or SCD patients’ own hematopoietic stem cells to allow autologous transplantation.
Among currently available engineered endonucleases, the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) adapted from an element of the bacteria immune system greatly facilitate the mammalian genome editing because of easy manipulation, low cost, and great flexibility (16–18).
In this study, we used RNA-guided Staphylococcus aureus Cas9 nuclease (SaCas9)-mediated genome editing to excise a 13-kb segment of the β-globin gene locus in bone marrow CD34+ hematopoietic stem and progenitor cells (HSPCs) to resemble the naturally occurring HPFH Sicilian deletion (HPFH5). After differentiation into erythroid cells, the cells with the deletion showed higher γ-globin gene expression than the cells without deletion. We conclude that this approach, applicable to homozygous SCD and most β-thalassemia, shows great promise for their treatment.
Results
Designing and Screening Guide RNAs for Generation of Deletional Sicilian HPFH5 by S. aureus Cas9 Nuclease.
There are eight common types of deletional HPFH that have been defined at the molecular level (12, 13, 19–24) (Fig. S1A). Studies analyzing the correlation between phenotype and genotype of patients with different deletional HPFHs have revealed that the elevated HbF levels are associated with the loss of repressive γδ-intergenic sequences in the β-globin locus (25). In particular, a critical area located 5′ to the δ-globin gene was defined as an important binding site for BCL11A that mainly has repressive activity for γ-globin expression (24, 26). The smallest HPFH deletion is the 7.2-kb Corfu deletion extending from the γδ-intergenic region upstream of the δ gene to the 5′ end of the structural δ-globin gene (27) (Fig. S1A). Two patients with homozygous Corfu deletion showed up-regulated γ-globin expression, which was not in the heterozygous parents (27, 28). The inconsistency of γ-globin expression between the homozygous and heterozygous status may suggest that importing the 3′ HS enhancer to the proximity of γ-globin gene is critical for increasing γ-globin expression. For these reasons, we first focused on the Sicilian HPFH5, in which the 12.9-kb deletion starts at 3.2 kb upstream of the δ gene and ends at the 3′ flanking region of the β-globin gene (29). The deletion abolishes the entire putative BCL11A binding sites upstream of the δ globin gene and extends to just upstream of the 3′ β-globin gene enhancer, bringing the latter close to the Aγ-globin gene (Fig. S1B). The deletion not only can increase the γ-globin expression but also can reduce mutant β-globin production, which may be of particular importance for SCD.
Fig. S1.

Deletion type of hereditary persistence of fetal hemoglobin (HPFH). (A) Schematic representation of genomic deletion in common deletion types of HPFH and their corresponding fetal hemoglobin (HbF) level according to published data. The structure of the β-globin cluster. The corresponding genes from γ-globin to the 3′ DNase I hypersensitive site (3′HS-1) are shown on the Top panel. The 3′ break point in HPFH1 to HPFH 4 is beyond the 3′HS-1 site. The corresponding HbF level to each heterozygote deletion is shown on the right. (B) Scheme of normal β-globin locus (Upper) and naturally occurring Sicilian HPFH-5 (Lower). The 12.9-kb deletion from the 5′ δ-globin gene to 3′ of the β-globin gene abolishes the BCL11A binding sites located on the γδ-intergenic region and brings the 3′ β-enhancer and 3′ HS-1 site closer to the γ-globin gene.
To improve on the efficient cleavage sequences in both the 5′ and 3′ breakpoint regions, we chose S. aureus Cas9 nuclease (SaCas9) for genome editing. We reasoned that (i) it is smaller than classical Streptococcus pyogenes (SpCas9), thus increasing the efficiency of gene delivery, and (ii) it requires a longer requisite protospacer-adjacent motif (PAM) (NNGRRT) than SpCas9 does for Cas9 nuclease recognition (NNGRRT vs. NGG, respectively), thus reducing the number of off-target sites in the genome.
We manually chose, by BLAST, four targeting sites each from the 5′ and 3′ break points at the deletion that show less homology with other genome sites (Fig. 1A). The eight guide RNAs (gRNAs), L1 to L4 and R1 to R4 (Table S1) corresponding to the 5′ and 3′ breakpoints, respectively, were constructed into pX601 (Addgene) that contains both SaCas9 and gRNA in a single vector. The constructs were individually transfected into 293T cells to determine the efficiency of indel generation by nuclease-induced double stand break (DSB) and subsequent repaired by nonhomologous end joining (NHEJ) using the T7 endonuclease 1 assay. Two gRNAs, L2 and L3 and R2 and R3 from each side of break points, were found to induce higher indel rates than the others (Fig. 1B). We then determined the efficiency of excision of the approximate 13-kb fragments by combining the left (L) with the right (R) gRNAs using PCR amplification by a pair of primers chosen from 5′ and 3′ junction areas. The combinations of 5′ and 3′ gRNA pairs (L2/R2 or L3/R3) were most efficient in making a clear deletion in 293T cells (Fig. 1C). Subcloning and sequencing of the PCR products from L2/R2 further confirmed that the 3′ and 5′ ends of the upstream and downstream DSBs after the 13-kb deletions were perfectly connected or with one base pair deletion at the junction (Fig. 1D). These results indicate that the chosen gRNAs can induce DSB, causing an ∼13-kb deletion and subsequent rejoining of the two ends.
Fig. 1.

Targeting clinically relevant HPFH5 deletions by S. aureus CRISPR-Cas9 in human cells. (A) Schematic of gRNAs targeting deletions in the β-globin locus to mimic the naturally occurring Sicilian HPHF deletion. The gRNAs were chosen from 4.5 kb upstream of HBD and 1.13 kb downstream of HBB. Black downward arrowheads, putative BCL11A binding sites. Blue arrowheads and red arrowheads, forward and reverse primers flanking each gRNA-targeting sequence used for amplifying fragments for the T7 endonuclease 1 assay (T7E1 assay). The 5′ break point gRNAs are labeled with “L”; the 3′ break point gRNAs are labeled with “R”; the “13kb” represents the length of the HPFH-5 deletion depicted in Fig. S1A. (B) Representatives of the T7E1 assay of gRNA targeting the β-globin locus in 293T cells. The percentage of indels is indicated under each gRNA. The size of each amplification and the size of cleavage product by T7 assay are listed in Table S3. (C) Dual gRNA-mediated deletion examined by PCR using the primer set of the most left and the most right ones depicted in A. The sizes of each PCR product are listed in Table S3. (D) Representative Sanger-sequencing results of two DSB junction areas from subcloning of the PCR product of L2/R2 transfectants aligned with WT junction sequences. Red arrows indicate the DSB cleavage sites (3 bp from PAM sequences NNGRRT). In between the two gRNA cleavage sites, there is an ∼13-kb fragment that was deleted in all five clones examined. The red sequences in the 5′ junction and green sequences in the 3′ junction are represented as sequences that gRNA, L2, and R2 used for targeting.
Table S1.
Oligonucleotides for gRNAs targeting β-globin locus
| gRNA | Targeting sequences-PAM (NNGRRT) | Oligonucleotide used to make gRNA construct |
| L1 | gtccactggattcagtgagctaGTGGGT | 5′-CACCgtccactggattcagtgagcta-3′ |
| 5′-AAACtagctcactgaatccagtggac-3′ | ||
| L2 | gcagaatgtacatgcgactgaAAGGGT | 5′-CACCgcagaatgtacatgcgactga-3′ |
| 5′-AAACtcagtcgcatgtacattctgc-3′ | ||
| L3 | tgggaggtatactaaggactcTAGGGT (C) | 5′-CACCgtgggaggtatactaaggactc-3′ |
| 5′-AAACgagtccttagtatacctcccac-3′ | ||
| L4 | cacaagaatagggccacatttGTGAGT (C) | 5′-CACCgcacaagaatagggccacattt-3′ |
| 5′-AAACaaatgtggccctattcttgtgc-3′ | ||
| R1 | gtggagtcaaggctgagagatgCAGGAT (C) | 5′-CACCgtggagtcaaggctgagagatg-3′ |
| 5′-AAACcatctctcagccttgactccac-3′ | ||
| R2 | tgccttattcatccctcagaaAAGGAT | 5′-CACCgtgccttattcatccctcagaa-3′ |
| 5′-AAACttctgagggatgaataaggcac-3′ | ||
| R3 | gtccttccaaagcagactgtgaAAGAGT (C) | 5′-CACCgtccttccaaagcagactgtga-3′ |
| 5′-AAACtcacagtctgctttggaaggac-3′ | ||
| R4 | gtcctctctcccagtcaaattCTGAAT | 5′-CACCgtcctctctcccagtcaaatt-3′ |
| 5′-AAACaatttgactgggagagaggac-3′ |
(C), target sequences in the complementary strand; L, 5′ breakpoint gRNA labeling; R, 3′ breakpoint labeling. Bold letters are the PAM sequences.
Modifying CD34+ Hematopoietic Stem and Progenitor Cells with Deletion.
To investigate whether the effective cleavage of the 13-kb fragment can be achieved in primary cells and induced pluripotent stem cells (iPSCs), gRNAs L2 and R2 or L3 and R3 were transfected into mobilized CD34+ HSPCs, iPSCs, and primary erythroblasts derived from peripheral blood mononuclear cells. Various combinations of the left and right gRNAs could induce deletions efficiently in all three types of cells (Fig. 2A). To analyze only the cells that received both the 5′ breakpoint (left) and 3′ breakpoint (right) gRNA-Cas9 constructs, we used a self-cleaving 2A peptide to connect a GFP reporter gene or an mCherry reporter gene to the Cas9 gene in left or right gRNA-Cas9 constructs, respectively. These constructs would facilitate the monitoring of the gRNA-Cas9 delivery by observing simultaneous GFP and mCherry expression. GFP and mCherry expression were confirmed in 293T cells and coexpressed in CD34+ HSPCs after cotransfecting both left and right constructs (Fig. 2B). By the T7E1 assay, both fluorescence marker-conjugated Cas9s showed comparable nuclease activity compared with the original constructs without fluorescence markers inserted. Using this approach, only the cells that expressed both left and right gRNA-Cas9 constructs simultaneously were isolated by sorting out the cells with two fluorescence markers expression.
Fig. 2.

Dual gRNA-mediated deletion in primary cells. (A) Detection of dual gRNA-mediated deletions in primary erythroblast (Primary Ery), iPSCs, and CD34+ HSPCs 72 h after transfection. Red arrowhead, WT; black arrow, deletion. NC, negative control (cells transfected with pX601 without gRNA); NT, nontransfected. (B) Modification of the Cas9 constructs for the enrichment of transfected cells. The 293T, transfected individually with Cas9 conjugated with GFP or mCherry, and HSPCs, cotransfected with both conjugated Cas9s, are shown by the green and red fluorescence signals. (Scale bars, 1 μm.)
Because the L2 and R3 construct demonstrated the most efficient cleavage activity in the T7E1 assay, we used these two gRNAs for subsequent genome modification in CD34+ HSPCs.
The scheme of introducing the L2-GFP and R3-mCherry combination into bone marrow CD34+ HSPCs is depicted in Fig. S2. We transfected the two constructs into bone marrow CD34+ cells, sorted out the GFP+/mCherry+ double positive cells by fluorescence-activated cell sorting, and seeded them on semisolid methylcellulose cultures to form clonal colonies. After 14 d of culture, the erythroid colonies from semisolid culture were picked, and each colony was divided to two halves, one for genomic DNA extraction and the other for RNA extraction. A variety of hematopoietic colonies were formed (Fig. 3A). PCR was performed on the differentiated erythroid colonies to detect the 13-kb deletion using the primer set chosen to detect the 5′ and 3′ junction regions (Fig. 3 B, Top line). About 31% (10/32) of the colonies had deletion at least in one allele (Fig. 3 B, Upper gel image). The PCR for 3′ breakpoint junction also was performed, and we found variable-size PCR products as well as same-size PCR products as the not-transfected sample in the tested clones (Fig. 3 B, Lower image). Eight of the same-size PCR products were further examined by Sanger sequencing and revealed a mixed histogram after the cleavage sites (Fig. S3), indicating that all of the tested clones had 3′ junctions altered by NHEJ. This result further confirmed that the chosen gRNA guided nuclease could reach to 100% target-site cleavage. When these two PCR results were analyzed together, two clones with homozygous deletion and eight clones with heterozygous deletion were identified. These results indicated that the chosen pairs of gRNAs could efficiently induce targeted HPFH5 deletion in transfected CD34+ HSPCs. To detect off-targeting, when these two gRNAs were BLAST searched for human genome sequences, very limited numbers of homology sequences were found (Table S2). We tested these potential off-target sites using the T7E1 assay in the clones that showed deletion. No off-target cleavage was detected at these sites (Fig. 3C), suggesting that the chosen gRNAs with SaCas9 could catalyze specific deletion at the targeted sites without affecting other genomic sites.
Fig. S2.

Schematic representation of procedures used for modifying CD34+ HSPCs with HPFH deletion.
Fig. 3.

Dual gRNAs mediated clinical relevant HPFH5 deletion in CD34+ HSPCs. (A) Representative images of burst-forming unit-erythroid (BFU-E); colony-forming unit-granulocyte and macrophage (CFU-GM); colony forming unit-granulocytes, erythroid, monocyte/macrophage, megakaryocyte (CFU-GEMM) in the semisolid medium culture. (B) Detection of deletion in colonies from semisolid cultured GFP+/mCherry+ colonies by PCR using the primer sets shown in the Upper line. The varying sizes of PCR products in the Lower panel indicate NHEJ in those colonies that have only a single double strand break (DSB) site. The clones positive for P1-P4 and P3-P4 amplifications indicate heterozygous deletion. The clone positive for P1-P4, but negative for P3-P4, indicates homozygous deletion. Non-deletion, no 13-kb deletion. (C) Shown is the T7E1 assay for detection of potential off-site targets that are listed in Table S2. L2 and R3 are the on-target controls. The faint bands in GRIK4 are not the expected sizes for corresponding gRNA-targeting cleavage products, indicating that these are derived from nonspecific cleavage of the PCR product.
Fig. S3.

Detection of NHEJ by Sanger sequencing in the clones which shown same size of PCR product. (A) Sequence alignment of representative clones with WT PCR products. PCR products with same-size fragments amplified from the 3′ breakpoint of eight clones showed mixed signal when aligned with WT sequences. The red line indicates the targeting site. The asterisks indicate matched nucleotides. (B) Histogram of Sanger sequencing further confirmed that the mixed signal started from the cleavage site indicated by the red arrow.
Table S2.
List of potential off-target sites for both 5′ and 3′gRNAs
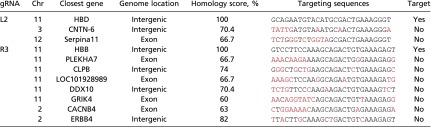 |
Mis-matched sequences are indicated by red. Chr, chromosome.
Elevated γ-Globin Gene Expression in Modified CD34+ with HPFH5 Deletion.
To analyze whether the deletion could increase γ-globin gene expression seen in naturally occurring HPFH, globin mRNA expression analysis was performed in the erythroid colonies differentiated from clones with and without the 13-kb deletion, based on the result from genomic DNA screening. Compared with the clones without deletions as control, the β-globin gene expression was decreased in the heterozygous deletions and absent in the homozygous deletions (P < 0.0001) (Fig. 4A). There is more variability in the γ-globin gene expression in the clones with or without deletions, probably due to the variable stages of erythroid differentiation of individual clones. However, both the heterozygous and homozygous deletion clones showed statistically higher γ-globin gene expression than the clones without deletions (P < 0.01) (Fig. 4B). The ratio of γ-globin gene to combined β- and γ-globin gene expression was significantly higher in the deletion clones than in the clones without deletion (P < 0.0001) and, as expected, reached the ratio of 1 in the clones with homozygous deletions (Fig. 4C). These results indicate that bone marrow-derived CD34+ HSPCs modified with HPFH deletion by CRISPR-Cas9 can produce elevated γ-globin expression like the naturally occurring HPFH.
Fig. 4.

Elevated γ-globin expression in erythroid cells differentiated from CD34+ HSPCs modified with HPFH5 deletion by real-time RT-PCR. (A) Comparison of β-globin mRNA expression normalized to α-globin expression in erythroid colonies with and without 13-kb deletion based on the result from the genomic DNA screen in Fig. 3B. (B) Comparison of γ-globin expression in erythroid colonies with heterozygous and homozygous deletion to the clones without deletion. (C) The ratio of γ-globin expression to combined β- and γ-globin expression in clones with and without deletions. All data represent the mean plus or minus SEM. Statistically significant differences are indicated as follows: **P < 0.01; ***P < 0.0001 as determined by the Student unpaired t test. For all figures: filled circles, clones without deletion (Non-del); filled squares, clones with heterozygous deletion Del (Het); filled triangle, clones with homozygous deletion Del (Hom). HBA, α-globin mRNA; HBB, β-globin mRNA; HBG, γ-globin mRNA.
Discussion
This study was based on a long-known observation that individuals compound heterozygous for β-thalassemia or SCD with deletional HPFH have pancellular HbF distribution, resulting in minimal hematological abnormalities and mild clinical manifestation. The high level of fetal hemoglobin prevents polymerization of HbS in SCD and partially compensates for the adult β-globin deficiency in β-thalassemia (1, 30, 31). Therefore, we made use of this clinical observation and modified CD34+ HSPCs to have part of the β-globin locus removed and repaired the genome by nonhomology end joining (NHEJ) to generate a genotype mimicking HPFH that produced a high level of γ-globin expression when differentiated into erythroid cells. By delivering target site-specific SaCas9, we were able to modify 31% of adult bone marrow CD34+ HSPCs with the 13-kb HPFH5 deletion that gave significantly increased γ-globin expression on differentiation into erythroid cells in vitro when compared to cells without the deletion.
Our study provides proof of concept for developing a potential new approach to autologous transplantation therapy to treat homozygous β-thalassemia and SCD. Single nucleotide mutations have been corrected by nucleases facilitating homologous recombination (32–34). However, a number of different nucleases or gRNAs will have to be designed for different mutations. In contrast, the same gRNA pair could be used in our approach for practically all of the β-globin gene mutations. This approach could overcome some of the problems associated with other approaches proposed for autologous stem cell therapies. Mutant gene correction through the homology-directed repair (HDR) pathway with introduced homologous donor templates is vastly inefficient compared with NHEJ (35, 36). Although the corrected clones can be enriched using selectable markers, the selection requires repeated cell cultures, which will affect the stemness of the HSPCs without going through iPSC generation. Furthermore, the barrier associated with differentiation of iPSCs into transplantable and β-globin–producing HSPCs has not yet been overcome (37). Fetal hemoglobin elevation has also been achieved by deleting the erythroid enhancer of the transcription factor BCL11A (38). To be effective, both alleles may be required to be deleted. By contrast, HPFH genotype editing in homozygous SCD and β-thalassemia is required in only one allele. One other approach proposed for reactive γ-globin expression is to force change in chromatin looping (39), but it will be difficult to achieve this effect in vivo. Currently, all these approaches have not yet been tested in clinical trials. Lentiviral delivery of a corrective β-globin gene has met with early successes, but the long-term effect of multiple lentiviral insertion into the genome is not yet known. By contrast, individuals who are compound heterozygous for HPFH and SCD or β-thalassemia are known to have mild clinical manifestation. Therefore, in a way, nature has already given a clinical trial demonstrating the efficacy and safety of this approach.
Methods
Construction of gRNA Plasmids.
The oligonucleotides used to construct the gRNAs in this study are listed in Table S1. A pair of oligonucleotides for each targeting site with 5′-CACC and 3′-AAC overhang were synthesized by Integrated DNA Technologies. The pair of oligonucleotides were treated with T4 polynucleotide kinase (PNK) (NEB), annealed, and then cloned into the BsaI site of the plasmid pX601-AAV-CMV::NLS-SaCas9-NLS-3xHAbGHpA; U6::BsaI-sgRNA (61591; Addgene), which contains both Cas9 and gRNA, using the T4 ligase from the Rapid Dephos & Ligation Kit (Roche). For constructing the fluorescence-conjugated Cas9 vector, the T2A sequences were incorporated into the 3′ primers and 5′ primers listed in Table S3 for amplifying Cas9 3′ ends and GFP or mCherry 5′ ends, respectively. The HindIII-BamHI fragment of T2A3′-Cas9 and the BamH1-EcoR1 fragment of 5′ T2A-GFP or 5′ T2A mCherry were ligated into HindIII-EcoR1–digested pX601 to complete the PX601-GFP and pX601-mCherry constructs.
Table S3.
Primer information for the studies
| Primer | Sequences | Product size, bp | Application |
| L-F1 | ccattatcagttactcaacctagaat | 691 | T7E1 assay for L1 (226 & 463bp) & L2 (411 & 278bp) |
| L-R1 | ctagagacacattctaagtgtgacat | ||
| L-F2 | gtgaatccaagagtgtgatgaataca | 806 | T7E1 Assay for L3 (325 & 481bp) |
| L-R2 | gaatgtcacacttagaatgtgtctct | ||
| L-F3 | gatctgttcttgtatgttctgttcca | 897 | T7E1 Assay for L4 (727 & 170bp) |
| L-R3 | ctctgttggtgacactgtacaatagt | ||
| R-F1 | gaagggccttgagcatctggatt | 1,145 | T7E1 assay for R1 (381 & 764bp), R2 (258 & 847bp), R3 (655 & 490bp) & R4 (1002 & 143bp) |
| R-R1 | ggtgtaagacaagggtctgattt | ||
| L-F1 | ccattatcagttactcaacctagaat | 14,740 (no-deletion) | Detection of deletion |
| R-R1 | ggtgtaagacaagggtctgattt | 995 (L1-R1 deletion) | |
| 1,270 (L2-R2 deletion) | |||
| 1,630 (L3-R3 deletion) | |||
| 2,820 (L4-R4 deletion) | |||
| P1 | atgggggcatcaaagtatca | ∼199 | Detection of L2-R3 deletion |
| P4 | aagttaggactgagaagaatttgaaagg | ||
| P3 | gactgtcctgtgagcccttctt | 152 | Detection of No-deletion |
| P4 | aagttaggactgagaagaatttgaaagg | ||
| HBA-S | gcc ctg gag agg atg ttc | 101 | Real-time PCR for HBA expression |
| HBA-AS | ttc ttg ccg tgg ccc tta | ||
| HBB-S | tga gga gaa gtc tgc cgt tac | 87 | Real-time PCR for HBB expression |
| HBB-AS | acc acc agc agc ctg ccc a | ||
| HBG-S | ggt tat caa taa gct cct agt cc | 134 | Real-time PCR for HBG expression |
| HBG-AS | aca acc agg agc ctt ccc a | ||
| Cas9-HindIII 5′ -S | gccccgtcgtgaagagaagcttcatcc | 1,887 | Fragment of cas9-T2A |
| Cas9-3′T2A5′-AS | gcagggatcctctgccctcctttttcttttttgcctggccggcctt | ||
| T2A3′-Egfp-5′-S | cagaggatccctgctaacatgtggtgacgtcgaggagaatcctggcccaatggtgagcaagggcgaggagctgttc | 747 | Fragment of T2A-GFP |
| EGFP-EcoR1-AS | cgtagaattcggccgctttacttgtacagctcgtccatgccga | ||
| T2A3′-mCherry5′-S | cagaggatccctgctaacatgtggtgacgtcgagg | 766 | Fragment of T2A-mCherry |
| agaatcctggcccaatggtgagcaagggcgaggaggataaca | |||
| mCherry –EcoR1-AS | cgtagaattcttacttgtacagctcgtccatgccgccggt | ||
| Primer for off-target analysis | |||
| HBD-F | ccattatcagttactcaacctagaat | 691 | For L2 gRNA on-target |
| HBD-R | ctagagacacattctaagtgtgacat | ||
| CNTN-6-F | gaacaatagagccaccctacaaatagt | 470 | For L2 gRNA off-target |
| CNTN-6-R | cttcatctcctagagtaaaggtatcct | ||
| Serpina11-F | cagccagcatccttctgctgtgacact | 590 | For L2 gRNA off-target |
| Serpina11-R | ctaacacatggaaggtgcttagtagat | ||
| HBB-F | gaagggccttgagcatctggatt | 1,145 | For R3 gRNA on-target |
| HBB-R | ggtgtaagacaagggtctgattt | ||
| PLEKHA7-F | gggcgtggtggcacatgcttgt | 680 | For R3 gRNA off-target |
| PLEKHA7-R | gggagagagatgaggagcagca | ||
| CLPB-F | gactcacagttcctcattgct | 735 | For R3 gRNA off-target |
| CLPB-R | ctaggtattctcctgccttct | ||
| LOC101928989-F | gggaatatgatccaaaggagcaa | 554 | For R3 gRNA off-targte |
| LOC101928989-R | cttattggcctttctcctcat | ||
| DDX10-F | gttcatgtcccatcactatgat | 648 | For R3 gRNA on-target |
| DDX10-R | gtatgttgtgtctttgtcctcact | ||
| GRIK4-F | gaatgtggtcaacaaagcttca | 463 | For R3 gRNA on-target |
| GRIK4-R | ccagccattctcctgcctca | ||
| CACNB4-F | gcaacactcttaggaaggtctt | 651 | For R3 gRNA on-target |
| CACNB4-R | gcctttgaagaagatagtggaat | ||
| HECW2-F | ccttctcctatacagtaagcca | 656 | For R3 gRNA on-target |
| HECW2-R | ccctaaggataatgatagata | ||
| ERBB4-F | gggcaaagatgaagtcagtgttt | 444 | For R3 gRNA on-target |
| ERBB4-R | cggttcaagtctcactgattt |
Cell Cultures and Transfection.
Human 293T cells were cultured in DMEM (Thermo Scientific) supplemented with 10% (vol/vol) FBS (HyClone), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Transfection to 293T cells was performed in 24-well plates using 1 μg of plasmid DNA (if a pair of plasmid DNAs were for transfection, 0.5 μg for each was used) mixed with TurboFect transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. Peripheral blood mobilized CD34+ HSPCs and bone marrow CD34+ HSPCs were purchased from AllCells and cultured in StemSpan SFEM (Stemcell Technologies) supplemented with human cytokines, 100 ng/mL SCF, 100 ng/mL Tpo, 100 ng/mL Flt3 ligand and small molecules, 1 μM StemRegenin 1, and 0.5 μM MU729. Erythroblasts were dedifferentiated from peripheral blood mononuclear cells (MNCs) by culturing MNCs in StemSpan SFEM (Stemcell Technologies) supplemented with 50 ng/mL SCF, 10 ng/mL IL3, 40 ng/mL IGF1, 2 U/mL Epo, and 1 mM dexamethasone for 9 d. The iPSCs were cultured in mTeSR E8 medium (Stemcell Technologies). All of the primary cell transfections were conducted in Nucleofector II (Lonza) using various amounts of DNA in corresponding programs (U08 for CD34+ HSPCs; A023 for iPSCs; T016 for erythroblasts).
T7 Endonuclease 1 Assay.
The transiently transfected various gRNAs incorporating plasmids (px601 or pX601-T2AGFP, pX601-T2AmCherry) in 293T cells were harvested at 48 h after transfection. The genomic DNAs were isolated using a PureLink Genomic DNA mini kit (Thermofisher). The corresponding PCR products with the gRNA cleavage site in the center were amplified using the primers listed in Table S3. The PCR products were purified and denatured and then reannealed to allow heteroduplex DNA formation, followed finally by T7 endonuclease 1 digestion according to the manufacturer’s instructions. The digested DNAs were run on 1% agarose gel. The densities of cut and uncut bands were calculated using Image J software. We used Fcut = (cut1 + cut 2)/(uncut + cut1 + cut2) and Indel% = 1 − √(1 − Fcut) formulas to get the efficiency of cleavage activity.
Cell Sorting and Clonal Analysis.
For targeting HPFH deletion, the bone marrow CD34+ HSPCs were thawed and cultivated in StemSpan SFEM medium supplemented with cytokines overnight before transfection. After the transfection, the CD34+ cells were plated in the new StemSpan SFEM medium with cytokines and recovered for 24 h. Cells were harvested and suspended in medium, and the GFP and mCherry double-positive cells were sorted using Avalon S3 (PropeLab).
Six hundred positive cells were seeded in 1.5 mL of methylcellulose (MethoCult H4435; Stemcell Technologies) on a 35-mm dish and cultured for 2 wk at 37 °C in a 5% CO2 incubator. The erythroid colonies were individually picked and divided into two portions. For DNA extraction, the colonies were lysed in 20 μL of lysis buffer containing 10 mM Tris⋅HCl (pH 7.6), 50 mM NaCl, 6.25 mM MgCl2, 0.045% Nonidet P-40, 0.45% Tween 20, and 20 μg of proteinase K (40). Samples were incubated at 56 °C for 2 h and followed by proteinase K inactivation at 95 °C for 20 min. Then, 20 μL of water with RNase A was added to each sample. Then, 2 μL from each sample was used for PCR for detection of modification. The primers for PCR are listed in Table S3. The PCR products were analyzed by agarose gel electrophoresis.
Globin Gene Expression Analysis.
Total RNA was extracted from cells collected from methycellulose culture using an Arcturus PicoPure RNA isolation kit following the protocol provided by the manufacturer. RNA was eluted in final 15 μL of RNase-free water, and 8 μL of RNA was used for generation of cDNA by reverse transcription with random hexamers and SuperScript III (Life Technologies) in a 20-μL reaction following the manufacturer’s instructions. Then, 0.6 μL of cDNA reaction was used for each reaction of the real-time quantitative PCR (RT-PCR). RT-PCR was performed on a DNA Engine OPTICON (Bio-Rad) using FastStart Universal SYBR Green Master Mix (Roche). The data analysis used the Qgene program. β− and γ-globin gene expression levels were normalized with endogenous control α-globin gene expression levels. All qPCR primers are listed in Table S3.
Statistics Analysis.
All of the data are expressed as means ± SE. Differences between deletion and no-deletion colonies were evaluated by unpaired Student’s t test using GraphPad Prism software. Statistical significance was defined as P < 0.05.
Off-Target Analysis for CRISPR-Cas9–Mediated Genome Modification.
Potential off-target sites were determined by using a BLAST search in the National Center for Biotechnology Information (NCBI) database of the human genome. The primers for amplifying the off-target sites resulted in 500-bp to 700-bp amplicons centered near the off-target sites. The corresponding primers are listed in Table S3. The DNAs from all of the clones with deletion were pooled together, and the off-target sites were PCR-amplified. The T7E1 assay was used to detect off-target cleavages.
Acknowledgments
We acknowledge the cell sorting service provided by the Diabetes Research Center (DRC) of the University of California, San Francisco supported by DRC Grant NIH P30 DK063720. This work is supported by NIH Grant P01DK088760, in part by Grant R01AI102825, and Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1612075113/-/DCSupplemental.
References
- 1.Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: Prospects for new therapies for the β-globin disorders. Blood. 2012;120(15):2945–2953. doi: 10.1182/blood-2012-06-292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raja JV, Rachchh MA, Gokani RH. Recent advances in gene therapy for thalassemia. J Pharm Bioallied Sci. 2012;4(3):194–201. doi: 10.4103/0975-7406.99020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negre O, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum Gene Ther. 2016;27(2):148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bank A, Dorazio R, Leboulch P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann N Y Acad Sci. 2005;1054:308–316. doi: 10.1196/annals.1345.007. [DOI] [PubMed] [Google Scholar]
- 5.Persons DA. Hematopoietic stem cell gene transfer for the treatment of hemoglobin disorders. Hematology Am Soc Hematol Educ Program. 2009;2009:690–697. doi: 10.1182/asheducation-2009.1.690. [DOI] [PubMed] [Google Scholar]
- 6.Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulad F, et al. Safe mobilization of CD34+ cells in adults with β-thalassemia and validation of effective globin gene transfer for clinical investigation. Blood. 2014;123(10):1483–1486. doi: 10.1182/blood-2013-06-507178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conley CL, Weatherall DJ, Richardson SN, Shepard MK, Charache S. Hereditary persistence of fetal hemoglobin: A study of 79 affected persons in 15 Negro families in Baltimore. Blood. 1963;21:261–281. [PubMed] [Google Scholar]
- 9.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 10.Boyer SH, et al. Inheritance of F cell frequency in heterocellular hereditary persistence of fetal hemoglobin: An example of allelic exclusion. Am J Hum Genet. 1977;29(3):256–271. [PMC free article] [PubMed] [Google Scholar]
- 11.Akinsheye I, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huisman TH, et al. Hereditary persistence of fetal hemoglobin: Heterogeneity of fetal hemoglobin in homozygotes and in conjunction with -thalassemia. N Engl J Med. 1971;285(13):711–716. doi: 10.1056/NEJM197109232851303. [DOI] [PubMed] [Google Scholar]
- 13.Ringelhann B, et al. Homozygotes for the hereditary persistence of fetal hemoglobin: The ratio of G gamma to A gamma chains and biosynthetic studies. Biochem Genet. 1977;15(11-12):1083–1096. doi: 10.1007/BF00484499. [DOI] [PubMed] [Google Scholar]
- 14.Bank A. Regulation of human fetal hemoglobin: New players, new complexities. Blood. 2006;107(2):435–443. doi: 10.1182/blood-2005-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(Suppl 4):S512–S521. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 16.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 20.Kutlar A, et al. Heterogeneity in the molecular basis of three types of hereditary persistence of fetal hemoglobin and the relative synthesis of the G gamma and A gamma types of gamma chain. Biochem Genet. 1984;22(1-2):21–35. doi: 10.1007/BF00499284. [DOI] [PubMed] [Google Scholar]
- 21.Motum PI, Hamilton TJ, Lindeman R, Le H, Trent RJ. Molecular characterisation of Vietnamese HPFH. Hum Mutat. 1993;2(3):179–184. doi: 10.1002/humu.1380020305. [DOI] [PubMed] [Google Scholar]
- 22.Saglio G, et al. Italian type of deletional hereditary persistence of fetal hemoglobin. Blood. 1986;68(3):646–651. [PubMed] [Google Scholar]
- 23.Ojwang PJ, et al. Gene deletion as the molecular basis for the Kenya-G gamma-HPFH condition. Hemoglobin. 1983;7(2):115–123. doi: 10.3109/03630268309048641. [DOI] [PubMed] [Google Scholar]
- 24.Sankaran VG, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katsantoni EZ, et al. Persistent gamma-globin expression in adult transgenic mice is mediated by HPFH-2, HPFH-3, and HPFH-6 breakpoint sequences. Blood. 2003;102(9):3412–3419. doi: 10.1182/blood-2003-05-1681. [DOI] [PubMed] [Google Scholar]
- 26.Sankaran VG, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 27.Galanello R, et al. Deletion delta-thalassemia: The 7.2 kb deletion of Corfu delta beta-thalassemia in a non-beta-thalassemia chromosome. Blood. 1990;75(8):1747–1749. [PubMed] [Google Scholar]
- 28.Chakalova L, et al. The Corfu deltabeta thalassemia deletion disrupts gamma-globin gene silencing and reveals post-transcriptional regulation of HbF expression. Blood. 2005;105(5):2154–2160. doi: 10.1182/blood-2003-11-4069. [DOI] [PubMed] [Google Scholar]
- 29.Camaschella C, et al. A new hereditary persistence of fetal hemoglobin deletion has the breakpoint within the 3′ beta-globin gene enhancer. Blood. 1990;75(4):1000–1005. [PubMed] [Google Scholar]
- 30.Platt OS, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 31.Weatherall DJ. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–255. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 32.Ma N, et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free β-thalassemia induced pluripotent stem cells. J Biol Chem. 2013;288(48):34671–34679. doi: 10.1074/jbc.M113.496174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie F, et al. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24(9):1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu P, et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci Rep. 2015;5:12065. doi: 10.1038/srep12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoban MD, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125(17):2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Risueño RM, et al. Inability of human induced pluripotent stem cell-hematopoietic derivatives to downregulate microRNAs in vivo reveals a block in xenograft hematopoietic regeneration. Stem Cells. 2012;30(2):131–139. doi: 10.1002/stem.1684. [DOI] [PubMed] [Google Scholar]
- 38.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527(7577):192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng W, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158(4):849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Burg M, et al. EU-supported EuroChimerism Consortium Project QLRT-2001-01485 Standardization of DNA isolation from low cell numbers for chimerism analysis by PCR of short tandem repeats. Leukemia. 2011;25(9):1467–1470. doi: 10.1038/leu.2011.118. [DOI] [PubMed] [Google Scholar]