

Abstract
Objective:
To investigate the effect of enriching mild cognitive impairment (MCI) clinical trials using combined markers of amyloid pathology and neurodegeneration.
Methods:
We evaluate an implementation of the recent National Institute for Aging–Alzheimer's Association (NIA-AA) diagnostic criteria for MCI due to Alzheimer disease (AD) as inclusion criteria in clinical trials and assess the effect of enrichment with amyloid (A+), neurodegeneration (N+), and their combination (A+N+) on the rate of clinical progression, required sample sizes, and estimates of trial time and cost.
Results:
Enrichment based on an individual marker (A+ or N+) substantially improves all assessed trial characteristics. Combined enrichment (A+N+) further improves these results with a reduction in required sample sizes by 45% to 60%, depending on the endpoint.
Conclusions:
Operationalizing the NIA-AA diagnostic criteria for clinical trial screening has the potential to substantially improve the statistical power of trials in MCI due to AD by identifying a more rapidly progressing patient population.
Anatomic and pathophysiologic changes in Alzheimer disease (AD) begin years before the emergence of clinical dementia.1–3 Recent revisions to AD diagnostic criteria have included explicit references to biomarkers for differential diagnosis in the study of subjects for clinical research in both AD and presymptomatic stages.4–9 In particular, the National Institute for Aging–Alzheimer's Association (NIA-AA) criteria propose positivity on both amyloid and neurodegeneration biomarkers (A+ and N+, respectively).6,10
Recent negative phase III clinical trials of antiamyloid therapies did not consider biomarkers for inclusion and recruited a significant portion of amyloid-negative subjects who did not progress substantially on primary endpoints.11–14 More recent phase II/III trials in prodromal or mild AD implemented enrichment strategies based on amyloid markers.15,16
The value of MRI measures of neurodegeneration for trial enrichment has been shown both in isolation and in combination with other measurements,17–25 has been endorsed by regulatory agencies,26 and more recently has been emphasized by a post hoc analysis of the failed (A+) Gantenerumab SCarlet RoAD study, showing a treatment effect in a multibiomarker-enriched subpopulation.15,27
This article presents an operationalization of the NIA-AA guidelines for mild cognitive impairment (MCI) due to AD4,6 for enriching clinical trials with the concept of a dual (A+N+) biomarker strategy.10,28 Applying established cut points19,29,30 to define biomarker-positive subpopulations, this study compares the performance of combining both biomarkers (A+N+) compared to screening based on either alone and the resulting improvements in sample size and screen fail fraction and presents total trial time and cost.
METHODS
Study population.
The present study was performed on 274 participants with MCI and 444 healthy controls from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://www.loni.usc.edu/ADNI).31
For the MCI cohort, we selected all participants with amnestic MCI (labeled MCI in ADNI-1 and L-MCI in ADNI-2) who met the following inclusion criteria: 1.5T (ADNI-1) or 3T (ADNI-2) MRI scan available at baseline, baseline assessment of amyloid status (from CSF or AV-45 amyloid PET) available, and clinical scores (Mini-Mental State Examination [MMSE], 13-point Alzheimer's Disease Assessment Scale Cognitive Subscale [ADAS-Cog13]) available at baseline and at month 24.
For the healthy control cohort, used as a normative reference population to define a hippocampal volume (HV) cut point, all participants who had an MRI available at baseline from both ADNI-1 and ADNI-2 were used. We have previously demonstrated minimal differences in HVs calculated from 1.5T and 3T scans on the same participants.32
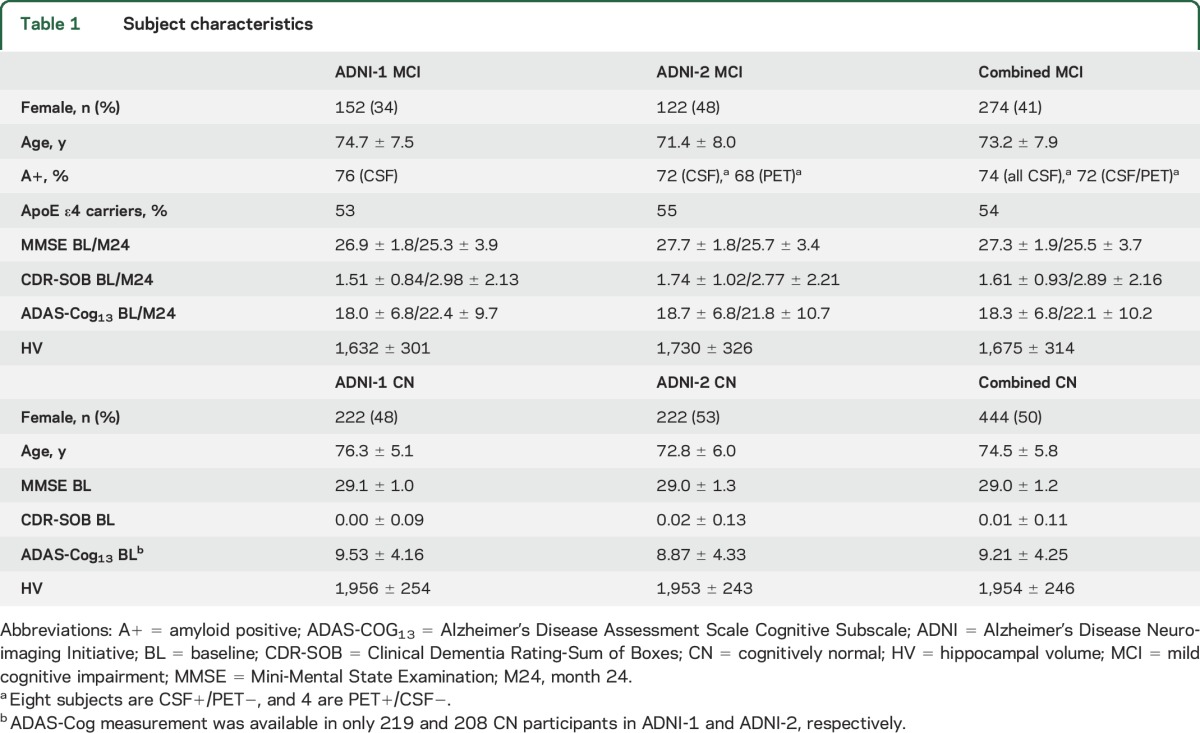
An overview of the included participants, together with key characteristics, is presented in table 1. A more detailed description of ADNI and its study design and the image acquisition parameters are given in appendix e-1 at Neurology.org.
Table 1.
Subject characteristics
Biomarker measures.
Amyloid deposition measured through PET imaging and CSF analyses of β-amyloid (Aβ) levels has been shown to be highly concordant and to show a similar diagnostic accuracy.33 Consequently, the selection of either marker is driven mainly by geographic availability and cost, as well as patient and doctor preferences.33 For the purpose of identifying participants as amyloid positive or negative, we used CSF Aβ42 for participants from ADNI-1 and florbetapir-PET for participants from ADNI-2; cut points derived from both biomarkers have been shown to perform consistently.30,33 Specifically, ADNI-1 participants were considered A+ with Aβ42 levels ≤192 pg/mL, while ADNI-2 participants were considered A+ with a florbetapir-PET standardized uptake value ratio >1.11 for the mean of 6 predefined regions of interest relative to the whole cerebellum. Appendix e-2 presents results from the ADNI-2 cohort on the concordance between amyloid positivity from the 2 biomarkers and its impact on patient selection. However, our cost estimates are based on the assumption that PET imaging is used to determine amyloid positivity for our simulated clinical trial scenarios.
HV was calculated with the learning embeddings for atlas propagation (LEAP) algorithm34 incorporated into the CE-marked medical device Assessa (www.assessa.com). HV was adjusted for age and head size as measured by intracranial volume, and left and right hippocampi were averaged for each subject (details are presented in appendix e-3). After a detailed validation of HV cut points,19 participants were considered N+ if their adjusted HV was smaller than the 25th percentile of the age-matched healthy subject cohort.
Combinatorial enrichment strategies.
We examined the following enrichment strategies: NIA-AA I (A+), enrichment by abnormal amyloid alone (neurodegeneration status undetermined); NIA-AA II (N+), enrichment by low HV alone (amyloid status undetermined); and NIA-AA III (A+N+), enrichment by both amyloid and HV. For comparison, the performance of the following 4 enrichment strategies is presented in appendix e-4: exploratory I, enrichment by ApoE ε4 status alone (ε4 carriers only included); exploratory II, enrichment based on a clinical functional deficit (Functional Assessment Questionnaire [FAQ] >0); exploratory III, enrichment based on a deficit in global cognition beyond that specified as part of the diagnostic criteria (ADAS-Cog13 >15); and exploratory IV, enrichment based on a more stringent definition of memory deficit (Rey Auditory Verbal Learning Test [RAVLT] <35).
Experiments.
The effect of biomarker-based enrichment was assessed with respect to the 2-year change in widely used clinical scales of global cognition, MMSE and ADAS-Cog13, in terms of 2 key trial characteristics: the rate and homogeneity of clinical progression in the included trial cohort and the additional screen failure fraction due to the biomarker selection criteria. As a joint measurement of rate and homogeneity of clinical progression, we defined a signal-to-noise ratio (SNR) as the mean 2-year change on the clinical scale in the included group divided by the SD of the 2-year change. On the basis of representative values from multisite trial operations, we also estimated how the change and interplay of these key measurements influence required sample sizes, the number of participants who need to undergo screening to obtain the required sample size, and indicative overall trial duration and cost.19 Calculations of the latter take into account the tradeoff between increased screen failures and the reduced number of participants needed to be randomized. Sample size calculations were performed for a 25% reduction in the worsening of either MMSE or ADAS-Cog13 at 80% power and 5% significance. Detailed models for sample size, number needed to screen, and trial time and cost are presented in appendix e-3. Although amyloid positivity is defined from CSF (ADNI-1 participants) and PET imaging (ADNI-2 participants) in this work, the use of PET imaging is assumed in the cost calculation.
RESULTS
All 3 NIA-AA–based enrichment strategies yielded increased 2-year SNRs for both MMSE and ADAS-Cog13 (figure 1A) compared to the unenriched population. Enrichment with HV (N+) showed the smallest improvement, followed by enrichment based on amyloid positivity (A+), whereas enrichment based on both biomarkers (A+N+) provided the overall best enrichment performance. SNR was increased by 18% (MMSE) and 25% (ADAS-Cog13) by N+ enrichment, 31% and 38%, respectively, by A+ enrichment, and 45% and 57%, respectively, by A+N+ enrichment.
Figure 1. Trial characteristics for different enrichment strategies.

Signal-to-noise ratio (SNR; A), screen fail fraction (B), and required sample sizes (C) for trials using different enrichment strategies. A+ = amyloid positive; ADAS-Cog13 = Alzheimer's Disease Assessment Scale Cognitive Subscale; MMSE = Mini-Mental State Examination; N+ = neurodegeneration positive.
The biomarker screen fail fractions were 28% for enrichment by A+ or N+ alone and 45% for the combined A+N+ enrichment (figure 1B). However, the required sample size to enroll was substantially reduced, by 26% (MMSE) and 33% (ADAS-Cog13) for N+ enrichment, by 40% and 50% for A+ enrichment, and by 53% and 60% for A+N+ enrichment (figure 1C). In terms of overall estimated trial duration and cost, all 3 enrichment strategies resulted in quicker, cheaper clinical trials (table 2). That is, the gain due to the reduced sample size more than compensated for the increased screen fail rate. Performing N+ enrichment before A+ enrichment leads to a further reduction in trial cost (table 2).
Table 2.
Screen failure fraction (SFF), 2-year change on clinical outcome measures (mean ± SD), 2-year signal-to-noise ratio (SNR: mean/SD), number of participants who need to undergo screening (NNS), trial cost, and overall trial duration

Two-year change on both clinical scales showed a nominally more rapid progression in the selected cohort relative to the unenriched cohort for all 3 enrichment methods, and this was statistically significant with the use of the A+ and A+N+ enrichment schemes for both scales (p < 0.05; figure 2). The excluded group showed substantially slower clinical progression rates for both scales using all 3 enrichment schemes (p < 0.01). When shorter 6-month and 12-month trials were simulated, SNR was increased for both measures by amounts similar to that found for the 24-month trial (appendix e-5).
Figure 2. Time course graphs for different enrichment strategies.

Change in Mini-Mental State Examination (MMSE; A) and Alzheimer's Disease Assessment Scale Cognitive Subscale (ADAS-Cog 13; B) for the unenriched sample (dashed black line), enriched sample (solid blue line), and excluded sample (solid green line). Whiskers present SE. Significance of the difference between included and excluded groups and the unenriched sample is shown as **p < 0.05 and *p < 0.01. A+ = amyloid positive; N+ = neurodegeneration positive.
Detailed results for the comparator enrichment strategies, based on ApoE ε4 genotype, cognition (ADAS-Cog13), function (FAQ), and memory (RAVLT), are presented in appendix e-4. Briefly, enrolling ApoE ε4 carriers (a strong risk factor for amyloid positivity) yielded an improvement in SNR of 24% (MMSE) and 28% (ADAS-Cog13) at a screen failure rate of 46%. Enriching with ADAS-Cog13 >15 improved the SNR by 40% on MMSE at the cost of an additional 31% screen failure rate, while FAQ-based enrichment gave an SNR increase of 40% (MMSE) and 20% (ADAS-Cog13) with an associated screen failure rate of 30%. Enriching with RAVLT <35 gave an SNR increase of 23% (MMSE) and 36% (ADAS-Cog13) at the cost of an additional 32% screen failure rate. A corresponding reduction in sample sizes was observed with all 4 of these alternative, non–biomarker-based enrichment strategies.
DISCUSSION
These results demonstrate the effect of using the current NIA-AA research diagnostic criteria as an enrichment strategy for amnestic MCI clinical trials. Compared to previous work that looked at multibiomarker enrichment, we explicitly examined the effects of these diagnostic criteria using specific, established cut points on both measures. With a view to operationalizing these criteria, we also assessed their effect on trial design by exploring the changes in a set of practical trial characteristics, including time and cost, using parameters from recent trials. Although these input values may vary, our findings of a substantial relative improvement in trial parameters should be relatively robust to realistic differences in these inputs and the assumptions underlying our model.
The presented results show that enrichment with either biomarker individually (A+ or N+) increases the SNR of the change in clinical endpoint measures and reduces the required sample sizes and the projected trial cost. When combined enrichment (A+N+) is applied, the SNR values increase and the required sample sizes decrease relative to no enrichment by 45% to 60%, depending on the clinical endpoint. The mean 2-year change in ADAS-Cog13 in the enriched population was increased from 3.9 points (unenriched) to 5.0 to 6.5 points, and the mean decrease in MMSE increased from 1.8 points (unenriched) to 2.2 to 2.8 points. These results show that when a trial with fixed power is designed, biomarker enrichment does not increase the number of participants who need to be screened because the reduction in the required sample size outweighs the increased screen failure rate. Projected trial durations were overall predicted to be no longer than in the unenriched scenario. These results were robust to trial duration because simulations with shorter follow-up times resulted in very similar relative characteristics of the enriched population (appendix e-5).
Several recent trials have enrolled amyloid-positive participants only. Our results show that using HV-based enrichment in addition further reduces sample sizes by ≈20% and cost by ≈25% when the cheaper exclusion criterion (MRI) is administered first because PET imaging is then required in fewer participants. Because an MRI scan is typically acquired at screening for other reasons, the added operational complexity of automated and rapid computation of a HV estimate is minimal but can lead to significant savings in trial cost. Automatically derived HV provides robust enrichment with relatively little sensitivity to the cut point used.19 To confirm this in the present study, we compared enrichment with a cut point at the 40th percentile (as an alternative to the 25th percentile used above) of HV in the healthy control population. Screen failure rate changes from 28% to 24%, yet the assessed trial characteristics change by <10% (details are given in appendix e-2). While HV is an important downstream biomarker of AD and therefore can help to identify patients close to accelerated clinical decline,1 it is not directly linked to Aβ, one of the hallmarks in the amyloid cascade hypothesis of AD.35 HV alone therefore cannot replace an amyloid marker to accurately select patients in amyloid targeting therapies but can complement it to help to identify subpopulations more likely to progress on primary trial endpoints. Even when not defining a specific exclusion criterion on HV, it can be a valuable stratification or subgrouping measure as shown by the recently presented post hoc analysis of the MCI Gantenerumab SCarlet RoAD study in which all the enrolled participants were amyloid positive but a significant treatment effect was observed only in the subgroup of participants predicted to progress more rapidly with a model that used HV and clinical scales.15
Our cost analysis assumed that PET imaging was used to determine amyloid status. Although some reports suggest that CSF levels of Aβ become abnormal earlier in the disease stage,33 the results presented in appendix e-6 confirm previous publications that showed high concordance between amyloid markers from CSF and PET.33,36 A recent systematic comparison of PET and CSF concludes that choices can be based on other factors such as availability, cost, and doctor/patient preferences because both have equally high diagnostic accuracy.36 While a CSF analysis can potentially be incorporated more easily with other biomarkers, it is highly invasive and requires careful standardization; PET imaging, on the other hand, requires highly advanced instruments and is less available in clinical practice in some countries.36
In a separate analysis, this article shows how alternative enrichment strategies that avoid imaging measurements can also increase the longitudinal progression rate of a patient population thus selected. The proposed models were applied to enrichment with ApoE e4 status and measures of global cognition and function that are widely used in clinical trials of mild AD and have furthermore been shown to perform well in the prodromal stage of the disease.37 The results using ApoE e4 status and measurements of cognition, memory, and function (appendix e-4) show an enrichment performance similar to that obtained with the 2 imaging biomarkers and following the NIA-AA criteria.
A limitation of the presented work is its validation on the ADNI study alone. Even though inclusion and exclusion criteria in ADNI were defined to be comparable to those in clinical trials, it is unclear how well the recruited participants compare to a typical clinical trial population, let alone a real-world treatment population. However, preliminary results from current clinical studies using biomarker enrichment (e.g., aducanumab) and the post-hoc analysis of the SCarlet RoAD study discussed above provide additional evidence for the benefit that single-biomarker and multibiomarker enrichment strategies can have.
Integrating different biomarkers, clinical measurements, and patient attributes into integrated disease progression models represents a next step toward a more detailed understanding of disease progression and the efficient use of available measurements for trial inclusion, as well as the selection of suitable patients once treatment is available.
Supplementary Material
GLOSSARY
- A+
amyloid positive
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAS-Cog13
Alzheimer's Disease Assessment Scale Cognitive Subscale
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- FAQ
Functional Assessment Questionnaire
- HV
hippocampal volume
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- N+
neurodegeneration positive
- NIA-AA
National Institute for Aging–Alzheimer's Association
- RAVLT
Rey Auditory Verbal Learning Test
- SNR
signal-to-noise ratio
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Robin Wolz: study concept and design, collation of data, analysis and interpretation of data. Adam J. Schwarz: study concept and design, interpretation of data, critical revision of manuscript for intellectual content. Katherine R. Gray: collation of data, analysis of data. Peng Yu: study concept and design, analysis and interpretation of data. Derek L.G. Hill: study concept and design, critical revision of manuscript for intellectual content, study supervision.
STUDY FUNDING
Data collection and sharing for this project were funded by the ADNI (NIH grant U01 AG024904) and Department of Defense ADNI (Department of Defense award No. W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc; Cogstate; Eisai Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co, Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The study is cofunded by Innovate UK (Formerly Technology Strategy Board).
DISCLOSURE
R. Wolz, K. Gray, and D. Hill are employees and shareholders of IXICO. A. Schwarz is an employee and shareholder of Eli Lilly. P. Yu is a former employee of Eli Lilly. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vinters H. Emerging concepts in Alzheimer's disease. Annu Rev Pathol 2015;10:291–319. [DOI] [PubMed] [Google Scholar]
- 3.Beach T, Schneider J, Sue L, et al. Theoretical impact of florbetapir (18F) amyloid imaging on diagnosis of Alzheimer dementia and detection of preclinical cortical amyloid. J Neuropathol Exp Neurol 2014;73:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging and the Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol 2014;13:614–629. [DOI] [PubMed] [Google Scholar]
- 6.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wharton S, Brayne C, Savva G, et al. Epidemiological neuropathology: the MRC Cognitive Function and Aging Study experience. J Alzheimers Dis 2011;25:359–372. [DOI] [PubMed] [Google Scholar]
- 9.Beach T, Monsell S, Phillips L, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol 2012;71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR Jr, Wiste HJ, Knopman DS, et al. Rates of β-amyloid accumulation are independent of hippocampal neurodegeneration. Neurology 2014;82:1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vellas B, Carrillo MC, Sampaio C, et al. EU/US/CTAD Task Force Members. Designing drug trials for Alzheimer's disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement 2013;9:438–444. [DOI] [PubMed] [Google Scholar]
- 12.Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programmes targeting the amyloid hypothesis for Alzheimer's disease. Ann Neurol 2014;76:185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampel H, Frank R, Broich K, et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov 2010;9:560–574. [DOI] [PubMed] [Google Scholar]
- 14.FDA. Guidance for Industry Alzheimer's Disease: Developing Drugs for the Treatment of Early Stage Disease. Washington, DC: US Food and Drug Administration; 2012. [Google Scholar]
- 15.Lasser R, Ostrowitzki S, Scheltens P, et al. Efficacy and safety of gantenerumab from the phase 3 SCarlet RoAD trial, a study of gantenerumab in patients with prodromal AD. Paper presented at: CTAD, November 5–7, 2015; Barcelona, Spain.
- 16.Sevigny J, Chiao P, Williams L, et al. Randomized, placebo-controlled, phase 1B study of the anti–beta-amyloid antibody aducanumab (BIIB037) in patients with prodromal or mild Alzheimer's disease: interim results. Presented at: CTAD, November 5–7, 2015; Barcelona, Spain.
- 17.Vos S, van Rossum I, Verhey F, et al. Prediction of Alzheimer disease in subjects with amnestic and nonamnestic MCI. Neurology 2013;80:1124–1132. [DOI] [PubMed] [Google Scholar]
- 18.Samtani M, Raghavan N, Novak G, Nandy P, Narayan V. Disease progression model for Clinical Dementia Rating-Sum of Boxes in mild cognitive impairment and Alzheimer's subjects from the Alzheimer's Disease Neuroimaging Initiative. Neuropsychiatr Dis Treat 2014;10:929–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu P, Sun J, Wolz R, et al. Operationalizing hippocampal volume as an enrichment biomarker for amnestic mild cognitive impairment trials: effect of algorithm, test-retest variability, and cut point on trial cost, duration, and sample size. Neurobiol Aging 2014;35:808–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenzi M, Donohue M, Paternicò D, et al. Enrichment through biomarkers in clinical trials of Alzheimer's drugs in patients with mild cognitive impairment. Neurobiol Aging 2010;31:1443–1451. [DOI] [PubMed] [Google Scholar]
- 21.Kohannim O, Hua X, Hibar DP, et al. Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiol Aging 2010;31:1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu P, Dean RA, Hall SD, et al. Enriching amnestic mild cognitive impairment populations for clinical trials: optimal combination of biomarkers to predict conversion to dementia. J Alzheimers Dis 2012;32:373–385. [DOI] [PubMed] [Google Scholar]
- 23.van Rossum IA, Vos SJ, Burns L, et al. Injury markers predict time to dementia in subjects with MCI and amyloid pathology. Neurology 2012;79:1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holland D, McEvoy L, Desikan R, Dale A. Enrichment and stratification for predementia Alzheimer disease clinical trials. PLoS One 2012;7:e47739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macklin EA, Blacker D, Hyman BT, Betensky RA. Improved design of prodromal Alzheimer's disease trials through cohort enrichment and surrogate endpoints. J Alzheimers Dis 2013;36:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill D, Schwarz A, Isaac M, et al. CAMD/EMA biomarker qualification of hippocampal volume for enrichment of clinical trials in pre-dementia stages of Alzheimer's disease. Alzheimers Dement 2014;10:421–429. [DOI] [PubMed] [Google Scholar]
- 27.Delor I, Charoin JE, Gieschke JE, Retout S, Jacqmin P. Modeling Alzheimer's disease progression using disease onset time and disease trajectory concepts applied to CDR-SOB scores from ADNI. CPT Pharmacometrics Syst Pharmacol 2013;2:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack CR, Knopman DS, Weigand SD, et al. An operational approach to NIA-AA criteria for preclinical Alzheimer's disease. Ann Neurol 2012;71:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw L, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's Disease Neuroimaging Initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weigand S, Vemuri P, Wiste H, et al. Transforming CSF Aβ42 measures into calculated Pittsburgh Compound B (PIBcalc) units of brain Aβ amyloid. Alzheimers Dement 2011;7:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller S, Weiner M, Thal L, et al. The Alzheimer's Disease Neuroimaging Initiative. Neuroimaging Clin N Am 2005;15:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolz R, Schwarz AJ, Yu P, et al. Robustness of automated hippocampal volumetry across magnetic resonance field strengths and repeat images. Alzheimers Dement 2014;10:430–438. [DOI] [PubMed] [Google Scholar]
- 33.Blennow K, Mattsson N, Schoell M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer's disease. Trends Pharmacol Sci 2015;36:297–309. [DOI] [PubMed] [Google Scholar]
- 34.Wolz R, Aljabar P, Hajnal J, Hammers A, Rueckert D. LEAP: learning embeddings for atlas propagation. Neuroimage 2010;49:1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ittner LM, Götz J. Amyloid-β and tau: a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci 2011;12:67–72. [DOI] [PubMed] [Google Scholar]
- 36.Palmqvist S, Zetterberg H, Mattsson N, et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015;85:1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wessels A, Siemers E, Yu P, et al. A combined measure of cognition and function for clinical trials: the integrated Alzheimer's disease rating scale (iADRS). J Prev Alzheimers Dis 2015;2:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.