

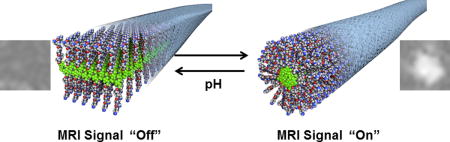
Abstract
Magnetic resonance imaging (MRI) is a noninvasive imaging modality that provides excellent spatial and temporal resolution. The most commonly used MR probes face significant challenges originating from the endogenous 1H background signal of water. In contrast, fluorine MRI (19F MRI) allows quantitative probe imaging with zero background signal. Probes with high fluorine content are required for high sensitivity, suggesting nanoscale supramolecular assemblies containing 19F probes offer a potentially useful strategy for optimum imaging as a result of improved payload. We report here on supramolecular nanostructures formed by fluorinated peptide amphiphiles containing either glutamic acid or lysine residues in their sequence. We identified molecules that form aggregates in water which transition from cylindrical to ribbon-like shape as pH increased from 4.5 to 8.0. Interestingly, we found that ribbon-like nanostructures had reduced magnetic resonance signal, whereas their cylindrical counterparts exhibited strong signals. We attribute this drastic difference to the greater mobility of fluorinated tails in the hydrophobic compartment of cylindrical nanostructures compared to lower mobility in ribbon-like assemblies. This discovery identifies a strategy to design supramolecular, self-assembling contrast agents for 19F MRI that can spatially map physiologically relevant changes in pH using changes in morphology.
Keywords: fluorine, magnetic resonance imaging (MRI), peptide amphiphile, fluorous, pH response, nanofiber, self-assembly
Graphical abstract
Magnetic resonance imaging (MRI) is an invaluable clinical and research tool used to noninvasively produce three-dimensional images of living tissues with excellent spatiotemporal resolution.1 MR probes capable of detecting specific biological events, including changes in pH,2,3 ion concentration,4–6 and the activity of enzymes7 have been developed to expand MRI’s capabilities. Responsive MR agents are often based on modulating magnetic effects on the 1H nuclei of water.8,9 The challenge of detecting MR contrast agents that modulate the 1H signal in biological systems where the background water signal is large10 has led to the development of hyperpolarization techniques11–16 and improvement in contrast agent design.17,18 One of the strategies to improve MRI has been to develop probes based on nuclei other than 1H. 19F is particularly attractive nucleus because of its large gyromagnetic ratio and natural abundance, resulting in an innate MR signal comparable to 1H that is not obscured by endogenous proton signal of biological systems.19–23
Despite improvements in MR sensitivity, 19F probes must present in millimolar quantities of fluorine atoms to be detectable.24 Recently reported 19F probes achieve these concentrations by employing nanoscale chemical strategies such as design of emulsions,20,25 dendrimers,26 and micellar structures.19,27,28 In addition to higher fluorine payloads, micellar constructs can be designed to respond to chemical stimuli, switching between MR signal “on” and “off” states.29 One specific mechanism is silencing the 19F MR signal by shortening the T2 (spin–spin) relaxation time with aggregate formation or interaction with a paramagnetic species.30–32 Aggregation-based T2 quenching works by increasing local molecular correlation times, resulting in rapid spin–spin relaxation and signal reduction. We hypothesized that 19F-containing nanoscale supramolecular assemblies that are more highly structured than spherical micelles could have MR signal response to stimuli based on differences in their internal dynamics.
Peptide amphiphiles (PAs) contain a short peptide sequence covalently grafted to a hydrophobic tail and can be programmed to generate one-dimensional assemblies such as cylinders and ribbons.33–35 PAs have a modular design that has been functionalized with an array of biomolecular epitopes and bioactive substituents for any number of applications in regenerative medicine,36–38 drug delivery,39–42 and as Gd(III)-based MRI contrast agents.43–45 These nanostructures exhibit structural responses to thermal annealing,46 light,47 pH,35,48,49 and enzymatic activity.39 These structural responses to external stimuli introduce functions in PA biomaterials.
An important biomedical target is the development of systems that sense pH as an indicator of hypoxia in ischemic or cancerous tissue.50,51 In this context, it is possible to vary the number of charged amino acids in peptide amphiphiles in order to gain structural sensitivity to changes in pH.35,49,52 It follows that differences in local dynamics between different nanostructure geometries could result in a measurable 19F MRI signal response, but this is not characterized in the literature to our knowledge. The present work aims to explore hydrophobicity and charge to examine 19F MRI response to pH in peptide amphiphile aggregates. Using this design strategy, we have developed a series of peptide amphiphiles as responsive MR contrast agents. The series varies the hydrophobicity of the perfluoroalkyl tail by amidating either perfluorooctanoic acid or perfluoroheptanoic acid to the amino terminus of the peptide sequence and varies charge in the peptide sequence using either E2, K2, or K3 sequences.
RESULTS AND DISCUSSION
The fluorinated peptide amphiphiles (F-PAs) used in this study were designed to probe the effect of varying fluorophilic interactions and the repulsive effects of charge on NMR spectra and consequently on MRI signals. We expected that acidic and basic headgroups would exhibit NMR signals that varied as a function of pH. We designed a series of F-PAs with either glutamic acid or lysine headgroups to explore the responsiveness of the acidic and basic side chains to pH (Figure 1). β-sheet-promoting sequences like V2A2 are known to direct assembly into one-dimensional nanostructures.53,54 Short, C7 and C8 perfluoro chains were selected to balance hydrophobic collapse with solubility in organic solvents needed for synthesis. All of the peptides were synthesized using conventional solid-phase peptide synthesis techniques (see Supporting Information). Each perfluoroalkyl tail was added via the Schotten–Baumann reaction as the acyl chloride dissolved in chloroform with excess N,N-diisopropylethylamine. The final peptide amphiphiles were obtained in approximately 50% overall yield after HPLC purification.
Figure 1.
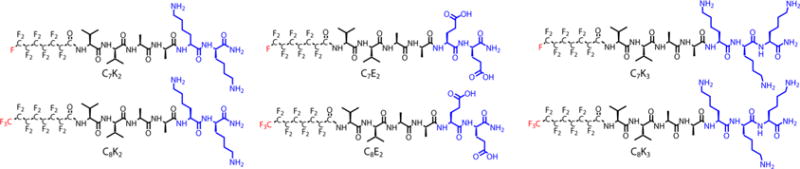
Chemical structures of peptide amphiphiles used in this report. The amino acid hydrogen bonding sequence V2A2 is kept constant while sequence charge (blue) and tail length (red) are varied. Throughout this report, compounds are referenced by their tail length (C7 or C8) and their charged residues (K2, K3, or E2).
In order to optimize amphiphile concentration for NMR detection, we obtained the 19F NMR spectra of each F-PA at concentrations of 1, 1.5, 2, and 3 mM and the signal-to-noise ratio (SNR) was assessed. MRI is innately less sensitive than NMR spectroscopy, because MRI is usually performed at lower field strengths and with much greater magnet bore size. Therefore, we targeted SNR values greater than 100 when performing NMR to ensure MRI detection of a given sample.
For 19F MR contrast agents to provide quantitative imaging information, peak intensity must increase predictably with concentration from a few hundred micromolar to several millimolar.24,32 The NMR spectra (Figure 2) of the agents in Figure 1 indicate clearly that the SNR is sensitive to the length of the F-PA tail segment. For example, C7E2 produced sharp NMR peaks at each concentration tested, indicative of high tail mobility. The terminal CF3 group peak at −80.8 ppm maintained a peak width of 9–10 Hz at over 100 SNR at each concentration tested. For C8E2, the CF3 signal at −80.8 ppm and 103 SNR broadened and shifted to −83.0 ppm and 11 SNR above 1.5 mM (Figure 2), indicating a transition from high to low mobility. This trend of high SNR for C7 and low SNR for C8 peptides held for all sequences studied. Comparing positively charged headgroups, C8K2 produced high (>100) SNR only at 1 mM, whereas C7K2 produced high SNR spectra up to 1.5 mM concentration. C7K3 exhibited sufficient signal for imaging (greater than 100 SNR) over the range of concentrations tested, whereas C8K3 produced low SNR peaks (SNR < 10) through the concentrations tested. Based on these observations, the C7 F-PAs are superior 19F MRI candidates compared to the C8 F-PAs because the C8 compounds have low SNR at the range of concentrations tested, while C7 compounds had high SNR. Moving forward, we considered only C7 compounds.
Figure 2.
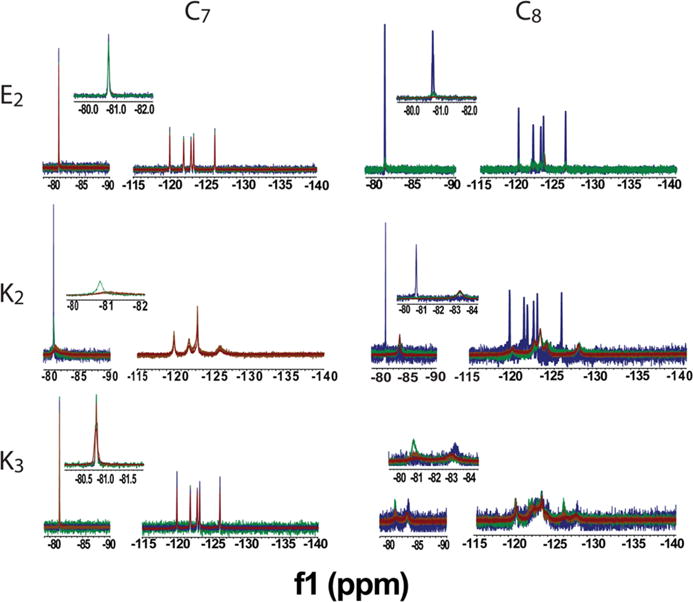
Comparison of NMR spectra for each F-PA as a function of peptide concentration: 1.0 mM (blue), 1.5 mM (green), 2.0 mM (orange) and 3.0 mM (red). C7E2 and C7K3 maintain sharp peaks at the higher concentrations needed to obtain 19F MR images. The other conjugates exhibited sharp spectral features only at concentrations lower than 2 mM or always exhibited broad features. Measurements conducted at pH 6.5.
Negatively charged glutamic acid residues were compared with positively charged lysine residues to determine if charge had an influence on the effect of concentration on signal intensity. The C7E2 spectral signal was unaffected by increasing peptide concentration through the range tested. C7K2 experienced significant peak broadening at concentrations above 1.5 mM, with SNR in the NMR spectra remaining less than 15 at 2 mM. The increased intramolecular repulsion of adding an additional lysine residue in the C7K3 sequence led to a nanostructure with an NMR signal that is linear with respect to concentration. On the basis of these concentration studies, C7E2 and C7K3 showed the most promise as contrast agents, and a concentration of 2 mM was chosen for all future experiments.
To determine pH response profiles of the F-PAs, pH titration 19F NMR experiments were conducted. Solutions were allowed to equilibrate overnight at room temperature before measurement, and were pH-adjusted between measurements by addition of dilute NaOH and HCl solutions (allowed to equilibrate at least 15 min between measurements; for each pH value a 1D 19F NMR spectra was acquired). For both C7E2 and C7K3, peak intensity could be modulated reversibly by pH as measured against the trifluoroethanol (TFE) internal standard (Figure 3A and B). Throughout these titrations, pH values corresponding to higher peptide charged states were expected to cause greater intramolecular repulsion, which in turn would increase intramolecular distance, leading to longer T2 relaxation times and greater integrated signal.
Figure 3.
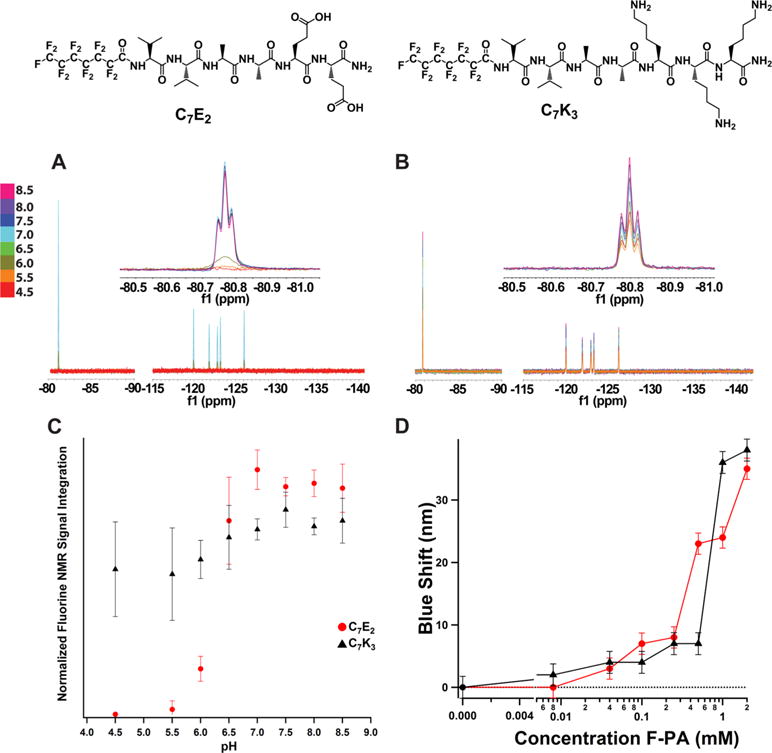
19F NMR pH response of (A) C7E2 and (B) C7K3. Both C7E2 and C7K3 line shapes were found to be robust to pH changes during titration. (C) C7K3 and C7E2 both showed an increase in NMR signal in response to pH starting around 5.5. C7E2 signal reached maximum at pH 6.5, whereas C7K3 signal increased slowly through the pH values tested. (D) Nile red fluorescence assay of C7E2 and C7K3. Concentration of F-PA is plotted versus blue shift of the Nile red florescence peak (measured at 658 nm). Dotted line is fluorescence maximum of Nile red in water. Both compounds exhibit detectable blue shifts (and therefore critical aggregation concentrations) at or below 0.1 mM. This implies that, at the 2 mM concentration and pH 7.5, less than 5% of compound is in the monomeric state.
Because both C7K3 and C7E2 exhibited MR signal that increased as a function of pH, they represent the best candidate compounds as pH-responsive 19F MRI agents. The transition from NMR-inactive to NMR-active occurred across a narrower pH range for C7E2, rising from baseline to 100% entirely in the pH window 5.5–7.0 (Figure 3C). The same transition was more gradual and much smaller for C7K3, approximating a linear relationship between pH 5.5 and pH 8.5 and rising from 80% activation to 100% over that range. pH response reinforces C7E2 as the best candidate F-PA contrast agents since this compound exhibits signal enhancement in the biologically relevant pH window. Interestingly, both C7E2 and C7K3 exhibit an increase in signal with pH despite of their opposite charge. This unexpected correlation between pH and 19F signal led us to hypothesize that local tail environment, perhaps as a function of nanostructure morphology, was contributing to 19F NMR signal intensity in unexpected ways. pH titration analysis by 19F NMR for the other investigated compounds at 2.0 mM are shown in Figures S1–S4. Interestingly, C7K2, C8E2, and C8K2 were found to exhibit sharp, low intensity peaks at intermediate pH values (Figure S1, S3, and S4). Although this is not ideal as 19F NMR agents, because signal intensity is too low to be detected in most imaging experiments, it is interesting that intermediate charge states might produce high tail mobility nanostructures in solution. C7E2 was shown to exhibit similar signal variation when dissolved in Tris-buffered saline rather than saline (100 mM NaCl and 30 mM Tris) and pH adjusted (Figure S5A–B). The same phenomenon was not observed when that buffered solution was diluted 1:1 in fetal bovine serum (FBS). We believe that this loss of function is likely due to protein-assembly interactions that disrupt the nanostructures through nonspecific interactions, because the resulting NMR signal was broadened (Figure S5C). Future applications of these compounds in vivo will require optimization of application and processing techniques as well as assessment of the agent in solid tissue, which are beyond the scope of the current work.
In order to prove that the observed signal increase with pH for C7K3 and C7E2 did not arise from unassembled (i.e., monomeric) F-PA, a Nile red assay was carried out to obtain the critical micelle concentration (CMC) for C7E2 and C7K3 (Figure 3D). We chose pH 7.5 for the assay because it is relevant to biological media and both compounds show high signal intensity at this value. Both C7E2 and C7K3 were found to induce a blue shift in the Nile red florescence at a concentration below 0.1 mM, corresponding to a CMC less than or equal to 0.1 mM for both compounds. These data support an NMR signal contribution of less than 5% for monomers in solution, indicating the NMR signal increase observed as a result of greater pH is largely the result of the nanostructures formed by both compounds.
Cryogenic electron microscopy (Cryo-TEM) experiments were carried out to investigate the effect of the amphiphile structure on nanoscale aggregates. Sample preparation for Cryo-TEMs used identical buffer conditions described for NMR measurements but omitted the use of deuterated solvent and internal standard. Results from these experiments are shown in Figures 4 and S5. C7E2 formed nanoribbon structures at pH 4 and 7 with widths of 25 ± 6 nm (13 ribbons measured) and 19 ± 5 nm (20 ribbons measured), respectively, and sparse cylindrical nanofibers at pH 9 with diameters of 11 ± 2 nm (16 fibers measured). C7K3 formed only very short, sparse ribbons at both pH 4 and 7, and long cylindrical fibers and some ribbons at pH 9 with a diameter of 14 ± 3 nm (16 ribbons measured). These observations across the C7 series were consistent with relatively weak hydrophobic interactions, resulting in structures that respond strongly to the effect of charge at the peptide terminus and, therefore, pH (Figure 4). Interestingly, the strongest signals at various values of pH for C7K3 and C7E2 were observed for cylindrical nanofibers (a larger view of C7K3 nanostructures can be found in Figure S7). This suggests a relationship between nanostructure morphology and 19F MRI signal, with cylindrical nanostructures producing higher integration NMR signal peaks and therefore higher MRI signals.
Figure 4.

Representative Cryo-TEM images of perfluorinated peptide amphiphiles at pH 4, 7, and 9. Scale bar is 100 nm. C7K2 exhibits sparse, disordered structures at pH 4 (panel A), and ribbons at pH 7 (B) and 9 (C). C7E2 produces ribbon nanostructures at pH 4 (D) and 7 (E), transitioning to cylindrical fibers at pH 9 (F). Finally, C7K3 forms sparse ribbon structures at 4 (G) and 7 (H), transitioning to cylindrical fibers at pH 9 (I).
In contrast to the C7 series, the C8 series did not exhibit pH sensitivity at the concentration ranges tested, and signal-to-noise for the NMR signal of these compounds proved prohibitively low for MR imaging. All C8 compounds, differing by only one CF2 unit from the C7 series, exhibited flat nanostructures that did not change with pH and were more likely to show nanostructure aggregation (Figure S6). C8E2 produced aggregated ribbons at pH 4, consistent with the viscous and turbid solutions. At pH 7, ribbon nanostructures were observed, and sparse nanoribbons at pH 9. For C8K2 at pH 4, a mixture of ribbons and fibers was observed and at pH 7 and 9 these transitioned to only ribbon nanostructures. Consistent with results obtained with the C7 series, C8K2 showed broad NMR signals at low pH which lost intensity at high pH. To summarize, Cryo-TEM images indicate that the C7 series is significantly more sensitive to pH and also that cylindrical nanostructures correlate with enhanced NMR signals. This result indicates that a ribbon to fiber transition in these supramolecular aggregates may be responsible for the pH response observed.
Peptide secondary structure may play a role in restricting tail mobility and reducing the signal observed. To assess this possibility, a series of circular dichroism (CD) experiments were conducted to determine if a correlation exists between NMR signals and peptide secondary structure (Figures 5 and S8–S11). Each of the spectra shows the decay of a random coil signal appearing at 200 nm and the growth of a β-sheet signal as a negative peak at 220 nm as the nanostructure becomes less charged as a result of pH. For C7E2, β-sheet character increases with decreasing pH. For C7K3, β-sheet character increases with increasing pH. Despite this inversion, pH increase exhibits a positive correlation with 19F NMR signal in both instances (Figure 4). The pH trends in peptide secondary structure for these samples therefore imply that peptide secondary structure does not directly result in higher or lower 19F NMR or 19F MRI signal.
Figure 5.
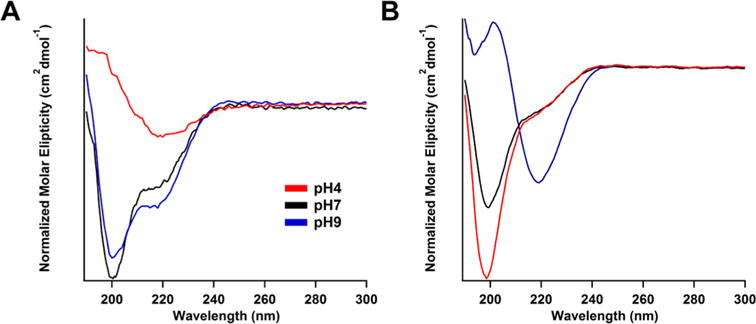
Circular dichroism spectra of (A) C7E2 and (B) C7K3 at different values of pH. ranges. β-sheet component increase for C7K3 coincides with NMR signal increase, whereas the opposite trend is observed for C7E2. This implies that β-sheet signal is not predictive of NMR signal enhancement. Spectra for other compounds investigated can be found in Figures S7–S10.
In order to examine MRI contrast agent properties in these compounds, C7E2 and C7K3 were imaged at pH 4, 6, 7, 8, and 9 using the same solution conditions as the NMR studies (Figure 6). The NMR signal peak corresponding to the terminal CF3 group of the perfluorocarbon tail was selected for imaging, due to its relatively high intensity. MRI signal for C7E2 was shown to increase until pH 7, as predicted by data shown in Figure 3. C7K3 was imaged, but imaging artifacts from the TFA counterions required dialysis prior to imaging. In these samples, measurable signals that showed pH response were observed, but the data did not fit the trend described in Figure 3 (Figure S12). One possible explanation is that the dialysis process changes the nature of the assembly and therefore the pH response profile.
Figure 6.
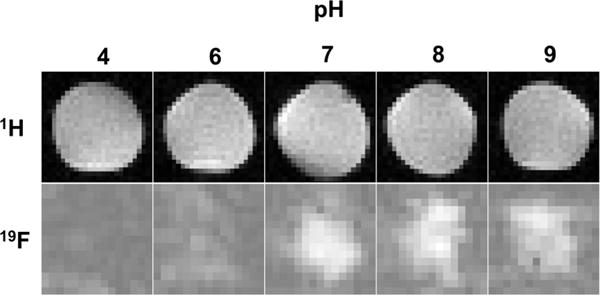
1H and 19F MRI solution images of C7E2 in 150 mM NaCl acquired at 9.4T and ambient temperature. Each image is 25 mm on edge. Sample preparation was identical to NMR experiments. Phantoms were imaged simultaneously with a trifluoroethanol standard to allow intensity comparison. Fluorine images increase in intensity as pH increases, reaching a maximum after pH 7. This is analogous with behavior observed in Figure 3.
The morphology of nanostructures formed by C7E2 and C7K3 molecules is controlled by the balance of repulsive and attractive intermolecular forces sensitive to the acid–base equilibrium of charged amino acids in each peptide sequence. Altering the peptide sequence by varying the nature of charged residues or hydrophobic tail structure results in loss of pH sensitivity. Morphologically, both C7E2 and C7K3 formed cylindrical nanofiber structures when producing NMR signal while molecules of the other compounds tested aggregated into ribbon morphologies. The implication is that the local mobility of perfluorinated tails is greater in cylindrical nanostructures than in nanoribbons. One hypothesis for this difference in dynamics between the two different types of supramolecular aggregates would be a denser packing of tail segments in the case of ribbon-like nanostructures, which possess less local curvature, and less dense packing in the hydrophobic domains of nanofibers, which possess more local curvature.
CONCLUSIONS
A series of self-assembling fluorinated peptide amphiphiles were designed to optimize 19F MRI signals in response to pH changes by varying their content of charged amino acids and number of fluorine atoms in hydrophobic segments. We found that the supramolecular aggregates formed by some of the amphiphiles undergo a morphological transition from low-curvature nanoscale ribbons to cylindrical nanofibers as pH increases that results in enhanced MR signals. This work suggests that morphological transitions in aggregates that affect the mobility of fluorinated segments represent a good strategy to design stimulus-responsive supramolecular 19F MRI contrast agents.
METHODS
Synthesis
Peptides were synthesized using standard solid-phase peptide synthesis techniques on an automated CSBio Synthesizer, model number CS136. Amino acids were obtained from Anaspec (Fremont, CA). DIC and Oxyma pure were obtained from ThermoFischer Sciences (Waltham, MA) and BAChem (Bubendorf, Switzerland), respectively. Dimethylformamide, dichloromethane, diisopropylethylamine, and 4-methylpiperidine were obtained and used without further purification from VWR. Synthesis was conducted on a rink amide resin on MBHA support (EMD Millipore, Billerica Massachusetts) at 0.5 mM scale. The perfluoroalkane tails were added on-resin via the Schotten-Baumann reaction. Acyl chlorides were obtained from Sigma-Aldrich. Acyl chloride (300 μL) was dissolved in 10 mL of solvent system-dried chloroform along with 600 μL of diisopropyl ethyl amine. The solution was directly added to a fritted shaker vessel along with the peptide on-resin and agitated for 30–45 min. The addition was repeated until ninhydrin test was negative. The peptide was cleaved from resin support in a solution of 95% trifluoroacetic acid, 2.5% water, and 2.5% triisopropyl silane over 3 h and precipitated in cold ether stored at −20 °C. The precipitate was purified using reverse-phase HPLC. Overall yield was typically 40%– 50%.
Purification
All peptides were purified using reverse-phase high pressure liquid chromatography on a Phenomenex Gemini C18 column (5 μm particle size) using a 2%–100% acetonitrile gradient program. To each solution was added 0.1% saturated NH4OH solution in water (for glutamic acid-containing sequences) or 0.1% trifluoroacetic acid (for lysine-containing sequences). System eluent was analyzed with an Agilent 6510 Q-TOF MS to identify product peaks. Acetonitrile was removed with rotary evaporation and water was removed via lyophilization. The resultant white, fluffy powder was stored at −20 °C.
General Conditions
Unless otherwise specified, all F–PA samples were analyzed at 2 mM concentration in 150 mM NaCl in Millipure water. All solutions were allowed to stand at room temperature for a period of not less than 16 h and not more than 24 h prior to NMR, CD, Cryo-EM, MRI, Nile red assay, and analytical HPLC.
Analytical HPLC
All lysine-based sequences were analyzed in acidic mobile phase and all glutamic acid–based sequences were analyzed in basic mobile phase. A Phenomenex Jupiter C18 and Phemomenex Gemini C18 column were used for acidic and basic condition separations, respectively, on an Agilent 6520 LC/MS system. Each column measured 1.0 × 150 mm, and contained a 5 μm particle size. A gradient of 5–95% acetonitrile over 30 min and 1 mL/min flow rate was employed. Mobile phases were doped with either 0.1% NH4OH solution (if basic) or 0.1% formic acid (if acidic).
Nuclear Magnetic Resonance Spectroscopy
All NMR spectra were acquired using a Varian 500 MHz NMR spectrometer at 25 °C. Spectra represent the average of 128 scans with a 30° flip angle and 1 s delay time. Precision, thin-walled Shigemi Tubes were used for NMR measurements. PA solutions were dissolved in 150 mM NaCl in Millipure water with 1.8 mM trifluoroethanol and 10% D2O as the lock solvent and aged overnight prior to measurement to promote structure stability. pH adjustment was conducted using a VWR SB21 pH meter between each measurement, using concentrated (0.1 or 0.01 M) solutions of NaOH and HCl in order to minimize concentration change through the pH adjustment process. Each sample at each pH point was equilibrated for 15 min in the scanner at 25 °C prior to spectral acquisition.
Critical Micelle Concentration Determination
Solutions of C7E2 and C7K3 were prepared as specified in the General Conditions section and adjusted to pH 7.5 using 0.1 M NaOH and HCl solutions. On the day of measurement, these solutions were serial diluted to triplicate sets of 1 mM, 0.5 mM, 0.25 mM, 0.1 mM, 0.04 mM, and 0.008 mM concentrations using a preadjusted 150 mM NaCl solution with a total volume of 1 mL. A solution of Nile red was prepared at 150 μM in ethanol. A micropipette calibrated to the density of ethanol was used to dispense 1 μL of Nile red solution in each sample. These samples were then agitated briefly and allowed to incubate at room temperature for 2 h. The solutions were then analyzed using a Hitachi F-4500 fluorimeter operating at a 550 nm excitation wavelength. Each acquisition was the average of three scans in a 5 × 5 mm quartz cuvette.
Circular Dichroism UV Spectroscopy
General conditions were used as described above. Measurements were performed with a AJ-715 Jasco circular dichroism spectrometer at 25 °C. Slit width was 2 nm and the scan speed was 100 nm/min. Samples were loaded into 0.1 mm path length plates. Each spectra is the average of three scans.
Cryo-TEM
Cryo-TEM samples were prepared according to General Conditions described above. Images were acquired using a JEOL 1230 microscope operating at 100 kV. Specimens were cast on plasma-cleaned 300 mesh copper grids with a lacey carbon support obtained from Electron Microscopy Sciences and frozen using a Vitrobot Mark IV plunging into liquid ethane. Images were acquired using a Gatan 831 CCD camera.
Magnetic Resonance Imaging (MRI)
MR images were acquired on a 9.4T Agilent (Varian) horizontal bore MRI scanner. For 1H images, a quad-volume coil of 63 mm diameter was used as both excitation and receiving RF device. The 1H channel was used to shim and to acquire the 1H images. After this, it was switched to a surface coil of 20 mm diameter for 19F images. For 19F MRI, a steady-state free procession (SSFP) sequence was used to selectively excite the desired 19F signal (ca. −85 ppm).29 19F MRI parameters: the repetition time (TR = 30 ms), the echo time (TE = 3.01 ms), the small flip angle (FA = 50), the number of transient (NT = 4096), the SINC excitation pulse length (Psinc = 1.5 ms), the matrix size = 32 × 32, which was zero-filling to 64 × 64 before Fourier transform, the field of view (FOV = 30 × 30 mm), and the slice thickness (THK = 11–15 mm), respectively.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from National Institutes of Health’s (NIH) National Heart, Lung, and Blood Institute under awards P01HL108795 and R01HL116577, the National Institute of Biomedical Imaging and Bioengineering under award R01 EB005866, and the National Institute of Craniofacial and Dental Research under award 5R01DE015920. Compound purification and characterization was performed in the peptide core at the Simpson Querrey Institute for BioNanotechnology (SQI), the Integrated Molecular Structure Education and Research Center (IMSERC), the Biological Imaging Facility (BIF) and Keck Biophysics facilities at Northwestern. We acknowledge A. Dean Sherry and Masaya Takahashi of the mouse MRI core facility and the Advanced Imaging Research Center at the University of Texas Southwestern Medical Center for facilitating 1H/19F MRI studies. The authors also thank Dr. Steve Wiegand (Argonne National Laboratory) for assistance with SAXS experiments, as well as Prof. Liam Palmer and Laura Lilley for helpful discussions, and Mark Seniw for the preparation of graphics.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b00267.
Additional NMR spectroscopy, cryogenic electron microscopy of all self-assemblies, additional circular dichroism spectroscopy of all compounds, physical characterization of compound purity (mass spectrometry and analytical HPLC traces), and additional 19F MRI images. (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Merbach ÂET. Eva The Chemistry of Contast Agents in Medical Magnetic Resonance Imaging. John Wiley & Sons; Chichester: 2001. [Google Scholar]
- 2.Garcia-Martin ML, Martinez GV, Raghunand N, Sherry AD, Zhang S, Gillies RJ. High Resolution pH Imaging of Rat Glioma Using pH-Dependent Relaxivity. Magn Reson Med. 2006;55:309–315. doi: 10.1002/mrm.20773. [DOI] [PubMed] [Google Scholar]
- 3.Raghunand N, Howison C, Sherry AD, Zhang S, Gillies RJ. Renal and Systemic pH Imaging by Contrast-Enhanced MRI. Magn Reson Med. 2003;49:249–257. doi: 10.1002/mrm.10347. [DOI] [PubMed] [Google Scholar]
- 4.Angelovski G, Gottschalk S, Milošević M, Engelmann J, Hagberg GE, Kadjane P, Andjus P, Logothetis NK. Investigation of a Calcium-Responsive Contrast Agent in Cellular Model Systems: Feasibility for Use as a Smart Molecular Probe in Functional MRI. ACS Chem Neurosci. 2014;5:360. doi: 10.1021/cn500049n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yordanov AT, Lodder AL, Woller EK, Cloninger MJ, Patronas N, Milenic D, Brechbiel MW. Novel Iodinated Dendritic Nanoparticles for Computed Tomography (CT) Imaging. Nano Lett. 2002;2:595–599. [Google Scholar]
- 6.Zhang S, Wu K, Sherry AD. A Novel pH-Sensitive MRI Contrast Agent. Angew Chem, Int Ed. 1999;38:3192–3194. [PubMed] [Google Scholar]
- 7.Louie AY, Huber MM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ. In Vivo Visualization of Gene Expression Using Magnetic Resonance Imaging. Nat Biotechnol. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 8.Heffern MC, Matosziuk LM, Meade TJ. Lanthanide Probes for Bioresponsive Imaging. Chem Rev. 2014;114:4496. doi: 10.1021/cr400477t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Major JL, Meade TJ. Bioresponsive, Cell-Penetrating, and Multimeric MR Contrast Agents. Acc Chem Res. 2009;42:893–903. doi: 10.1021/ar800245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalman FK, Woods M, Caravan P, Jurek P, Spiller M, Tircsó G, Kiraly R, Brücher E, Sherry aD. Potentiometric and Relaxometric Properties of a Gadolinium-Based MRI Contrast Agent for Sensing Tissue pH. Inorg Chem. 2007;46:5260–5270. doi: 10.1021/ic0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day SE, Kettunen MI, Gallagher FA, Hu D-E, Lerche M, Wolber J, Golman K, Ardenkjaer-Larsen JH, Brindle KM. Detecting Tumor Response to Treatment Using Hyperpolarized 13C Magnetic Resonance Imaging and Spectroscopy. Nat Med. 2007;13:1382–1387. doi: 10.1038/nm1650. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro MG, Ramirez RM, Sperling LJ, Sun G, Sun J, Pines A, Schaffer DV, Bajaj VS. Genetically Encoded Reporters for Hyperpolarized Xenon Magnetic Resonance Imaging. Nat Chem. 2014;6:629–634. doi: 10.1038/nchem.1934. [DOI] [PubMed] [Google Scholar]
- 13.Lippert AR, Keshari KR, Kurhanewicz J, Chang CJ. A Hydrogen Peroxide-Responsive Hyperpolarized 13C Mri Contrast Agent. J Am Chem Soc. 2011;133:3776–3779. doi: 10.1021/ja111589a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods JC, Choong CK, Yablonskiy DA, Bentley J, Wong J, Pierce JA, Cooper JD, Macklem PT, Conradi MS, Hogg JC. Hyperpolarized 3He Diffusion MRI and Histology in Pulmonary Emphysema. Magn Reson Med. 2006;56:1293–1300. doi: 10.1002/mrm.21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ardenkjær-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, Golman K. Increase in Signal-to-Noise Ratio of > 10,000 Times in Liquid-State NMR. Proc Natl Acad Sci U S A. 2003;100:10158–10163. doi: 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouchard L-S, Kovtunov KV, Burt SR, Anwar MS, Koptyug IV, Sagdeev RZ, Pines A. Para-Hydrogen-Enhanced Hyperpolarized Gas-Phase Magnetic Resonance Imaging. Angew Chem, Int Ed. 2007;46:4064–4068. doi: 10.1002/anie.200700830. [DOI] [PubMed] [Google Scholar]
- 17.Reineri F, Viale A, Giovenzana G, Santelia D, Dastrù W, Gobetto R, Aime S. New Hyperpolarized Contrast Agents for 13C-MRI from Para-Hydrogenation of Oligooxyethylenic Alkynes. J Am Chem Soc. 2008;130:15047–15053. doi: 10.1021/ja8059733. [DOI] [PubMed] [Google Scholar]
- 18.Bulte JWM. Hot Spot MRI Emerges from the Background. Nat Biotechnol. 2005;23:945–946. doi: 10.1038/nbt0805-945. [DOI] [PubMed] [Google Scholar]
- 19.Tirotta I, Mastropietro A, Cordiglieri C, Gazzera L, Baggi F, Baselli G, Bruzzone MG, Zucca I, Cavallo G, Terraneo G, Baldelli Bombelli F, Metrangolo P, Resnati G. A Superfluorinated Molecular Probe for Highly Sensitive in Vivo 19F-MRI. J Am Chem Soc. 2014;136:8524–8527. doi: 10.1021/ja503270n. [DOI] [PubMed] [Google Scholar]
- 20.Srinivas M, Boehm-Sturm P, Figdor CG, de Vries IJ, Hoehn M. Labeling Cells for in Vivo Tracking Using 19F MRI. Biomaterials. 2012;33:8830–8840. doi: 10.1016/j.biomaterials.2012.08.048. [DOI] [PubMed] [Google Scholar]
- 21.Ahrens ET, Bulte JWM. Tracking Immune Cells in Vivo Using Magnetic Resonance Imaging. Nat Rev Immunol. 2013;13:755–763. doi: 10.1038/nri3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morawski AM, Lanza GA, Wickline SA. Targeted Contrast Agents for Magnetic Resonance Imaging and Ultrasound. Curr Opin Biotechnol. 2005;16:89–92. doi: 10.1016/j.copbio.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Lanza GM, Winter PM, Caruthers SD, Hughes MS, Hu G, Schmieder AH, Wickline SA. Theragnostics for Tumor and Plaque Angiogenesis with Perfluorocarbon Nanoemulsions. Angiogenesis. 2010;13:189–202. doi: 10.1007/s10456-010-9166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Cabello J, Barnett BP, Bottomley PA, Bulte JWM. Fluorine (19F) MRS and MRI in Biomedicine. NMR Biomed. 2011;24:114–129. doi: 10.1002/nbm.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janjic JM, Srinivas M, Kadayakkara DKK, Ahrens ET. Self-Delivering Nanoemulsions for Dual Fluorine-19 MRI and Fluorescence Detection. J Am Chem Soc. 2008;130:2832–2841. doi: 10.1021/ja077388j. [DOI] [PubMed] [Google Scholar]
- 26.Criscione JM, Le BL, Stern E, Brennan M, Rahner C, Papademetris X, Fahmy TM. Self-Assembly of pH-Responsive Fluorinated Dendrimer-Based Particulates for Drug Delivery and Noninvasive Imaging. Biomaterials. 2009;30:3946–3955. doi: 10.1016/j.biomaterials.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Takaoka Y, Kiminami K, Mizusawa K, Matsuo K, Narazaki M, Matsuda T, Hamachi I. Systematic Study of Protein Detection Mechanism of Self-Assembling 19F NMR/MRI Nanoprobes toward Rational Design and Improved Sensitivity. J Am Chem Soc. 2011;133:11725–11731. doi: 10.1021/ja203996c. [DOI] [PubMed] [Google Scholar]
- 28.Takaoka Y, Sakamoto T, Tsukiji S, Narazaki M, Matsuda T, Tochio H, Shirakawa M, Hamachi I. Self-Assembling Nanoprobes That Display Off/On 19F Nuclear Magnetic Resonance Signals for Protein Detection and Imaging. Nat Chem. 2009;1:557–561. doi: 10.1038/nchem.365. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Huang G, Zhang S, Sagiyama K, Togao O, Ma X, Wang Y, Li Y, Soesbe TC, Sumer BD, Takahashi M, Sherry AD, Gao J. Multi-Chromatic Ph-Activatable 19F-MRI Nanoprobes with Binary On/Off pH Transitions and Chemical-Shift Barcodes. Angew Chem, Int Ed. 2013;52:8074–8078. doi: 10.1002/anie.201301135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chalmers KH, De Luca E, Hogg NHM, Kenwright AM, Kuprov I, Parker D, Botta M, Wilson JI, Blamire AM. Design Principles and Theory of Paramagnetic Fluorine-Labelled Lanthanide Complexes as Probes for 19F Magnetic Resonance: A Proof-of-Concept Study. Chem - Eur J. 2010;16:134–148. doi: 10.1002/chem.200902300. [DOI] [PubMed] [Google Scholar]
- 31.Harvey P, Chalmers KH, De Luca E, Mishra A, Parker D. Paramagnetic 19F Chemical Shift Probes That Respond Selectively to Calcium or Citrate Levels and Signal Ester Hydrolysis. Chem - Eur J. 2012;18:8748–8757. doi: 10.1002/chem.201200737. [DOI] [PubMed] [Google Scholar]
- 32.Tirotta I, Dichiarante V, Pigliacelli C, Cavallo G, Terraneo G, Bombelli FB, Metrangolo P, Resnati G. 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem Rev. 2015;115:1106–1129. doi: 10.1021/cr500286d. [DOI] [PubMed] [Google Scholar]
- 33.Hartgerink JD, Beniash E, Stupp SI. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 34.Cui H, Cheetham AG, Pashuck ET, Stupp SI. Amino Acid Sequence in Constitutionally Isomeric Tetrapeptide Amphiphiles Dictates Architecture of One-Dimensional Nanostructures. J Am Chem Soc. 2014;136:12461–12468. doi: 10.1021/ja507051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui H, Muraoka T, Cheetham AG, Stupp SI. Self-Assembly of Giant Peptide Nanobelts. Nano Lett. 2009;9:945–951. doi: 10.1021/nl802813f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui HG, Webber MJ, Stupp SI. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapiro MG, Westmeyer GG, Romero PA, Szablowski JO, Kuster B, Shah A, Otey CR, Langer R, Arnold FH, Jasanoff A. Directed Evolution of a Magnetic Resonance Imaging Contrast Agent for Noninvasive Imaging of Dopamine. Nat Biotechnol. 2010;28:264–270. doi: 10.1038/nbt.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Webber MJ, Han X, Prasanna Murthy SN, Rajangam K, Stupp SI, Lomasney JW. Capturing the Stem Cell Paracrine Effect Using Heparin-Presenting Nanofibres to Treat Cardiovascular Diseases. J Tissue Eng Regener Med. 2010;4:600–610. doi: 10.1002/term.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webber MJ, Newcomb CJ, Bitton R, Stupp SI. Switching of Self-Assembly in a Peptide Nanostructure with a Specific Enzyme. Soft Matter. 2011;7:9665–9672. doi: 10.1039/c1sm05610g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webber MJ, Matson JB, Tamboli VK, Stupp SI. Controlled Release of Dexamethasone from Peptide Nanofiber Gels to Modulate Inflammatory Response. Biomaterials. 2012;33:6823–6832. doi: 10.1016/j.biomaterials.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matson JB, Stupp SI. Drug Release from Hydrazone-Containing Peptide Amphiphiles. Chem Commun. 2011;47:7962–7964. doi: 10.1039/c1cc12570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conda-Sheridan M, Lee SS, Preslar AT, Stupp SI. Esterase-Activated Release of Naproxen from Supramolecular Nanofibres. Chem Commun. 2014;50:13757–13760. doi: 10.1039/c4cc06340f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Preslar AT, Parigi G, McClendon MT, Sefick SS, Moyer TJ, Haney CR, Waters EA, MacRenaris KW, Luchinat C, Stupp SI, Meade TJ. Gd(III)-Labeled Peptide Nanofibers for Reporting on Biomaterial Localization in Vivo. ACS Nano. 2014;8:7325–7332. doi: 10.1021/nn502393u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bull SR, Guler MO, Bras RE, Venkatasubramanian PN, Stupp SI, Meade TJ. Magnetic Resonance Imaging of Self-Assembled Biomaterial Scaffolds. Bioconjugate Chem. 2005;16:1343–1348. doi: 10.1021/bc050153h. [DOI] [PubMed] [Google Scholar]
- 45.Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI. Self-Assembled Peptide Amphiphile Nanofibers Conjugated to MRI Contrast Agents. Nano Lett. 2005;5:1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- 46.Zhang S, Greenfield MA, Mata A, Palmer LC, Bitton R, Mantei JR, Aparicio C, de la Cruz MO, Stupp SI. A Self-Assembly Pathway to Aligned Monodomain Gels. Nat Mater. 2010;9:594–601. doi: 10.1038/nmat2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muraoka T, Cui H, Stupp SI. Quadruple Helix Formation of a Photoresponsive Peptide Amphiphile and Its Light-Triggered Dissociation into Single Fibers. J Am Chem Soc. 2008;130:2946–2947. doi: 10.1021/ja711213s. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A, Haverick M, Stump K, Yang X, Tweedle MF, Goldberger JE. Fine-Tuning the pH Trigger of Self-Assembly. J Am Chem Soc. 2012;134:3647–3650. doi: 10.1021/ja211113n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moyer TJ, Finbloom JA, Chen F, Toft DJ, Cryns VL, Stupp SI. Ph and Amphiphilic Structure Direct Supramolecular Behavior in Biofunctional Assemblies. J Am Chem Soc. 2014;136:14746–14752. doi: 10.1021/ja5042429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalinowski L, Dobrucki LW, Meoli DF, Dione DP, Sadeghi MM, Madri Ja, Sinusas AJ. Targeted Imaging of Hypoxia-Induced Integrin Activation in Myocardium Early after Infarction. J Appl Physiol. 2008;104:1504–1512. doi: 10.1152/japplphysiol.00861.2007. [DOI] [PubMed] [Google Scholar]
- 51.Brown JM, Wilson WR. Exploiting Tumour Hypoxia in Cancer Treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 52.Appel R, Tacke S, Klingauf J, Besenius P. Tuning the pH-Triggered Self-Assembly of Dendritic Peptide Amphiphiles Using Fluorinated Side Chains. Org Biomol Chem. 2015;13:1030–1039. doi: 10.1039/c4ob02185a. [DOI] [PubMed] [Google Scholar]
- 53.Webber MJ, Tongers J, Newcomb CJ, Marquardt K-T, Bauersachs J, Losordo DW, Stupp SI. Supramolecular Nanostructures That Mimic VEGF as a Strategy for Ischemic Tissue Repair. Proc Natl Acad Sci U S A. 2011;108:13438–13443. doi: 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pashuck ET, Cui H, Stupp SI. Tuning Supramolecular Rigidity of Peptide Fibers through Molecular Structure. J Am Chem Soc. 2010;132:6041–6046. doi: 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.