

Abstract
Objective:
To evaluate the comparative safety and adjunctive efficacy of pregabalin and gabapentin in reducing seizure frequency in patients with partial-onset seizures based on prestudy modeling showing superior efficacy for pregabalin.
Methods:
The design of this comparative efficacy and safety study of pregabalin and gabapentin as adjunctive treatment in adults with refractory partial-onset seizures was randomized, flexible dose, double blind, and parallel group. The study included a 6-week baseline and a 21-week treatment phase. The primary endpoint was the percentage change from baseline in 28-day seizure rate to the treatment phase.
Results:
A total of 484 patients were randomized to pregabalin (n = 242) or gabapentin (n = 242). Of these, 359 patients (187 pregabalin, 172 gabapentin) completed the treatment phase. The observed median and mean in percentage change from baseline was −58.65 and −47.7 (SD 48.3) for pregabalin and −57.43 and −45.28 (SD 60.6) for gabapentin. For the primary endpoint, there was no significant difference between treatments. The Hodges-Lehman estimated median difference was 0.0 (95% confidence interval −6.0 to 7.0). Safety profiles were comparable and consistent with prior trials.
Conclusions:
The absence of the anticipated efficacy difference based on modeling of prior, nearly identical trials and the larger-than-expected response rates of the 2 antiepileptic drugs were unexpected. These findings raise questions that are potentially important to consider in future comparative efficacy trials.
ClinicalTrials.gov identifier:
Classification of evidence:
This study provides Class II evidence that for patients with partial seizures enrolled in this study, pregabalin is not superior to gabapentin in reducing seizure frequency. Because of the atypical response rates, the results of this study are poorly generalizable to other epilepsy populations.
Although placebo-controlled randomized trials are fundamental to drug development, they are not popular with participants, who may be reluctant to participate in studies in which there is a chance of not receiving active treatment. There is also an increased interest by payers in comparative-effectiveness drug trials in which 2 drugs are directly compared.1 When trials are performed with antiepileptic drugs (AEDs), no-difference outcomes are common and are typically interpreted as a demonstration of comparable efficacy.2,3 Trials that show a clear efficacy difference between therapies are more interpretable than trials that show no difference, but the ability of any AED to demonstrate superiority has not been commonly shown in add-on studies.2 Pregabalin and gabapentin are 2 AEDs with similar α-2-δ-ligand pharmacologies4 that Emax modeling of published clinical trial data5–7 showed had important different magnitudes of response for efficacy endpoints (figure e-1 at Neurology.org).8 Modeling has been used to help with decision-making in early drug development.6,8 However, this approach is relevant for comparative-efficacy trials for established therapies. Assumptions based on modeling8 (figure e-1) indicated that the expected difference between treatments would be 17% in favor of pregabalin. At the time this study was planned (2006–2007), gabapentin and pregabalin were part of the armamentarium available for the treatment of resistant focal epilepsy, and it was anticipated that clinicians and payers, among others, would value information comparing both drugs since they had similar mechanisms. We therefore designed an add-on trial providing active treatments in both arms, which, as hypothesized, would demonstrate clear superiority of pregabalin compared to gabapentin in patients with refractory partial-onset seizures (POSs).
METHODS
Study design.
This was a 2-arm, randomized, flexible-dose, double-blind, parallel-group, superiority study conducted in adult patients at 56 centers in Eastern and Western Europe, Asia, and South and Central America between February 2008 and July 2013 (EVENT: ClinicalTrials.gov NCT00537940). The study comprised 3 main phases: 6 weeks of baseline (screening), 9 weeks of double-blind dose escalation (titration), and 12 weeks of double-blind maintenance phase (21-week treatment phase) (figure 1). This design mirrored prior efficacy trials of both drugs, although to optimize safety and tolerability, there were 2 differences from prior trials. The first is that this study included 9 weeks of dose escalation compared to 0 to 4 weeks in earlier trials to potentially reduce discontinuations due to adverse events. Another difference is that uptitration was optional beyond a minimum dose of 100 mg 3 times daily (pregabalin) and 400 mg 3 times daily (gabapentin). After the 12-week double-blind maintenance phase, patients could enter a blinded continuation phase for a maximum of 2 years.9
Figure 1. Study design diagram.

This study began before the 2010 International League Against Epilepsy (ILAE) terminology for the organization of seizures.10 Therefore, the 1981 terminology11 was used. The equivalent 2010 terminology10 is also noted.
Standard protocol approvals, registrations, and patient consents.
The protocol adhered to the Good Clinical Practice guidelines of the Declaration of Helsinki and was approved by the independent review board or independent ethics committee of each site.
Patients.
Inclusion/exclusion criteria mirrored the prior modeled trials. Key criteria were age ≥18 to ≤80 years, a diagnosis of epilepsy with POSs (equivalent to the 2010 ILAE classification10 of focal seizures) that had been inadequately controlled with ≥2 to <5 prior AEDs, and receiving 1 or 2 standard AEDs (other than pregabalin or gabapentin) with a minimum of 4 POSs (regardless of secondary generalization) during the 6-week baseline phase with no 28-day POS-free period. In early 2010, the Epilepsy Study Consortium Inc. (ESCI) was introduced to review submitted information to confirm the POS diagnosis and to verify seizure classification across study sites.
Treatment.
Using a computer-generated randomization system, we randomized patients to either pregabalin or gabapentin (1:1). The method of treatment administration was double-blind, double-dummy, with patients assigned either active pregabalin and dummy gabapentin or active gabapentin and dummy pregabalin. Investigators escalated doses of pregabalin (150, 300, 450, and 600 mg/d) or gabapentin (300, 600, 1,200, 1,500, and 1,800 mg/d) during the 9-week dose-escalation phase to the highest effective dose provided that tolerability was acceptable. The minimum maintenance phase dose was 300 mg/d (pregabalin) and 1,200 mg/d (gabapentin) divided 3 times daily. The optimized dose was continued during the maintenance phase. Dose reduction of the study drug was permitted once (for tolerability). Maintenance of prestudy regimens of other AEDs was required; no dose changes were permitted.
Efficacy endpoints.
This study was very similar in the definition of efficacy endpoints to a concurrent study that compared pregabalin and levetiracetam.9 The primary efficacy endpoint was the percent change in 28-day seizure frequency during the 21-week treatment phase compared with baseline. We defined the key secondary efficacy endpoints as the proportion of participants with a 50% reduction (50% responder rate) or 75% reduction (75% responder rate) in 28-day seizure (all partial seizure) from baseline, with a reduction in the proportion of secondary generalized tonic-clonic (SGTC) from baseline (SGTC responder rate), and seizure-free in the last 28 days of treatment (seizure-free responder rate). SGTC seizures were equal to focal seizures developing into a bilateral, convulsive seizure in the 2010 ILAE classification. The proportion of SGTC was defined as the ratio of SGTC to all partial seizures.
Assessment methods.
Adverse events were monitored to assess safety and tolerability.
Statistical analyses.
On the basis of the primary efficacy parameter and simulations using previous studies, the estimated response for pregabalin ranged from 30% to 50% and for gabapentin from 20% to 30%.8 For >80% power to detect a difference of 10% in percent change in seizure frequency, a sample size of 482 randomized patients (241 per treatment arm) was calculated, assuming a 2-sided test with type I error rate of 5% and a common SD of 35%. Analyses of the primary endpoint and all secondary endpoints were based on the modified intent-to-treat (mITT) population. We defined the mITT population as all randomized patients who received ≥1 dose of study medication and for whom there were at least 28 days of usable seizure data reported during baseline and after baseline, similar to the concurrent study.9 The safety population included all randomized patients who received ≥1 dose of study medication. We used ranked analysis of covariance to assess treatment difference for the primary endpoint, which was percent change from baseline in 28-day seizure rate with the model including percent change from baseline as a response variable, treatment as the main effect, and baseline seizure count and cluster as covariates. In addition, we used Hodges-Lehmann estimation to estimate the median difference and 95% confidence interval (CI) for percent change from baseline in 28-day seizure rate. Logistic regression was used to assess 50% responder rate, with the model including 50% responder rate as the response variable, treatment as the main effect, and baseline seizure count and cluster as covariates. The Fisher exact test was used to assess 75% responder rate, SGCT responder rate, and last 28-day seizure-free responder rate.
An external data-monitoring committee carried out an unblinded interim analysis of the primary efficacy endpoint, which sought to determine whether the study could be concluded for superiority with fewer patients after ≈65% of patients had completed the maintenance phase. The data-monitoring committee recommended continuing as planned.
RESULTS
Patients.
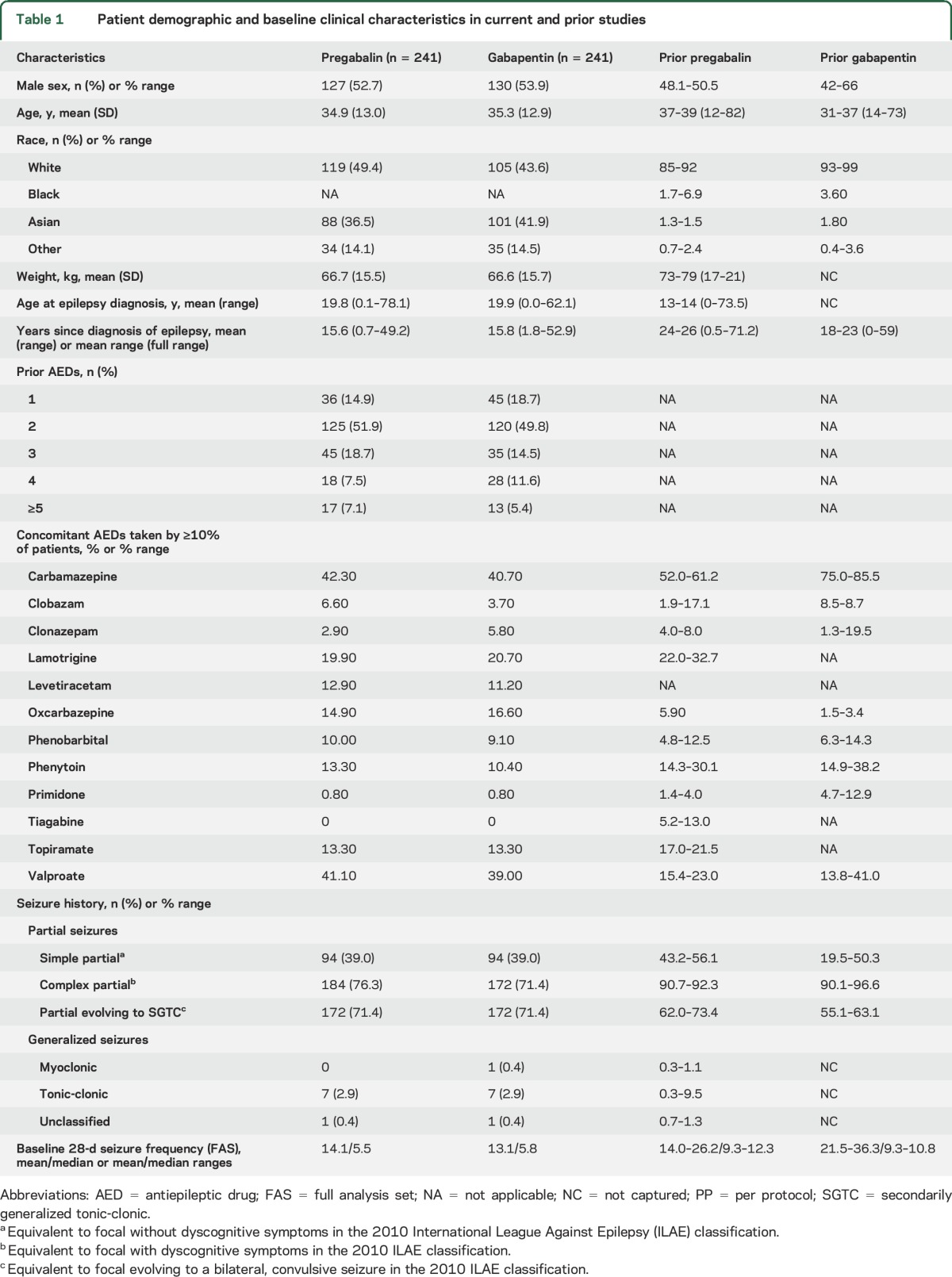
Of 561 patients screened, 484 patients were randomized (pregabalin n = 242; gabapentin n = 242), 482 (pregabalin n = 241; gabapentin n = 241) were treated, and 359 (pregabalin n =187, gabapentin n = 172) completed the maintenance phase of the study (figure 2). Both groups had comparable baseline clinical and demographic characteristics (table 1). The most common concomitant AEDs are listed in table 1. During the 21-week double-blind phase of the study, the median doses of pregabalin and gabapentin were 450 and 1,500 mg/d, respectively. A few patients who on ESCI review were rediagnosed with generalized seizures but who had already been randomized (table 1) were allowed to complete the study and remained in the mITT population (i.e., the primary analysis). They were excluded from the per protocol analysis.
Figure 2. CONSORT (Consolidated Standards of Reporting Trials) flow diagram of patient disposition and study populations.

Table 1.
Patient demographic and baseline clinical characteristics in current and prior studies
Efficacy analyses.
There was no significant difference between treatments for the results of the primary endpoint, percent change from baseline in 28-day seizure rate during the treatment phase. The Hodges-Lehman estimated median difference (95% CI) during the double-blind phase was 0.0 (−6.0 to 7.0; p = 0.87). Similarly, the observed median percentage change from baseline was −58.65 for pregabalin and −57.43 for gabapentin, and the mean percent change from baseline was −47.7 (SD 48.3) for pregabalin and −45.28 (SD 60.6) for gabapentin.
There were no significant differences between pregabalin and gabapentin for all secondary endpoints. The ≥50% responder rates were 56.3% (95% CI 50.0–62.6) and 58.3% (95% CI 52.1–64.6) for pregabalin and gabapentin, respectively, with an odds ratio of 0.92% (95% CI 0.64–1.33; p = 0.662). The ≥75% responder rates were 33.6% (95% CI 27.6–39.6) and 34.2% (95% CI 28.2–40.2) for pregabalin and gabapentin, respectively (p = 0.92). The last 28-day seizure-free rates were 30.8% (95% CI 24.1–37.3) and 34.1% (95% CI 27.2–41.0; p = 0.51). The SGTC proportion of responders was comparable with pregabalin (30.8%) and gabapentin (39.8%; p = 0.1881; table 2).
Table 2.
Secondary efficacy endpoints

Safety.
The type and occurrence of treatment-emergent adverse events (TEAEs) were representative of the known adverse event profiles of pregabalin and gabapentin. There were 142 patients (58.9%) in the pregabalin group and 129 patients (53.5%) in the gabapentin group with TEAEs (table 3) that occurred between the start of treatment and the end of the maintenance phase. The most common adverse events with an incidence ≥5% in either treatment were somnolence (n = 34 [14.1%] and n = 34 [14.1%]), dizziness (n = 22 [9.1%] and 20 [8.3%]), weight increase (19 [7.9%] and 13 [5.4%]), headache (17 [7.1%] and 20 [8.3%]), and dry mouth (12 [5.0%] and 8 [3.3%]) for pregabalin and gabapentin, respectively.
Table 3.
Treatment-emergent adverse events (TEAEs) during the treatment phase (all causalities)

Most TEAEs were mild to moderate in severity. Serious adverse events were reported in 6 patients (2.5%) each for pregabalin and gabapentin. Sixteen patients (6.6%) in the pregabalin group and 15 (6.2%) in the gabapentin group discontinued treatment as a result of a TEAE. Laboratory tests, ECGs, vital signs, or physical examinations did not demonstrate clinically significant changes.
DISCUSSION
Although prestudy modeling8 of extensive prior data predicted pregabalin to be superior to gabapentin by at least 10% in improving seizure frequency endpoints in patients with refractory POSs, this was not the observed outcome of this study. Compared with earlier trials, both pregabalin and gabapentin had higher 50% responder rates than expected, gabapentin more so, leading to a no-difference outcome. In phase 3 placebo-controlled add-on trials of new AEDs in refractory POSs, only 3 drugs, all in the highest-dose arm (topiramate, vigabatrin, and levetiracetam), have demonstrated ≥50% responder rates above 50%. Gabapentin had ≥50% responder rates below 26.4% in earlier placebo-controlled add-on studies.12 Drugs behaved differently from expected, and populations differed from those used for modeling, which raises questions about the loss of assay sensitivity for this trial design. Assay sensitivity is defined in the International Conference on Harmonization E-10 guideline as “a property of a clinical trial defined as the ability to distinguish an effective treatment from a less effective or ineffective treatment.”13 The E-10 guideline13 suggests that the presence of assay sensitivity can be determined if prior similarly designed trials (such as used in the modeling) regularly distinguished effective from noneffective treatments. Whereas this criterion appears to have been satisfied, the guidance also states that “in addition, the actual study population entered, the concomitant therapies actually used, etc., should be assessed to ensure that conduct of the study was, in fact, similar to the previous trials.” This requirement likely was not met for the current study.
This outcome may reflect that no difference exists between the efficacies of these drugs in this population. If this is the case, then both are also more efficacious in this population than observed in previously studied populations. Another possibility is that this is a failed trial, unable to distinguish relative clinical efficacy of the 2 drugs. The present study found that the gabapentin 50% responder rate was nearly twice as high as it had been in all prior studies, despite the fact that only 20% of patients reached the maximum 1,800-mg dose, and since dose escalation, which was extended to 9 weeks, was included in the efficacy calculation, only 57% of the study duration was at the maintenance dose. The differences between the prior studies and the present study, including a longer titration and the individualized option of limiting dose, should have, if anything, reduced the apparent efficacy. The lack of difference in seizure control is also surprising in that the median dosages of gabapentin (1,500 mg/d) and pregabalin (450 mg/d) for the study are biased in regard to expected efficacy and in relation to the maximum dosages listed in the product labels for gabapentin (3,600 mg/d) and pregabalin (600 mg/d). Nonetheless, since there was no placebo group, it is impossible to know definitively whether these results are true. Gabapentin when tested against placebo had a relatively weak effect (highest median percentage effect of 19.5%) in the original add-on trials. If the current results are taken at face value, this drug would now be demonstrating better responder rates for treatment-resistant patients than any other AED.12
The dropouts resulting from adverse events for both study drugs were similar within the current study (pregabalin 6.2%, gabapentin 7.1%) compared to the prior studies (pregabalin 16.2%, gabapentin 5.6%).
Because of the richness of the available prior data for both drugs, this study represents a unique opportunity to dissect the causes for the loss of assay sensitivity and to explain why past experience (as exemplified by modeled data) does not predict the future when shifting environmental factors and populations are at play. While the inclusion and exclusion criteria remained virtually unchanged over time, there were notable differences between the original gabapentin and pregabalin studies. The pivotal gabapentin and pregabalin studies were performed in the late 1980s and 1990s. All were completed in 15 to 30 months and were performed in Western Europe, North America, Australia, and South Africa. In contrast, the current study ran for >5 years (February 2008–July 2013) at 56 sites, with few in Western Europe and none in the United States or Canada. Table 1 lists patient characteristics that differed between the previous trials and the current trial. As an indication of the increase in available drugs, in the original gabapentin studies, only 3 to 5 concomitant AEDs were used commonly (taken by ≥10% of patients); a decade later, this rose to 5 to 7; and in the current study, it remained in this range, at 8, although 4 of them were different drugs (table 1). Another factor that may contribute to the mismatch of the original studies and the current study is the lower baseline 28-day seizure frequency in the current study (mean 13.1–14.1, median 5.5–5.8). The previous pregabalin and gabapentin studies had higher means (18.6–27.4 and 20.2–51.7, respectively) and medians (8.8–12.3 and 9.5–12.7, respectively). These differences could be due to referral source: large academic centers in the original trials vs clinical practice sites in the current trial. In the postmarketing environment, the balance or ratio of research and clinical practice investigational sites may change. Pivotal registration trials are more likely to be performed in epilepsy centers, while the postmarketing studies may have investigators with more general expertise. All of these factors, including differing time periods, countries (and demographic makeup of the patients enrolled), types of sites, seizure frequencies at baseline, and the types and number of concomitant AEDs, together may contribute to the divergence of the modeled prior data from the current study. Although the written protocols did not substantially differ, these factors changed the type of patients who enrolled in the trial.
Placebo-controlled arms were included in the earlier Pfizer pregabalin and gabapentin studies,5,7 and each drug was compared to placebo.14,15 Here, there was no placebo-controlled arm, and patients knew they were receiving active treatment; thus, all had reason to expect improvement. The fact that both treatment arms were active may have caused the patients, caregivers, and site staff to be biased to notice improvement.14,15 While seizures appear to be an objective event, they are still patient-reported, which may increase the likelihood of ascertainment bias. Methodologies to improve outcome assessment with technology such as seizure detection devices have been discussed15 and could be considered for future trials.
The patients’ response overall demonstrated more improvement than expected from modeling, which might suggest a combination of drug effect and placebo response. Placebo response has been climbing over time in epilepsy studies.12 In the absence of a placebo arm, it is very difficult to determine the influence of a placebo effect on the trial results. Of interest, the patients in the current study had a shorter history of epilepsy (≈15 years) than the original trials, which ranged from 18 to 26 years. In a lacosamide pooled analysis,16 shorter duration of epilepsy (with fewer background AEDs) increased the likelihood of placebo-associated improvement.
A negative clinical trial is one in which it is obvious that the drug under study did not work. The cause of a failed clinical trial is potentially more difficult to discern. Had the expectation been that these 2 drugs were the same rather than that they were different, this would not have been a failed trial. Here, both study drugs showed more improvement in seizure frequencies than expected. The results again raise the question of the impact of the changing environment of epilepsy studies, which are now often conducted in numerous sites and countries with differing background medical care. A future topic of discussion must be how to account for these changes in modern clinical trial design and how to conduct them without losing the benefit of prior knowledge.
These results suggest that designing trials that compare the efficacy of 2 active treatments is fraught with potential confounders that can mute the real differences that may exist in drug effect.17 This should be taken into consideration when a no-difference outcome is observed, regardless of whether prior trials have suggested the existence of assay sensitivity, if study populations have changed over time.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Jaap Mandema, PhD, for modeling work8 and Bhairavi (Bella) Pandya, PhD, for attention to detail during the conduct of the study.
GLOSSARY
- AED
antiepileptic drug
- CI
confidence interval
- ESCI
Epilepsy Study Consortium Inc.
- ILAE
International League Against Epilepsy
- mITT
modified intent-to-treat
- POS
partial-onset seizure
- SGTC
secondary generalized tonic-clonic
- TEAE
treatment-emergent adverse event
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
All authors contributed to the design/conduct of the study. M.A. was responsible for the statistical analysis, and D.F., H.B.P., J.F., L.K., M.A., and V.P. contributed to the interpretation of the data. All authors contributed to the drafting/revision of the manuscript and approved the final version.
STUDY FUNDING
This study was funded by Pfizer Inc.
DISCLOSURE
Dr. French is president of the Epilepsy Study Consortium. All consulting is done on behalf of the consortium, and fees are paid to the consortium. The NYU Comprehensive Epilepsy Center receives salary support from the consortium. Dr. French has acted as a consultant for Acorda, Biotie, Brabant Pharma, Eisai Medical Research, Glaxo Smith-Kline, GW Pharma, Impax, Johnson & Johnson, Marathon Pharmaceuticals, Marinus, Neusentis, Novartis, Pfizer, Sage, Sunovion, SK Life Sciences, Supernus Pharmaceuticals, Takeda, UCB, Upsher-Smith, Ultragenyx, Vertex, and Zynerba; has received grants and research support from Acorda, Alexza, LCGH, Eisai Medical Research, Lundbeck, Pfizer, SK Life Sciences, UCB, Upsher-Smith, and Vertex; and grants from the National Institute of Neurological Disorders and Stroke, Epilepsy Therapy Project, Epilepsy Research Foundation, and Epilepsy Study Consortium. Dr. Glue was an employee of Pfizer Inc at the time this research was designed and initiated. He has received research funding from Roche and Demerx and is a consultant to Kinex Pharma. Dr. Friedman is an investigator at NYU on studies for UCB Inc/Schwarz Pharma and receives salary support for consulting and clinical trial–related activities performed on behalf of the Epilepsy Study Consortium, a nonprofit organization. Dr. Friedman receives no personal income for these activities. NYU receives a fixed amount from the Epilepsy Study Consortium toward Dr. Friedman’s salary. Within the past year, the Epilepsy Study Consortium received payments for research services performed by Dr. Friedman from Alexza Pharmaceuticals, Acorda, Eisai Medical Research, Marinus, Pfizer, SK Life Science, and Upsher Smith. M. Almas is an employee of Pfizer Inc. Dr. Yardi received principal investigator fees from KEM Hospital for studies from Pfizer and UCB in the past year and from Abbott for an epidemiologic study. He has received travel support and advisory fees from UCB, Abbott Health Care, Abbott India, Ranbaxy, Sun Pharma, Torrent Pharma, Intas Pharma, and GSK for work on advisory boards, as well as work as a speaker, chairperson, or course director at conferences. Currently, he is a reviewer for Epilepsia and a member of the ILAE Task Force on Pediatric Epilepsy; both are honorary (noncompensated) positions. Drs. Knapp, Pitman, and Posner are employees of Pfizer Inc. We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Loscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 2011;52:657–678. [DOI] [PubMed] [Google Scholar]
- 2.Trinka E, Marson AG, Van Paesschen W, et al. KOMET: an unblinded, randomised, two parallel-group, stratified trial comparing the effectiveness of levetiracetam with controlled-release carbamazepine and extended-release sodium valproate as monotherapy in patients with newly diagnosed epilepsy. J Neurol Neurosurg Psychiatry 2013;84:1138–1147. [DOI] [PubMed] [Google Scholar]
- 3.Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007;369:1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uchitel OD, Di Guilmi MN, Urbano FJ, Gonzalez-Inchauspe C. Acute modulation of calcium currents and synaptic transmission by gabapentinoids. Channels 2010;4:490–496. [DOI] [PubMed] [Google Scholar]
- 5.Al-Bachari S, Pulman J, Hutton JL, Marson AG. Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev 2013:CD001415. [DOI] [PubMed] [Google Scholar]
- 6.Lalonde RL, Kowalski KG, Hutmacher MM, et al. Model-based drug development. Clin Pharmacol Ther 2007;82:21–32. [DOI] [PubMed] [Google Scholar]
- 7.Pulman J, Hemming K, Marson AG. Pregabalin add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev 2014:CD005612. [DOI] [PubMed] [Google Scholar]
- 8.Mandema J. Impact of prior knowledge on drug development decisions: case studies across companies. Available at: http://www.fda.gov/ohrms/dockets/ac/06/slides/2006-4248s2-2-QuantitativeSolutionsMandema.ppt. Accessed December 3, 2014.
- 9.Zaccara G, Almas M, Pitman V, Knapp L, Posner H. Efficacy and safety of pregabalin versus levetiracetam as adjunctive therapy in patients with partial seizures: a randomized, double-blind, noninferiority trial. Epilepsia 2014;55:1048–1057. [DOI] [PubMed] [Google Scholar]
- 10.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 11.Proposal for revised clinical and electroencephalographic classification of epileptic seizures: from the Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1981;22:489–501. [DOI] [PubMed] [Google Scholar]
- 12.Rheims S, Perucca E, Cucherat M, Ryvlin P. Factors determining response to antiepileptic drugs in randomized controlled trials: a systematic review and meta-analysis. Epilepsia 2011;52:219–233. [DOI] [PubMed] [Google Scholar]
- 13.International Conference on Harmonization. Choice of control group and related issues in clinical trials. In: ICH Harmonised Tripartite Guideline. Geneva, Switzerland: ICH; 2000. [Google Scholar]
- 14.Enck P, Klosterhalfen S, Weimer K, Horing B, Zipfel S. The placebo response in clinical trials: more questions than answers. Philos Trans R Soc Lond B Biol Sci 2011;366:1889–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman D, French JA. Clinical trials for therapeutic assessment of antiepileptic drugs in the 21st century: obstacles and solutions. Lancet Neurol 2012;11:827–834. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt D, Beyenburg S, D'Souza J, Stavem K. Clinical features associated with placebo response in refractory focal epilepsy. Epilepsy Behav 2013;27:393–398. [DOI] [PubMed] [Google Scholar]
- 17.Mancini M, Wade AG, Perugi G, Lenox-Smith A, Schacht A. Impact of patient selection and study characteristics on signal detection in placebo-controlled trials with antidepressants. J Psychiatr Res 2014;51:21–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.