

Abstract
Post-transplant lymphoproliferative disorder (PTLD) is an aggressive complication of solid organ and hematopoietic stem cell transplantation that arises in up to 20% of transplant recipients. Infection or reactivation of the Epstein-Barr virus (EBV), a ubiquitous human herpesvirus, in combination with chronic immunosuppression are considered as the main predisposing factors, however insight in PTLD biology is fragmentary. The study of PTLD is complicated by its morphological heterogeneity and the lack of prospective trials, which also impede treatment optimization. Furthermore, the broad spectrum of underlying disorders and the graft type represent important confounding factors. PTLD encompasses different malignant subtypes that resemble histologically similar lymphomas in the general population. Post-transplant diffuse large B-cell lymphoma (PT-DLBCL), Burkitt lymphoma (PT-BL) and plasmablastic lymphoma (PT-PBL) occur most frequently. However, in many studies various EBV+ and EBV- PTLD subtypes are pooled, complicating the interpretation of the results. In this review, studies of the gene expression pattern, the microenvironment and the genetic profile of PT-DLBCL, PT-BL and PT-PBL are summarized to better understand the mechanisms underlying post-transplantation lymphomagenesis. Based on the available findings we propose stratification of PTLD according to the histological subtype and the EBV status to facilitate the interpretation of future studies and the establishment of clinical trials.
Keywords: Epstein-Barr virus, Post-transplant lymphoproliferative disorder, Immunodeficiency, Diffuse large B-cell lymphoma, Burkitt lymphoma, Plasmablastic lymphoma
Core tip: At the moment different post-transplant lymphoproliferative disorders (PTLD) are grouped in broad categories (early, polymorphic, monomorphic and Hodgkin-like PTLD) and the Epstein-Barr virus (EBV) status is not taken into account. However, increasing evidence demonstrates that different malignant PTLD and EBV+ and EBV- lesions are clinically and biologically distinct, stressing the need for subtype-specific management. We propose that in future studies patients should be stratified according to the histological lymphoma subtype and the EBV status to minimize bias and to simplify the establishment and analysis of clinical trials.
INTRODUCTION
Despite the increasing incidence of cancer worldwide, only a limited number of cancer-causing factors have been identified. Viruses are amongst them: An estimated 15% of cancers are attributed to viral infections. One of the most widely spread oncogenic viruses is the Epstein-Barr virus (EBV), a gamma human herpesvirus with a seroprevalence of 90%-95% in adults. EBV, discovered in 1964[1], is best known as the cause of infectious mononucleosis (or kissing disease)[2]. EBV-driven lymphoproliferative disorders (LPD) are characterized by an EBV-driven immortalization of B-cells. In an otherwise healthy individual, development of such LPD is countered by a strong immune response [mainly of cytotoxic T-cells (CTL)], which ultimately resolves the infection. However, when the immune system is compromised [e.g., in acquired immunodeficiency syndrome (AIDS) patients or in organ transplant recipients under chronic immunosuppression] EBV-driven LPD may eventually progress to overt lymphoma.
During the last decades, the number of solid organ (e.g., kidney, heart, liver, etc.) and stem cell transplantations has increased significantly. In parallel, the risk of graft rejection has dropped thanks to the development of more potent immunosuppressive agents resulting in longer survival of transplant recipients. However, a major drawback of the chronically immunosuppressed status of these individuals is the development of a potentially fatal post-transplant lymphoproliferative disorder (PTLD) in up to 20% of transplant recipients[3]. PTLD is a relatively new disease entity that is now widely recognized. The first cases were described in renal transplant patients, shortly after the introduction of chronic immunosuppressive drugs in the 1960s[4]. Despite the strong association between EBV and PTLD (about 70% of PTLD are EBV-positive, EBV+), disease biology is not well understood[3]. The pathological presentation of PTLD is variable, ranging from a localized benign LPD to lymphoma associated with poor survival[5]. Treatment of PTLD patients is largely based on insights in lymphomagenesis in immunocompetent patients, in which there is no evident role for EBV in the majority of cases. For application of more adequate therapy it is indispensable to characterize PTLD more thoroughly.
The most common malignant PTLD subtype is post-transplant diffuse large B-cell lymphoma (PT-DLBCL), followed by Burkitt lymphoma (PT-BL) and plasmablastic lymphoma (PT-PBL). PT-BL and PT-PBL are aggressive, but poorly studied malignant PTLD subtypes. The number of reported cases is limited and most studies mainly focus on patient management[6-9].
In this review we summarize the available data on the genetic profile, the gene expression pattern and the microenvironment of these malignancies to better understand the mechanisms underlying post-transplantation lymphomagenesis. A literature search was performed for “PTLD” or “post-transplant lymphoproliferative disorder” with or without “diffuse large B-cell lymphoma”, “Burkitt lymphoma” or “plasmablastic lymphoma” and the available literature regarding PTLD pathogenesis was collected. For a review of the diagnosis and management of PTLD we refer the reader to[3,10].
DISCUSSION
EBV exploits the germinal center route of B-cell activation
During a normal humoral immune response, a circulating B-cell that encounters its cognate antigen becomes an activated blast with two possible faiths.
The B-cell can mature into a short-lived plasma cell that quickly produces IgM class antibodies with limited specificity (T-cell independent pathway). Alternatively, the B-cell may form a germinal center (GC) in a lymph node, mucosa-associated lymphoid tissue or spleen (T-cell dependent pathway). In the GC, the specificity of the B-cell’s antibody is enhanced by somatic hypermutation (SHM, random mutation of the antibody’s variable chain, IgV) and its functional versatility is altered by class switch recombination from IgM to IgG, IgE or IgA. Eventually, the B-cell matures into a plasma cell or a memory B-cell[11]. B-cells transiting the GC are germinal center B-cells (GCB). B-cells that have completed the GC reaction are called activated B-cells, non-GCB or post-germinal B-cells (Figure 1).
Figure 1.
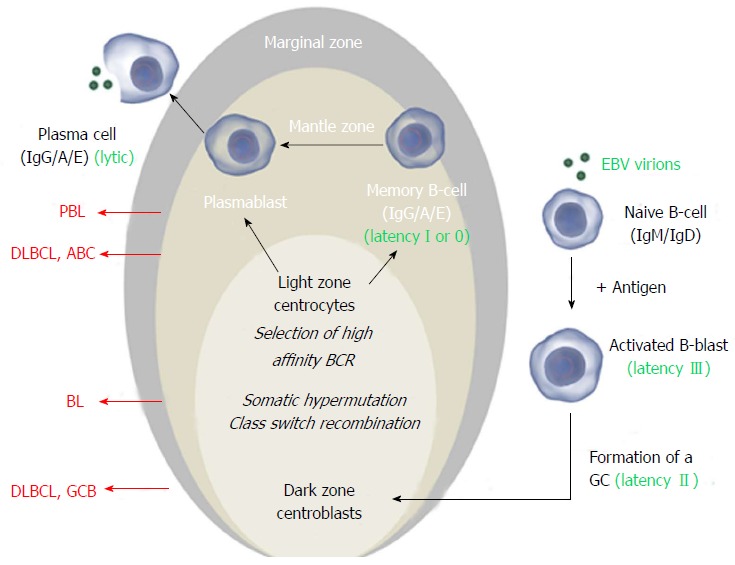
The Epstein-Barr virus exploits normal B-cell activation pathways. Activation of a naive B-cell (that expresses IgM and IgD on its surface) by its cognate antigen results in B-cell activation and differentiation into a memory B-cell or a plasma cell, most commonly via T-cell dependent activation. The antigen-activated B-cell enters a primary follicle in lymph node or spleen and forms a germinal center (GC), transforming the primary follicle into a secondary follicle. This structure is composed of three distinct regions. The marginal zone[1], which consists mainly of activated B-cells and GC-matured IgM+ B-cells, the mantle zone or corona[2], which comprises naïve and memory B-cells surrounds the GC[3]. The GC consists of a dark zone and a light zone. In the dark zone, the activated B-cells (centroblasts) proliferate and downregulate expression of IgM and IgD to allow somatic hypermutation (SHM) and class switch recombination (CSR), increasing the antibody’s affinity, specificity and functional versatility. In the light zone of the GC, the B-cells (centrocytes) with the best antibody are selected and ultimately mature into memory B-cells or plasma cells. Instead of IgM and IgD, these express high affinity IgG, IgA or IgE antibodies. Classically, Epstein-Barr virus (EBV) infects naïve B-cells that are stimulated to form a GC. In the activated blast, viral latency III (LMP1+/EBNA2+) is expressed and induces proliferation. In the GC, latency II (LMP1+/EBNA2-) is expressed and infected centroblasts presumably undergo SHM and CSR, involved in antibody maturation. After leaving the GC, they differentiate into plasma cells or (mainly) memory cells (latency I, EBNA1+ or latency 0, no expression of viral proteins). In vitro and in vivo, plasma cell differentiation results in activation of the EBV lytic cycle. In all stages, the viral DNA (circle in the nucleus) is maintained as an episome. Different stages of this process can give rise to malignancy resulting in different lymphoma subtypes that have features of their normal counterpart. Here the stages at which EBV+ and EBV- B-cell lymphoma may arise are shown for the most common subtypes. Images from www.somersault1824.com were used in this figure. PBL: Plasmablastic lymphoma; DLBCL: Diffuse large B-cell lymphoma; ABC: Activated B-cell; GCB: Germinal center B-cell.
According to the classic model, EBV infects naive B-cells and promotes formation of a GC. During GC transition, EBV proteins provide a selective advantage and stimulate differentiation to memory B-cells, the presumed reservoir of EBV. This process is enabled by coordinate expression of EBV proteins, primarily latent membrane proteins (LMP1, 2A-B) and EBV nuclear antigens (EBNA1, 2, 3A-C). Based on the pattern of expression, three different latency expression profiles are recognized[12]. These latency programs are associated with different stages of EBV B-cell infection and with particular lymphoproliferative disorders (Table 1 and Figure 1). EBV+ PT-DLBCL is classically associated with the most elaborate viral expression pattern, latency III. EBV+ PT-BL and PT-PBL on the other hand most frequently express the more restricted latency patterns I or II[13,14].
Table 1.
Epstein-Barr virus-driven lymphoproliferative disorders are linked with particular Epstein-Barr virus latency programs
| Latency | Expressed EBV gene products | Normal B-cell stage | Associated disease |
| III (growth) | EBER1-2, EBNA1-6, LMP1, LMP2A-B | Activated B lymphoblast | PT-DLBCL AIDS-related lymphoma Acute infectious mononucleosis |
| II (default) | EBER 1-2, EBNA1, LMP1- 2A | B-cell undergoing the GC reaction | PT-DLBCL Classical Hodgkin lymphoma |
| I | EBER 1-2 , EBNA1 | Memory B-cell | (PT-) Burkitt lymphoma (PT-) PBL |
EBER: Epstein-Barr virus-encoded RNA; EBNA: Epstein-Barr virus nuclear antigen; LMP: Latent membrane protein; PT-DLBCL: Post-transplant diffuse large B-cell lymphoma; PBL: Plasmablastic lymphoma; EBV: Epstein-Barr virus.
LMP1, a constitutively active mimic of CD40 (a crucial costimulatory factor in T-cell mediated B-cell activation), is regarded as the major oncogenic protein of EBV. LMP2A is a functional mimic of a B-cell receptor and provides survival signals to the B-cells. EBNA1 ensures replication of the viral genome during cell division. EBNA2 acts as a master transcriptional regulator of both viral and cellular genes[12]. Two viral miRNA clusters (BART-miRNAs and BHRF1 miRNAs) are differentially expressed depending on the particular viral latency program[15]. EBV-encoded RNA (EBER) 1 and 2 are the only gene products that are expressed throughout all latency and lytic phases of the viral cycle and represent the most reliable markers to determine EBV infection[16].
Key features of EBV latent proteins are shown in Figure 2A. For more details about the viral gene products we refer the reader to other reviews[17,18].
Figure 2.
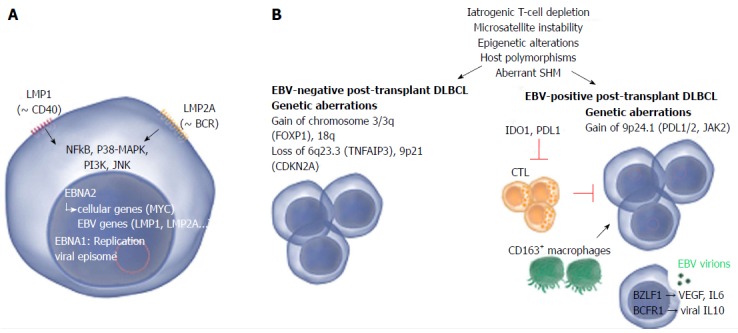
Common and distinct pathogenetic mechanisms in Epstein-Barr virus-positive and -negative post-transplant diffuse large B-cell lymphoma. A: Two Epstein-Barr virus (EBV) proteins that are thought to play a role in EBV-driven lymphomagenesis are LMP1 and LMP2A. LMP1 is analogous to CD40 and promotes cell transformation by inducing NF-κB, that in turn upregulates BCL-2, A20 and C-FLIP, all involved in blocking apoptosis. LMP2A mimics a chronically active B-cell receptor (BCR) and prevents BCR-mediated activation of EBV lytic replication. LMP2A also provides the necessary survival signals which can compensate for the loss of a functional BCR. Other pathways that are induced comprise janus kinase, p38-MAPK and PI3K signaling. Nuclear EBNA1 and EBNA2 are involved in replication of the viral episome and induction of viral as well as cellular genes respectively; B: The pathogenesis of EBV-positive and -negative lymphoma is marked by a number of common as well as distinct pathogenetic mechanisms. Mechanisms that contribute to both EBV-positive and -negative lymphoma involve iatrogenic T-cell suppression, microsatellite instability (resulting in accumulation of mutations), epigenetic alterations (mainly hypermethylation), host polymorphisms (in particular in genes encoding proteins involved in immunity), aberrant somatic hypermutation (SHM, resulting in accumulation of point mutations) and aberrant up- or down-regulation of host miRNAs which may substantially impact gene expression. EBV-negative PT-DLBCL is characterized by genetic aberrations found in EBV-negative DLBCL arising in the general population, e.g., alterations involving FOXP1. EBV-positive PT-DLBCL on the other hand harbors fewer genetic lesions. Gain of 9p24.1 (harboring PDL1/2, JAK2) has been detected and may contribute to tumor immune evasion. A minority of the EBV-positive cells actively produce viral particles. This lytic replication may promote lymphoma growth by expression of IL-6 and VEGF. Also viral IL-10 (vIL10) is expressed which contributes to suppression of anti-tumor responses by antagonizing IFN-γ. The expression of EBV proteins attracts cytotoxic T-cells (CTLs) to the site of the tumor however the question remains whether effective anti-tumor responses can be produced as also tolerant immune responses are induced. IDO1 (expressed in tumor cells and dendritic cells) and PDL1 (expressed in tumor cells and macrophages) suppress T-cells and may substantially impair the activity of CTLs. Also CD163+ macrophages (thought to be immunotolerant M2 macrophages) may play a role in immune evasion. Images from www.somersault1824.com were used in this figure. PT-DLBCL: Post-transplant diffuse large B-cell lymphoma; IL: Interleukin; IFN: Interferon; VEGF: Vascular endothelial growth factor; BCR: B-cell Receptor.
In vitro and in vivo, plasma cell differentiation of an EBV-infected B-cell is associated with activation of EBV lytic replication resulting in production of new viral particles[19]. The main activators of this process are viral ZEBRA/BZLF1 and BRLF1 proteins[20].
Although still highly debated, increasing evidence indicates that also the lytic program of EBV is of importance for B-cell transformation, the early stages in particular[21-23]. EBV lacking ZEBRA/BZLF1 and BRLF1 has significantly decreased transforming potential in vivo, associated with reduced expression of proliferation-promoting factors (IL-6, IL-10 and viral IL-10) (Figure 2B)[24]. Intriguingly, particular genetic variants of ZEBRA/BZLF1 and BRLF1 have been associated with lymphoma[25]. So far, few studies have examined lytic replication in human lymphoma biopsies[26]. In a recent report, EBV lytic replication in PTLD was associated with tumoral XBP-1 expression, early onset and short survival[27].
In the following sections, the pathogenesis of PT-DLBCL (Figure 2B), PT-BL and PT-PBL, the most common malignant PTLD subtypes, is discussed.
DLBCL
Cell of origin: DLBCL in the general population comprises at least two molecular subtypes: GCB derived and non-GCB derived DLBCL[28], thought to arise from normal GC and non-GC B-cells respectively (Figure 1). Both subtypes have been reported in the transplant setting[29]. The cell of origin is classically determined using a microarray-based surrogate set of three immunostainings (CD10, BCL6, MUM1)[30] and has prognostic implications: In the general population, GCB DLBCL has a better prognosis than non-GCB DLBCL[28]. Whether the same is true for post-transplant DLBCL is difficult to determine since the vast majority of EBV-associated cases are of non-GCB origin[26,31,32] (Figure 1). The induction of pathways like NF-κB signaling by EBV, which is highly characteristic for non-GCB DLBCL could explain this observation[33] (Figure 2A).
Another way to define the cell of origin is provided by genotypic analysis of SHM. A naïve pre-GC B-cell carries unmutated IgV, intraclonal heterogeneity reflects ongoing IgV SHM in GC centroblasts and a centrocyte/post-GC B-cell carries stable IgV mutations. Using this method the vast majority of EBV+ as well as EBV- PT-DLBCL were shown to carry IgV mutations indicating that PT-DLBCL derive mainly from GC and post-GC B-cells[26,29]. The few PT-DLBCL that do lack SHM are consistently EBV+ and arise early after transplantation. They may derive from naïve pre-GC B-cells or from B-cells that have transited the GC without completing the GC program[34-36].
Genetics: Genetic studies have demonstrated that PT-DLBCL has genomic aberrations in common with DLBCL arising in immunocompetent individuals (gains of 8q24 harboring MYC, 3q27 harboring BCL6, 18q21 harboring BCL2, 7q harboring CDK6; loss of 17p13 harboring TP53) but also bears distinct alterations (gain of 5p, loss of 4q, 17q, Xp)[37,38]. EBV+ and EBV- PTLD are rarely distinguished, but in one study EBV- PT-DLBCL was associated with gains of 7p, 7q and 11q24-q25 and del(4q25-q35)[39]. EBV+ PT-DLBCL on the other hand frequently harbored trisomies of chromosomes 9 and 11. It has been suggested that overall, EBV+ PT-DLBCL carries fewer (recurrent) genetic lesions than EBV- cases[37].
An aCGH study on a series of 21 non-GCB PT-DLBCL validated these findings[40]. Overall, EBV+ PT-DLBCL harbored fewer copy number alterations than EBV- cases. EBV+ and EBV- PT-DLBCL shared only one recurrent aberration (gain 12q21q21); the significance of this lesion is unclear. The most frequent genetic aberration detected in the EBV+ cases was gain of 9p24.1 that harbors PDL1, PDL2 and JAK2 and could contribute to PDL1 overexpression (Figure 2B). Notably, also in EBV+ DLBCL in elderly individuals (DLBCL-E) gain of 9p24.1 was among the most frequently detected lesions[41] suggesting that overlapping processes underlie the pathogenesis of EBV-driven lymphomas.
In contrast, EBV- PT- and IC-DLBCL shared many common aberrations (gain of chromosome 3/3q and 18q, and loss of 6q23.3/TNFAIP3 and 9p21/CDKN2A) characteristic for non-GCB DLBCL[42] suggesting EBV- PT-DLBCL and IC-DLBCL are biologically similar (Figure 2B).
SHM may also contribute to oncogenesis when it misfires and results in mutation of proto-oncogenes, like PIM1, PAX5, RhoH/TTF and MYC. Because primarily the 5’ regulatory region is targeted, aberrant SHM may alter the expression profile of the affected gene(s)[29]. In one study, aberrant SHM of PIM1, PAX5, RhoH/TTF and/or MYC was detected in 40% of PT-DLBCL, independently of the EBV status[29,43].
Microsatellite instability (MSI) is induced by loss of a gene involved in DNA mismatch repair accelerating the accumulation of mutations (mainly in microsatellite sequences). Interestingly, MSI seems restricted to immunodeficiency-related lymphomas and has been reported in a fraction of PTLD, unrelated to EBV status (in a series of 72 PT-DLBCL, 7% was microsatellite instable[44]). In colon carcinoma, MSI has been associated with an increased number of tumor-infiltrating lymphocytes (presumably because of the formation of neo-antigens which are then presented in MHC I on the surface of the tumor cell) suggesting that MSI lymphomas are more immunogenic than microsatellite stable tumors[45,46]. It is feasible that such immunogenic lymphomas are only tolerated in an immunocompromised host, accounting for the lack of MSI lymphomas in immunocompetent individuals.
Gene expression profile: Two early gene expression profiling studies of PTLD produced partly contradictory results, probably because of the small sample size and the different composition of the case series. Segregation of eight PT-DLBCL cases based on the EBV status in a study by Craig et al[32] could not be confirmed by a report of Vakiani et al[26], who suggested that PTLD was distinct from non-Hodgkin lymphoma in immunocompetent individuals. As a result, a number of key questions remained unresolved until recently. Are EBV+ and EBV- PTLD different or not? And how do these disease states relate to lymphoma in the general population?
Consistent with the study of Craig et al[32] a GEP study of 21 PT-DLBCL by our group pointed to a dominant role for cytotoxic antiviral immune signaling in EBV+ vs EBV- cases, implying that the presence of EBV in the tumor cells greatly affects the microenvironment[47].
Cytokines upregulated in EBV+ PT-DLBCL and associated with viral infection included CCL3, CCL4 and CCL8 involved in chemotaxis and/or activation of monocytes (CCL3, CCL4) and T-cells (CCL3, CCL8). Notably, CCL3 and CLL4 could also be part of an autocrine loop: In vitro, these cytokines were highly expressed by EBV+ lymphoblastoid cell lines (LCL) and promoted LCL proliferation and survival[48].
In contrast to Craig et al[32] we also detected enhanced immunotolerant signaling (PDL1, IDO1) in EBV+ vs EBV- PT-DLBCL (Figure 2B). These networks are likely induced to counter pro-inflammatory signaling. Upregulation of PDL1 is in line with in vitro studies that demonstrated a functional link between EBV and PDL1 expression in tumor cells[49], confirmed by histological studies of human EBV+ tumor biopsies[50]. IDO1 is involved in suppression of T-cells by degradation of tryptophan and was previously found overexpressed in EBV+ gastric carcinoma[51].
Notably, blockade of immune checkpoints (IDO1 or the PDL-PD1 axis) results in boosting of the immune response and has already shown promising results in clinical cancer trials[52]. This approach may be useful also in PTLD where it may increase the efficacy of adoptive T-cell therapy. However, because of the associated increased risk of graft rejection, the safety of checkpoint inhibitors in PTLD treatment requires further investigation.
EBV- PT-DLBCL represents the minority of PT-DLBCL cases, however there is some evidence that its incidence is increasing[53], potentially (partly) because of the overall longer survival of transplant recipients. The etiology of EBV- PTLD is unknown and therefore a major question is how these tumors relate to EBV- lymphomas in the general population.
A number of hypotheses have been raised to explain the etiology of EBV- PTLD.
The hit-and-run theory, based on in vitro data[54], states that after transformation EBV-infected B-cells may eventually lose (part of) the viral genome. However so far, there is no in vivo evidence supporting this theory[55,56].
Given the strong association between EBV and PTLD other infectious agents, e.g., HHV8 or cytomegalovirus (CMV) may be implicated in EBV- PTLD. However, PTLD cases in which HHV8 is detected are extremely rare[57,58] and because CMV does not infect B-cells it can only play an indirect role[59]. A study of AIDS-related lymphoma found only EBV to be significantly associated with pathogenesis, suggesting that also EBV- PTLD is probably not caused by an infectious agent[60].
Craig et al[32] suggested that EBV+ and EBV- monomorphic PTLD are biologically distinct and the results of our GEP analysis support this hypothesis. In the comparison of GEP data of EBV+ and EBV- PT-DLBCL, BCR signaling was upregulated in EBV- cases. As suggested by the authors, this finding could be the result of mimicked BCR signaling by LMP2A in EBV+ PT-DLBCL[32], however it could also be an artifact: Because of dominant immune signaling in EBV+ cases tumoral BCR signaling is seemingly upregulated in EBV- cases.
To gain more insight in the biology of EBV- PT-DLBCL, GEP profiles of EBV- PT and IC cases were compared. Only pathways involved in T-cell signaling were significantly differentially expressed and downregulated in PT compared to IC-DLBCL suggesting that the tumoral expression profiles are overall similar. Notably, decreased T-cell signaling explains why some cases of EBV- PT-DLBCL respond to RIS[61,62], which is generally more effective for EBV+ lesions. Therefore, restoration of the immune response in EBV- PTLD patients should remain one of the cornerstones of treatment.
Notably, gain of chromosome 3/3q (encoding FOXP1) in EBV- IC/PT-DLBCL had the strongest impact on gene expression (Figure 2B). Bio-informatics analysis of the gene set upregulated in this subgroup predicted that FOXP1, a master transcriptional regulator, regulates the expression of the majority of the genes (unreported data), suggesting FOXP1 is a major network hub in the pathogenesis of these cases. Because several studies support a central role of FOXP1 in non-GCB DLBCL pathogenesis[63] the downregulation of FOXP1 in EBV+ non-GCB PT-DLBCL is striking. Also following in vitro EBV infection of peripheral blood mononuclear cells FOXP1 is downregulated[64], indicating that FOXP1 expression is incompatible with EBV signaling. An interesting question is whether forced expression of FOXP1 in EBV+ non-GCB DLBCL cells is toxic for the tumor cells.
Microenvironment: The tumor microenvironment consists of the collection of stromal and immune cells that make up the cellular environment in which the tumor cells reside and has been shown to significantly influence prognosis in different lymphoma subtypes[65,66], also in PTLD. Particularly the infiltration of CTL has been associated with favorable prognosis (the EBV status was not taken into account). In the same study, the infiltration of regulatory T-cells (Treg), immune response modulators that prevent excessive immune activation, was limited in all PTLD cases[67]. This may be attributed to obstruction of Treg cell development by immunosuppressive agents. Analysis of the normal intestinal mucosa showed that liver transplant patients on a long-term combination regimen had significantly lower levels of Treg cells compared to healthy controls[68]. Although the scarcity of Treg cells in PTLD lesions may impede suppression of anti-tumor immune responses, also inhibition of B-cell proliferation by Treg cells is alleviated, potentially contributing to PTLD development[67]. A thorough review of the microenvironment of PTLD has not been performed but a study of AIDS-related DLBCL may give clues: Increased tumor vascularization and a higher number of infiltrating CTL were detected in EBV+ compared to EBV- cases[69].
Cell counts for different immune markers (manuscript submitted) performed on a series of PT-DLBCL showed increased infiltration of CD8+ CTL in part of the EBV+ compared to EBV- cases. CTL, probably attracted to the tumor site by the presence of EBV, expressed granzyme B suggesting they were activated (Figure 2B). In contrast, NK cells, critical cytotoxic effector cells in the early response to viral infection and tumor cells, were virtually absent in all biopsies, based on staining for NCAM1/CD56. However, this does not exclude a role for NK cells in PTLD. In a study involving pediatric transplant recipients, CD56high NK cells were abundant only in asymptomatic transplant recipients whereas in PTLD patients, the functionally impaired CD56dim/negative NK population was increased[70].
Tumor immune evasion is a major challenge for effective cancer treatment[71] and several reports have shown that such mechanisms also play a role in PTLD. Tumoral expression of PDL1, involved in T-cell suppression[72], as well as galectin-1, involved in apoptosis-induction of CTL among others[73], has been reported[51]. Also immunoregulatory M2 macrophages (marked by CD163 expression) may be part of a negative feedback loop to prevent excessive CTL-induced tissue damage[74]. M2 macrophages, which were significantly more abundant in EBV+ vs EBV- PT-DLBCL (manuscript submitted), are thought to contribute to tissue remodeling and tumor progression in contrast to classical pro-inflammatory M1 macrophages[75] (Figure 2B). These data are consistent with studies of EBV+ DLBCL-E and EBV+ Hodgkin lymphoma. Also in these malignancies, the presence of EBV has been associated with upregulation of CD163 expression[41,74].
It is not clear whether these cells are recruited to the tumor site or develop in situ. Studies have shown that the M2 phenotype can be induced by particular cytokines, among which IL-4 and IL-10[76]. We speculate that also EBV-encoded IL-10 contributes to M2 macrophage polarization in EBV+ PT-DLBCL[75]. Interestingly, M2 macrophages are themselves producers of IL-10 and may be the source of the high levels of IL-10 detected in PTLD patients[77].
In a prospective trial of Hodgkin lymphoma, increased tumor-associated macrophage infiltration was associated with inferior outcome[78]. An interesting question to be resolved is whether also in PTLD macrophages influence prognosis.
BL
Cell of origin: BL is a highly aggressive lymphoma characterized by a high mitotic rate and numerous tingible body macrophages (loaded with debris from apoptotic cells). Three clinical variants of BL are recognized: Endemic BL (with a high prevalence in equatorial Africa), sporadic BL (prevalent in Western countries) and immunodeficiency-associated BL [primarily affecting human immunodeficiency virus (HIV) infected patients, but also reported in transplant recipients]. The association with EBV is different for the three subtypes and strongest in the endemic variant (nearly 100% EBV+), followed by the immunodeficiency-associated variant (30%-80% EBV+) and sporadic BL (15%-20% EBV+). Notably, EBV+ Burkitt lymphoma is the EBV- transformed tumor with the most limited expression of viral proteins (typically only EBNA1 is expressed)[79].
BL is classically thought to arise from a GCB cell however analysis of the SHM patterns in a series of endemic, sporadic and AIDS-related BL suggested that BL may arise from different stages of B-cell differentiation, associated with the EBV status. EBV+ BL were highly mutated and may derive from a late antigen-selected GC B-cell or memory B-cell for EBV+ BL. EBV- BL on the other hand harbored only a limited number of mutations and may arise from an early centroblast[80].
Genetics: The hallmark of BL is the presence of translocations involving MYC [with IgH: t(8;14)(q24;q32)] which are also found in PTLD with Burkitt morphology[37]. It is highly debated whether MYC-translocation-negative BL is a form of true molecular BL[81]. In a recent study, an 11q aberration was detected in MYC-negative high-grade B-cell lymphomas resembling BL (both at the morphological as well as the molecular level, but without MYC rearrangement)[82]. In our series of IC- and PT-BL this peculiar 11q gain/loss was particularly frequent in PT cases lacking MYC translocation, suggesting a different pathogenesis of BL in different immune settings. However, a recent study demonstrated that 11q gain/loss and MYC translocation are not mutually exclusive[83]. It is possible that both aberrations have complementary effects: Integrated analysis of genomic and transcriptomic data of our series of MYC translocation-positive and –negative cases suggested that the 11q-gain/loss is a molecular variant of MYC rearrangement, affecting similar pathways.
Gene expression profile and microenvironment: In contrast to PT-DLBCL, the gene expression profile of EBV+ and EBV- BL is not significantly different, indicating that MYC signaling rather than the EBV status has the major impact on the expression profile[84]. BL lesions are composed of very little stromal infiltrate indicating that BL tumor cells are poorly immunogenic. Remarkably, even when BL cells express highly immunogenic EBV antigens EBNA3A, -3B, and -3C[85] or foreign antigens are introduced by a recombinant virus[86] they are not recognized by antigen-specific CTL clones. An in vitro study pointed to a crucial role of MYC. It was demonstrated that this oncogene negatively regulates NF-κB and interferon signaling by suppression of STAT1 resulting in decreased immunogenicity[87].
PBL
Cell of origin: PBL is an aggressive terminally differentiated variant of DLBCL that has many morphological and immunophenotypic characteristics in common with a plasmablast (a B-cell in the final stages of plasma cell differentiation). PBL typically arises in the oral cavity of HIV+ patients[88] but has also been reported in immunocompetent individuals[89] and transplant recipients[8].
In a series of AIDS-related PBL (10/12 were EBV+), evidence of somatic hypermutation was found in only 4/10 analyzed cases suggesting histogenetic heterogeneity of PBL[90].
Genetics: Currently, very little is known about the molecular-genetic basis that drives PBL. One study showed that up to 47% of EBV+ AIDS-related PBLs are marked by MYC translocations[91]. Array-comparative genomic hybridization involving 16 PBL demonstrated that, despite the high degree of immunophenotypical similarity between PBL and plasma cell myeloma (PCM)[92], the genomic aberration pattern of PBL is more similar to DLBCL than to PCM[93].
Gene expression profile and microenvironment: A gene expression profiling study reported that PBL was more similar to extraosseous plasmacytoma than to DLBCL[94] reflecting the plasma cell immunophenotypical features of these malignancies. No significant differences were found between EBV+ and EBV- PBL, however this may be related to the small sample size.
Reanalysis of our gene expression data (3 EBV+ PT-PBL vs 20 EBV+ PT-DLBCL, fold change 2, FDR < 0.05[95]) confirmed enhanced MYC signaling and demonstrated unfolded protein response endoplasmic reticulum stress signaling in PBL (unreported data). These findings provide an explanation for the success of bortezomib treatment in PBL case reports[96,97] and suggest that BET bromodomain inhibitors may represent a potential new therapeutic strategy, as has been successfully demonstrated in experimental models of multiple myeloma[98].
As for EBV+ DLBCL, EBV+ PBL may be associated with a tolerant microenvironment. In a recent clinicopathological analysis of 82 PBL arising in HIV+ and HIV- patients particularly EBV+ tumors highly expressed PDL1-PD1 in both malignant cells and microenvironment[99].
CONCLUSION
The findings presented in this review underscore the heterogeneous nature of PTLD and could serve as a basis to revise the current PTLD classification. We propose that within the group of monomorphic PTLD, the different histological lymphoma entities (DLBCL, BL, PBL) should be distinghed. We suggest that also the EBV status should be included to further stratify PTLD patients in future studies and clinical trials.
ACKNOWLEDGMENTS
The authors would like to acknowledge the other members of the “Leuven PTLD consortium”, in particular Bittoun E, Dierickx D, Finalet Ferreiro J, Gheysens O, Marcelis L, Verhoef G and Wlodarska I.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest.
Manuscript source: Invited manuscript
Specialty type: Transplantation
Country of origin: Belgium
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): D, D
Grade E (Poor): 0
Peer-review started: April 24, 2016
First decision: June 6, 2016
Article in press: August 18, 2016
P- Reviewer: Govil S, Kin T, Ramsay MA, Salvadori M, Taheri S S- Editor: Gong XM L- Editor: A E- Editor: Wu HL
References
- 1.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet. 1964;1:702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 3.Dharnidharka VR, Webster AC, Martinez OM, Preiksaitis JK, Leblond V, Choquet S. Post-transplant lymphoproliferative disorders. Nat Rev Dis Primers. 2016;2:15088. doi: 10.1038/nrdp.2015.88. [DOI] [PubMed] [Google Scholar]
- 4.Murray JE, Wilson RE, Tilney NL, Merrill JP, Cooper WC, Birtch AG, Carpenter CB, Hager EB, Dammin GJ, Harrison JH. Five years’ experience in renal transplantation with immunosuppressive drugs: survival, function, complications, and the role of lymphocyte depletion by thoracic duct fistula. Ann Surg. 1968;168:416–435. doi: 10.1097/00000658-196809000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swerldow S, Webber SA, Chadburn A, Ferry JA. Post-transplant lymphoproliferative disorders. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 343–349. [Google Scholar]
- 6.Picarsic J, Jaffe R, Mazariegos G, Webber SA, Ellis D, Green MD, Reyes-Múgica M. Post-transplant Burkitt lymphoma is a more aggressive and distinct form of post-transplant lymphoproliferative disorder. Cancer. 2011;117:4540–4550. doi: 10.1002/cncr.26001. [DOI] [PubMed] [Google Scholar]
- 7.Akar Özkan E, Özdemir BH, Akdur A, Deniz EE, Haberal M. Burkitt lymphoma after transplant: an aggressive lymphoproliferative disease. Exp Clin Transplant. 2014;12 Suppl 1:136–138. [PubMed] [Google Scholar]
- 8.Borenstein J, Pezzella F, Gatter KC. Plasmablastic lymphomas may occur as post-transplant lymphoproliferative disorders. Histopathology. 2007;51:774–777. doi: 10.1111/j.1365-2559.2007.02870.x. [DOI] [PubMed] [Google Scholar]
- 9.Apichai S, Rogalska A, Tzvetanov I, Asma Z, Benedetti E, Gaitonde S. Multifocal cutaneous and systemic plasmablastic lymphoma in an infant with combined living donor small bowel and liver transplant. Pediatr Transplant. 2009;13:628–631. doi: 10.1111/j.1399-3046.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- 10.Dierickx D, Tousseyn T, Gheysens O. How I treat posttransplant lymphoproliferative disorders. Blood. 2015;126:2274–2283. doi: 10.1182/blood-2015-05-615872. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation-an update. Curr Opin Immunol. 2016;39:59–67. doi: 10.1016/j.coi.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Thorley-Lawson DA. EBV the prototypical human tumor virus--just how bad is it? J Allergy Clin Immunol. 2005;116:251–261; quiz 262. doi: 10.1016/j.jaci.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 13.Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323–2330. doi: 10.1182/blood-2014-10-567479. [DOI] [PubMed] [Google Scholar]
- 14.Gong JZ, Stenzel TT, Bennett ER, Lagoo AS, Dunphy CH, Moore JO, Rizzieri DA, Tepperberg JH, Papenhausen P, Buckley PJ. Burkitt lymphoma arising in organ transplant recipients: a clinicopathologic study of five cases. Am J Surg Pathol. 2003;27:818–827. doi: 10.1097/00000478-200306000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Cai X, Schäfer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, Raab-Traub N, Cullen BR. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006;2:e23. doi: 10.1371/journal.ppat.0020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwakiri D, Takada K. Role of EBERs in the pathogenesis of EBV infection. Adv Cancer Res. 2010;107:119–136. doi: 10.1016/S0065-230X(10)07004-1. [DOI] [PubMed] [Google Scholar]
- 17.Kempkes B, Robertson ES. Epstein-Barr virus latency: current and future perspectives. Curr Opin Virol. 2015;14:138–144. doi: 10.1016/j.coviro.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elgui de Oliveira D, Müller-Coan BG, Pagano JS. Viral Carcinogenesis Beyond Malignant Transformation: EBV in the Progression of Human Cancers. Trends Microbiol. 2016;24:649–664. doi: 10.1016/j.tim.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv Cancer Res. 2007;97:81–109. doi: 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- 21.Whitehurst CB, Li G, Montgomery SA, Montgomery ND, Su L, Pagano JS. Knockout of Epstein-Barr virus BPLF1 retards B-cell transformation and lymphoma formation in humanized mice. MBio. 2015;6:e01574–e01515. doi: 10.1128/mBio.01574-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones RJ, Seaman WT, Feng WH, Barlow E, Dickerson S, Delecluse HJ, Kenney SC. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int J Cancer. 2007;121:1274–1281. doi: 10.1002/ijc.22839. [DOI] [PubMed] [Google Scholar]
- 23.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. 2011;85:165–177. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. 2005;79:13993–14003. doi: 10.1128/JVI.79.22.13993-14003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Jia Y, Wang Y, Wang X, Sun Z, Luo B. Sequence analysis of EBV immediate-early gene BZLF1 and BRLF1 in lymphomas. J Med Virol. 2014;86:1788–1795. doi: 10.1002/jmv.23911. [DOI] [PubMed] [Google Scholar]
- 26.Vakiani E, Basso K, Klein U, Mansukhani MM, Narayan G, Smith PM, Murty VV, Dalla-Favera R, Pasqualucci L, Bhagat G. Genetic and phenotypic analysis of B-cell post-transplant lymphoproliferative disorders provides insights into disease biology. Hematol Oncol. 2008;26:199–211. doi: 10.1002/hon.859. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez-Farre B, Rovira J, Martinez D, Valera A, Garcia-Herrera A, Marcos MA, Sole C, Roue G, Colomer D, Gonzalvo E, et al. In vivo intratumoral Epstein-Barr virus replication is associated with XBP1 activation and early-onset post-transplant lymphoproliferative disorders with prognostic implications. Mod Pathol. 2014;27:1599–1611. doi: 10.1038/modpathol.2014.68. [DOI] [PubMed] [Google Scholar]
- 28.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 29.Capello D, Rossi D, Gaidano G. Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol. 2005;23:61–67. doi: 10.1002/hon.751. [DOI] [PubMed] [Google Scholar]
- 30.Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–282. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- 31.Kinch A, Baecklund E, Backlin C, Ekman T, Molin D, Tufveson G, Fernberg P, Sundström C, Pauksens K, Enblad G. A population-based study of 135 lymphomas after solid organ transplantation: The role of Epstein-Barr virus, hepatitis C and diffuse large B-cell lymphoma subtype in clinical presentation and survival. Acta Oncol. 2014;53:669–679. doi: 10.3109/0284186X.2013.844853. [DOI] [PubMed] [Google Scholar]
- 32.Craig FE, Johnson LR, Harvey SA, Nalesnik MA, Luo JH, Bhattacharya SD, Swerdlow SH. Gene expression profiling of Epstein-Barr virus-positive and -negative monomorphic B-cell posttransplant lymphoproliferative disorders. Diagn Mol Pathol. 2007;16:158–168. doi: 10.1097/PDM.0b013e31804f54a9. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, Karube K, Yamamoto K, Takizawa J, Tsuzuki S, Yatabe Y, Kanda T, Katayama M, Ozawa Y, Ishitsuka K, et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014;105:537–544. doi: 10.1111/cas.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capello D, Cerri M, Muti G, Berra E, Oreste P, Deambrogi C, Rossi D, Dotti G, Conconi A, Viganò M, et al. Molecular histogenesis of posttransplantation lymphoproliferative disorders. Blood. 2003;102:3775–3785. doi: 10.1182/blood-2003-05-1683. [DOI] [PubMed] [Google Scholar]
- 35.Bräuninger A, Spieker T, Mottok A, Baur AS, Küppers R, Hansmann ML. Epstein-Barr virus (EBV)-positive lymphoproliferations in post-transplant patients show immunoglobulin V gene mutation patterns suggesting interference of EBV with normal B cell differentiation processes. Eur J Immunol. 2003;33:1593–1602. doi: 10.1002/eji.200323765. [DOI] [PubMed] [Google Scholar]
- 36.Timms JM, Bell A, Flavell JR, Murray PG, Rickinson AB, Traverse-Glehen A, Berger F, Delecluse HJ. Target cells of Epstein-Barr-virus (EBV)-positive post-transplant lymphoproliferative disease: similarities to EBV-positive Hodgkin’s lymphoma. Lancet. 2003;361:217–223. doi: 10.1016/S0140-6736(03)12271-4. [DOI] [PubMed] [Google Scholar]
- 37.Djokic M, Le Beau MM, Swinnen LJ, Smith SM, Rubin CM, Anastasi J, Carlson KM. Post-transplant lymphoproliferative disorder subtypes correlate with different recurring chromosomal abnormalities. Genes Chromosomes Cancer. 2006;45:313–318. doi: 10.1002/gcc.20287. [DOI] [PubMed] [Google Scholar]
- 38.Poirel HA, Bernheim A, Schneider A, Meddeb M, Choquet S, Leblond V, Charlotte F, Davi F, Canioni D, Macintyre E, et al. Characteristic pattern of chromosomal imbalances in posttransplantation lymphoproliferative disorders: correlation with histopathological subcategories and EBV status. Transplantation. 2005;80:176–184. doi: 10.1097/01.tp.0000163288.98419.0d. [DOI] [PubMed] [Google Scholar]
- 39.Rinaldi A, Capello D, Scandurra M, Greiner TC, Chan WC, Bhagat G, Rossi D, Morra E, Paulli M, Rambaldi A, et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. Br J Haematol. 2010;149:569–577. doi: 10.1111/j.1365-2141.2010.08125.x. [DOI] [PubMed] [Google Scholar]
- 40.Ferreiro JF, Morscio J, Dierickx D, Vandenberghe P, Gheysens O, Verhoef G, Zamani M, Tousseyn T, Wlodarska I. EBV-Positive and EBV-Negative Posttransplant Diffuse Large B Cell Lymphomas Have Distinct Genomic and Transcriptomic Features. Am J Transplant. 2016;16:414–425. doi: 10.1111/ajt.13558. [DOI] [PubMed] [Google Scholar]
- 41.Yoon H, Park S, Ju H, Ha SY, Sohn I, Jo J, Do IG, Min S, Kim SJ, Kim WS, et al. Integrated copy number and gene expression profiling analysis of Epstein-Barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2015;54:383–396. doi: 10.1002/gcc.22249. [DOI] [PubMed] [Google Scholar]
- 42.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerri M, Capello D, Muti G, Rambaldi A, Paulli M, Gloghini A, Berra E, Deambrogi C, Rossi D, Franceschetti S, et al. Aberrant somatic hypermutation in post-transplant lymphoproliferative disorders. Br J Haematol. 2004;127:362–364. doi: 10.1111/j.1365-2141.2004.05203.x. [DOI] [PubMed] [Google Scholar]
- 44.Duval A, Raphael M, Brennetot C, Poirel H, Buhard O, Aubry A, Martin A, Krimi A, Leblond V, Gabarre J, et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci USA. 2004;101:5002–5007. doi: 10.1073/pnas.0400945101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saeterdal I, Bjørheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, Nesland JM, Eriksen JA, Møller M, Lindblom A, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA. 2001;98:13255–13260. doi: 10.1073/pnas.231326898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Smedt L, Lemahieu J, Palmans S, Govaere O, Tousseyn T, Van Cutsem E, Prenen H, Tejpar S, Spaepen M, Matthijs G, et al. Microsatellite instable vs stable colon carcinomas: analysis of tumour heterogeneity, inflammation and angiogenesis. Br J Cancer. 2015;113:500–509. doi: 10.1038/bjc.2015.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morscio J, Dierickx D, Ferreiro JF, Herreman A, Van Loo P, Bittoun E, Verhoef G, Matthys P, Cools J, Wlodarska I, et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am J Transplant. 2013;13:1305–1316. doi: 10.1111/ajt.12196. [DOI] [PubMed] [Google Scholar]
- 48.Tsai SC, Lin SJ, Lin CJ, Chou YC, Lin JH, Yeh TH, Chen MR, Huang LM, Lu MY, Huang YC, et al. Autocrine CCL3 and CCL4 induced by the oncoprotein LMP1 promote Epstein-Barr virus-triggered B cell proliferation. J Virol. 2013;87:9041–9052. doi: 10.1128/JVI.00541-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, Neuberg D, Shipp MA. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18:1611–1618. doi: 10.1158/1078-0432.CCR-11-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ouyang J, Juszczynski P, Rodig SJ, Green MR, O’Donnell E, Currie T, Armant M, Takeyama K, Monti S, Rabinovich GA, et al. Viral induction and targeted inhibition of galectin-1 in EBV+ posttransplant lymphoproliferative disorders. Blood. 2011;117:4315–4322. doi: 10.1182/blood-2010-11-320481. [DOI] [PubMed] [Google Scholar]
- 51.Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR, Strong AL, Lehman TA, Seddon MB, Lin Z, et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. 2013;9:e1003341. doi: 10.1371/journal.ppat.1003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luskin MR, Heil DS, Tan KS, Choi S, Stadtmauer EA, Schuster SJ, Porter DL, Vonderheide RH, Bagg A, Heitjan DF, et al. The Impact of EBV Status on Characteristics and Outcomes of Posttransplantation Lymphoproliferative Disorder. Am J Transplant. 2015;15:2665–2673. doi: 10.1111/ajt.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimizu N, Tanabe-Tochikura A, Kuroiwa Y, Takada K. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J Virol. 1994;68:6069–6073. doi: 10.1128/jvi.68.9.6069-6073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ambrosio MR, Rocca BJ, Ginori A, Mourmouras V, Amato T, Vindigni C, Lazzi S, Leoncini L. A look into the evolution of Epstein-Barr virus-induced lymphoproliferative disorders: a case study. Am J Clin Pathol. 2015;144:817–822. doi: 10.1309/AJCP2G0VKTKPNPRR. [DOI] [PubMed] [Google Scholar]
- 56.Gallagher A, Perry J, Freeland J, Alexander FE, Carman WF, Shield L, Cartwright R, Jarrett RF. Hodgkin lymphoma and Epstein-Barr virus (EBV): no evidence to support hit-and-run mechanism in cases classified as non-EBV-associated. Int J Cancer. 2003;104:624–630. doi: 10.1002/ijc.10979. [DOI] [PubMed] [Google Scholar]
- 57.Chen W, Huang Q, Zuppan CW, Rowsell EH, Cao JD, Weiss LM, Wang J. Complete absence of KSHV/HHV-8 in posttransplant lymphoproliferative disorders: an immunohistochemical and molecular study of 52 cases. Am J Clin Pathol. 2009;131:632–639. doi: 10.1309/AJCP2T4IIIZKBHMI. [DOI] [PubMed] [Google Scholar]
- 58.Theate I, Michaux L, Squifflet JP, Martin A, Raphael M. Human herpesvirus 8 and Epstein-Barr virus-related monotypic large B-cell lymphoproliferative disorder coexisting with mixed variant of Castleman’s disease in a lymph node of a renal transplant recipient. Clin Transplant. 2003;17:451–454. doi: 10.1034/j.1399-0012.2003.00098.x. [DOI] [PubMed] [Google Scholar]
- 59.Zallio F, Primon V, Tamiazzo S, Pini M, Baraldi A, Corsetti MT, Gotta F, Bertassello C, Salvi F, Rocchetti A, et al. Epstein-Barr virus reactivation in allogeneic stem cell transplantation is highly related to cytomegalovirus reactivation. Clin Transplant. 2013;27:E491–E497. doi: 10.1111/ctr.12172. [DOI] [PubMed] [Google Scholar]
- 60.Arvey A, Ojesina AI, Pedamallu CS, Ballon G, Jung J, Duke F, Leoncini L, De Falco G, Bressman E, Tam W, et al. The tumor virus landscape of AIDS-related lymphomas. Blood. 2015;125:e14–e22. doi: 10.1182/blood-2014-11-599951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leblond V, Davi F, Charlotte F, Dorent R, Bitker MO, Sutton L, Gandjbakhch I, Binet JL, Raphael M. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol. 1998;16:2052–2059. doi: 10.1200/JCO.1998.16.6.2052. [DOI] [PubMed] [Google Scholar]
- 62.Nelson BP, Nalesnik MA, Bahler DW, Locker J, Fung JJ, Swerdlow SH. Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity? Am J Surg Pathol. 2000;24:375–385. doi: 10.1097/00000478-200003000-00006. [DOI] [PubMed] [Google Scholar]
- 63.van Keimpema M, Grüneberg LJ, Mokry M, van Boxtel R, Koster J, Coffer PJ, Pals ST, Spaargaren M. FOXP1 directly represses transcription of proapoptotic genes and cooperates with NF-κB to promote survival of human B cells. Blood. 2014;124:3431–3440. doi: 10.1182/blood-2014-01-553412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price AM, Tourigny JP, Forte E, Salinas RE, Dave SS, Luftig MA. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κB activation. J Virol. 2012;86:11096–11106. doi: 10.1128/JVI.01069-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greaves P, Clear A, Coutinho R, Wilson A, Matthews J, Owen A, Shanyinde M, Lister TA, Calaminici M, Gribben JG. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of classical Hodgkin lymphoma is predictive of outcome. J Clin Oncol. 2013;31:256–262. doi: 10.1200/JCO.2011.39.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koch K, Hoster E, Unterhalt M, Ott G, Rosenwald A, Hansmann ML, Engelhard M, Hiddemann W, Klapper W. The composition of the microenvironment in follicular lymphoma is associated with the stage of the disease. Hum Pathol. 2012;43:2274–2281. doi: 10.1016/j.humpath.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 67.Richendollar BG, Tsao RE, Elson P, Jin T, Steinle R, Pohlman B, Hsi ED. Predictors of outcome in post-transplant lymphoproliferative disorder: an evaluation of tumor infiltrating lymphocytes in the context of clinical factors. Leuk Lymphoma. 2009;50:2005–2012. doi: 10.3109/10428190903315713. [DOI] [PubMed] [Google Scholar]
- 68.Verdonk RC, Haagsma EB, Jonker MR, Bok LI, Zandvoort JH, Kleibeuker JH, Faber KN, Dijkstra G. Effects of different immunosuppressive regimens on regulatory T-cells in noninflamed colon of liver transplant recipients. Inflamm Bowel Dis. 2007;13:703–709. doi: 10.1002/ibd.20087. [DOI] [PubMed] [Google Scholar]
- 69.Liapis K, Clear A, Owen A, Coutinho R, Greaves P, Lee AM, Montoto S, Calaminici M, Gribben JG. The microenvironment of AIDS-related diffuse large B-cell lymphoma provides insight into the pathophysiology and indicates possible therapeutic strategies. Blood. 2013;122:424–433. doi: 10.1182/blood-2013-03-488171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, Metes D. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol. 2012;42:541–550. doi: 10.1002/eji.201141832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HM, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35 Suppl:S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 72.Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, Yu H, Fletcher CD, Freeman GJ, Shipp MA, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–3473. doi: 10.1158/1078-0432.CCR-13-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 74.Barros MH, Hassan R, Niedobitek G. Tumor-associated macrophages in pediatric classical Hodgkin lymphoma: association with Epstein-Barr virus, lymphocyte subsets, and prognostic impact. Clin Cancer Res. 2012;18:3762–3771. doi: 10.1158/1078-0432.CCR-12-0129. [DOI] [PubMed] [Google Scholar]
- 75.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 76.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hinrichs C, Wendland S, Zimmermann H, Eurich D, Neuhaus R, Schlattmann P, Babel N, Riess H, Gärtner B, Anagnostopoulos I, et al. IL-6 and IL-10 in post-transplant lymphoproliferative disorders development and maintenance: a longitudinal study of cytokine plasma levels and T-cell subsets in 38 patients undergoing treatment. Transpl Int. 2011;24:892–903. doi: 10.1111/j.1432-2277.2011.01282.x. [DOI] [PubMed] [Google Scholar]
- 78.Tan KL, Scott DW, Hong F, Kahl BS, Fisher RI, Bartlett NL, Advani RH, Buckstein R, Rimsza LM, Connors JM, et al. Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: a correlative study from the E2496 Intergroup trial. Blood. 2012;120:3280–3287. doi: 10.1182/blood-2012-04-421057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat Rev Microbiol. 2008;6:913–924. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- 80.Bellan C, Lazzi S, Hummel M, Palummo N, de Santi M, Amato T, Nyagol J, Sabattini E, Lazure T, Pileri SA, et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood. 2005;106:1031–1036. doi: 10.1182/blood-2005-01-0168. [DOI] [PubMed] [Google Scholar]
- 81.Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C, van Rijk A, Nyagol J, Byakika B, Lazzi S, et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008;216:440–450. doi: 10.1002/path.2410. [DOI] [PubMed] [Google Scholar]
- 82.Salaverria I, Martin-Guerrero I, Wagener R, Kreuz M, Kohler CW, Richter J, Pienkowska-Grela B, Adam P, Burkhardt B, Claviez A, et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood. 2014;123:1187–1198. doi: 10.1182/blood-2013-06-507996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Havelange V, Ameye G, Théate I, Callet-Bauchu E, Lippert E, Luquet I, Raphaël M, Vikkula M, Poirel HA. The peculiar 11q-gain/loss aberration reported in a subset of MYC-negative high-grade B-cell lymphomas can also occur in a MYC-rearranged lymphoma. Cancer Genet. 2016;209:117–118. doi: 10.1016/j.cancergen.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 84.Piccaluga PP, De Falco G, Kustagi M, Gazzola A, Agostinelli C, Tripodo C, Leucci E, Onnis A, Astolfi A, Sapienza MR, et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood. 2011;117:3596–3608. doi: 10.1182/blood-2010-08-301556. [DOI] [PubMed] [Google Scholar]
- 85.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–1104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 86.Frisan T, Zhang QJ, Levitskaya J, Coram M, Kurilla MG, Masucci MG. Defective presentation of MHC class I-restricted cytotoxic T-cell epitopes in Burkitt’s lymphoma cells. Int J Cancer. 1996;68:251–258. doi: 10.1002/(SICI)1097-0215(19961009)68:2<251::AID-IJC19>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 87.Schlee M, Hölzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, Eick D, Marinkovic D, Wirth T, Rosenwald A, et al. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. Int J Cancer. 2007;120:1387–1395. doi: 10.1002/ijc.22372. [DOI] [PubMed] [Google Scholar]
- 88.Delecluse HJ, Anagnostopoulos I, Dallenbach F, Hummel M, Marafioti T, Schneider U, Huhn D, Schmidt-Westhausen A, Reichart PA, Gross U, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413–1420. [PubMed] [Google Scholar]
- 89.Huang X, Zhang Y, Gao Z. Plasmablastic lymphoma of the stomach with C-MYC rearrangement in an immunocompetent young adult: a case report. Medicine (Baltimore) 2015;94:e470. doi: 10.1097/MD.0000000000000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaidano G, Cerri M, Capello D, Berra E, Deambrogi C, Rossi D, Larocca LM, Campo E, Gloghini A, Tirelli U, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622–628. doi: 10.1046/j.1365-2141.2002.03872.x. [DOI] [PubMed] [Google Scholar]
- 91.Boy SC, van Heerden MB, Babb C, van Heerden WF, Willem P. Dominant genetic aberrations and coexistent EBV infection in HIV-related oral plasmablastic lymphomas. Oral Oncol. 2011;47:883–887. doi: 10.1016/j.oraloncology.2011.06.506. [DOI] [PubMed] [Google Scholar]
- 92.Vega F, Chang CC, Medeiros LJ, Udden MM, Cho-Vega JH, Lau CC, Finch CJ, Vilchez RA, McGregor D, Jorgensen JL. Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles. Mod Pathol. 2005;18:806–815. doi: 10.1038/modpathol.3800355. [DOI] [PubMed] [Google Scholar]
- 93.Chang CC, Zhou X, Taylor JJ, Huang WT, Ren X, Monzon F, Feng Y, Rao PH, Lu XY, Fabio F, et al. Genomic profiling of plasmablastic lymphoma using array comparative genomic hybridization (aCGH): revealing significant overlapping genomic lesions with diffuse large B-cell lymphoma. J Hematol Oncol. 2009;2:47. doi: 10.1186/1756-8722-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chapman J, Gentles AJ, Sujoy V, Vega F, Dumur CI, Blevins TL, Bernal-Mizrachi L, Mosunjac M, Pimentel A, Zhu D, et al. Gene expression analysis of plasmablastic lymphoma identifies downregulation of B-cell receptor signaling and additional unique transcriptional programs. Leukemia. 2015;29:2270–2273. doi: 10.1038/leu.2015.109. [DOI] [PubMed] [Google Scholar]
- 95.Morscio J, Dierickx D, Nijs J, Verhoef G, Bittoun E, Vanoeteren X, Wlodarska I, Sagaert X, Tousseyn T. Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent, and posttransplant patients: single-center series of 25 cases and meta-analysis of 277 reported cases. Am J Surg Pathol. 2014;38:875–886. doi: 10.1097/PAS.0000000000000234. [DOI] [PubMed] [Google Scholar]
- 96.Hirosawa M, Morimoto H, Shibuya R, Shimajiri S, Tsukada J. A striking response of plasmablastic lymphoma of the oral cavity to bortezomib: a case report. Biomark Res. 2015;3:28. doi: 10.1186/s40364-015-0053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fernandez-Alvarez R, Gonzalez-Rodriguez AP, Rubio-Castro A, Gonzalez ME, Payer AR, Alonso-Garcia A, Rodriguez-Villar D, Dominguez-Iglesias F, Sancho JM. Bortezomib plus CHOP for the treatment of HIV-associated plasmablastic lymphoma: clinical experience in three patients. Leuk Lymphoma. 2015 doi: 10.3109/10428194.2015.1050666. Jun 18; Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 98.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laurent C, Fabiani B, Do C, Tchernonog E, Cartron G, Gravelle P, Amara N, Malot S, Palisoc MM, Copie-Bergman C, et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: a clinical and pathological study in 82 patients. Haematologica. 2016;101:976–984. doi: 10.3324/haematol.2016.141978. [DOI] [PMC free article] [PubMed] [Google Scholar]