

Abstract
Various polymerization mechanisms have been developed to prepare peptide-immobilized poly(ethylene glycol) (PEG) hydrogels, a class of biomaterials suitable for studying cell biology in vitro. Here, we report a visible light mediated thiol-acrylate photopolymerization scheme to synthesize dually degradable PEG-peptide hydrogels with controllable crosslinking and degradability. We systematically evaluated the influence of immobilized mono-thiol pendant peptide on the crosslinking of these hydrogels. We further proposed methods to modulate hydrogel crosslinking, including adjusting concentration of co-monomer or altering the design of multifunctional peptide crosslinker. Due to the formation of thioether ester bonds, these hydrogels were hydrolytically degradable. If the di-thiol peptide linkers used were susceptible to protease cleavage, these thiol-acrylate hydrogels could be designed to undergo partial proteolysis. The differences between linear and multi-arm PEG-acrylate (i.e., PEGDA vs. PEG4A) were also evaluated. Finally, we explored the use of the mixed-mode thiol-acrylate PEG4A-peptide hydrogels for in situ encapsulation of hepatocellular carcinoma cells (Huh7). The effects of matrix stiffness and integrin binding motif (e.g., RGDS) on Huh7 cell growth and HIPPO pathway activation were studied using PEG4A-peptide hydrogels. This visible light polymerized thiol-acrylate hydrogel system represents an alternative to existing light-cured hydrogel platforms and should be useful in many biomedical applications.
Introduction
Peptide-immobilized poly(ethylene glycol) (PEG) hydrogels have emerged as a powerful class of biomaterials for studying cell biology in vitro and for facilitating tissue regeneration in vivo.[1–3] Since PEG contains no biological motifs for cell or protein recognition, the incorporation of peptides (as receptor ligand or protease substrate) renders the otherwise inert PEG network ‘bioactive’ or ‘biomimetic’ such that cell attachment and protease-mediated matrix cleavage are promoted.[4–6] Various chemistries have been exploited to immobilize peptides, either as pendant ligands or as part of gel crosslinks, within a crosslinked hydrogel network.[4] In particular, acrylated or thiolated peptides can be copolymerized with PEG-derivatives (i.e., acrylate, methacrylate, norbornene, etc.) through ultraviolet (UV) light initiated chain-growth, step-growth, or mixed-mode photopolymerization.[4, 7–10] Alternatively, visible light initiated reaction can be used to create chemically crosslinked and peptide-immobilized hydrogels.[11–14] Most visible light induced gelation schemes utilize a photosensitizer (e.g., eosin-Y or rose bengal), a co-initiator (e.g., ethanolamine, TEOA), and a co-monomer (e.g., N-vinylpyrrolidone, NVP).[12, 14] The use of TEOA might not be desirable in some biological applications because it is a strong base. Other amine-based molecules, such as riboflavin [15], have been used as a co-initiator to initiate radical propagation in vinyl monomers and hydrogel crosslinking.
Our group has reported a thiol-based co-initiation scheme where bi-functional thiols (e.g., dithiothreitol, DTT) or bis-cysteine peptides were used as dual-purpose co-initiator and gel crosslinker to initiate thiol-vinyl photopolymerization and gelation.[16–18] Mechanistically, hydrogens on thiols are abstracted by the light excited eosin-Y, creating thiyl radicals that can transfer to unsaturated C=C bond on vinyl monomers and macromers. The generated carbonyl radicals can propagate across many other vinyl moieties.[19, 20] The later reaction forms hydrolytically stable poly(acrylate-co-NVP) kinetic chains. Alternatively, the carbonyl radicals can abstract other hydrogens from separate thiols to regenerate thiyl radicals and form a hydrolytically labile thioether ester bonds if an acrylate-terminated macromer is used (e.g., PEGDA or multi-arm PEG-acrylate). Adjusting thiol concentration in the system can yield hydrogel network with controlled structure, mechanics, and degradability without altering the contents of macromer and co-monomer. Importantly, macromer components required in this crosslinking scheme (e.g., PEG-acrylates, peptides, NVP, eosin-Y) are all commercially accessible, making this modular crosslinking method ideal for a diverse array of hydrogel-based tissue engineering applications. These visible light and thiol-initiated hydrogels can be modularly engineered to be non-degradable (using acrylamide-terminated macromer and non-degradable linker) or susceptible to hydrolytic (using acrylate-terminated macromers such as PEG-acrylate) and enzymatic (using bis-cysteine protease-sensitive peptides) degradations.[16, 17] The use of multi-functional thiol (e.g., bi-functional DTT or tetra-functional PEG-thiol) is essential in visible light initiated thiol-acrylate gelation, as mono-functional thiol is inefficient in initiating gel crosslinking. Careful selection of multi-functional thiol concentration is especially important in thiol-acrylate photopolymerization because thiol moiety serves not only as a co-initiator, but also as a chain-transfer agent that decreases the degree of polymerization. Indeed, our previous work has revealed a parabolic relationship between hydrogel mechanics and thiol concentration.[16] The initial increase in thiol concentration leads to increased initiation efficiency in the thiol-acrylate polymerization, whereas excess amount of thiol would cause significant chain-transfer events that decrease molecular weight of crosslinks and the modulus of the hydrogel.
Pendant peptides bearing mono-cysteine can be easily co-polymerized in the hydrogel network during network crosslinking with high incorporation efficiency (>90%).[16] While introducing pendant peptides in a PEG-based hydrogel imparts bioactive features into the otherwise inert network, one should carefully evaluate the influence of added peptides on the biophysical properties of the hydrogel, namely permeability, crosslinking density, and degradability.[21, 22] During our exploration of this attractive visible light initiated gel crosslinking, we found that the co-polymerization of pendant thiol peptide significantly impact the degree of network crosslinking. Several approaches could be used to improve the crosslinking of pendant peptide immobilized thiol-acrylate hydrogels, such as increasing concentration of co-monomer NVP, decreasing macromer chain-length, or increasing macromer functionality.[16, 17] One common feature of these approaches is the increase of monomer molarity, which accelerates propagation of the polymerization. These approaches also shift the crosslinking towards chain-growth polymerization that decreases the hydrolytic degradability of the resulting hydrogels.[20]
In order to exploit visible light cured thiol-acrylate hydrogels for studying cell fate in 3D, it is essential to understand the crosslinking and degradation of these gels. In this work, we aimed to improve the crosslinking efficiency of thiol-acrylate hydrogels through designing peptides with higher functionality. We first evaluated the impact of mono-functional pendant motifs on gel properties. Bioactive motif (e.g., RGDS) was incorporated in the thiol-acrylate network not through pendant peptide, but as part of the gel crosslinkers. In addition to characterizing the hydrolytic degradability of these hydrogels, we also assessed the proteolytic degradation of these unique visible light cured thiol-acrylate hydrogels. Finally, through in situ encapsulation and culture of hepatocellular carcinoma cells Huh7, we demonstrated the cytocompatibility of this system. We also found that Huh7 cells encapsulated in 3D hydrogel had significantly lower expression of CTGF gene, an indicative gene up-regulated by the HIPPO pathway effector YAP that controls cell proliferation, differentiation, tissue development and tumorigenesis.
Materials & Methods
Materials
Linear and four-arm raw PEGs were purchased from Sigma-Aldrich and JenKem Technology USA, respectively. Eosin-Y disodium salt was purchased from MP Biomedical. N-vinylpyrrolidone (NVP) and dithiothreitol (DTT) were obtained from Acros Organics and Thermo Fisher, respectively. Reagents for solid phase peptide synthesis (Fmoc-amino acids, Fmoc-Rink amide-MBHA resin, HBTU, etc.) were purchased from Chempep or Anaspec. 1-Hydroxybenzotriazole (HOBT) hydrate was acquired from AK Scientific. All reagents for cell culture (e.g., Dulbecco’s modified Eagle’s medium (DMEM), Dulbecco’s phosphate-buffered saline (DPBS), trypsin, fetal bovine serum (FBS), and 1× Antibiotic-Antimycotics) as well as live/dead staining kit were purchased from Life Technologies. AlamarBlue cell viability/metabolic indicator was procured from AbD Serotec. All other chemicals were acquired from Sigma-Aldrich unless noted otherwise.
Synthesis of PEGDA, PEG4A, and peptides
PEG-diacrylate (PEGDA) and PEG-tetra-acrylate (PEG4A) were synthesized using the same protocol published previously.[16] All PEGDA macromers were characterized by 1H NMR (AVANCE Bruker 500) and the degree of functionalization was at least 95%. All peptides were built on Fmoc-Rink amide-MBHA resin using standard solid phase peptide synthesis in a microwave peptide synthesizer (CEM Discover) using protocols reported previously.[23, 24] Preparative reverse phase HPLC (RP-HPLC, PerkinElmer Flaxer System) was used to purify the peptides (>90% purity).
Gel modulus measurements – Gel stiffness and hydrolytic degradation
Storage moduli (G’) and loss moduli (G’’) were determined using a Bohlin CVO digital rheometer as described previously.[16, 17] Prior to the measurements, hydrogels were formed in between two glass slides separated by 1mm thick Teflon spacers. Hydrogel discs (8 mm in diameter) were punched out from the gel slabs and incubated in pH 7.4 PBS at 37 °C. Storage (or shear) moduli of the hydrogels were measured at predetermined time periods to reveal gel stiffness and hydrolytic degradation as a function of time. Rheometry was performed in oscillatory strain-sweep (0.1–5%) mode using a parallel plate geometry (8 mm).
To assess hydrolytic degradation of the hydrogels over time, gel moduli were measured at pre-determined time periods and plotted as a function of incubation time. Shear modulus of the hydrogels at day 2 was used as the starting modulus (i.e., G’0) for normalization to determine the rate of hydrolytic gel degradation, which was modeled using the pseudo-first order degradation kinetics as reported previously.[16, 17, 24]
Proteolytic degradation of thiol-acrylate hydrogels
CGGYC, a peptide substrate for α-chymotrypsin (Worthington Biochemical),[23] was used to crosslink thiol-acrylate PEGDA and PEG4A hydrogels were prepared as previously described.[16, 17] Following gelation, hydrogels were incubated in PBS for 24 hours prior to the proteolytic degradation study. Thiol-acrylate PEG hydrogels were incubated in α-chymotrypsin solution prepared in PBS at desired concentrations at room temperature for a pre-determined period of time. Gel mass before and after incubation in chymotrypsin solution were measured gravimetrically to determine mass change as a function of time.
Cell culture, encapsulation, and viability assays
Hepatocellular carcinoma-derived Huh7 cells were maintained in high glucose DMEM containing 10% FBS and standard Antibiotic-Antimycotics (Life Technologies) and cultured in a 5% CO2, 37°C tissue culture incubator. In preparation for encapsulation, three millions of Huh7 cells were seeded in a 10cm plastic culture plate 24 hours prior to the experiment. To encapsulate these freshly seeded cells, cells were trypsinized and dissociated into single cells. Required volume of cell solution was mixed in pre-polymer solutions to a final cell density of 5 × 106 cells/mL. Twenty micro-liters (20 µL) of cell-containing pre-polymer solutions were transferred to a silicon mold, followed by visible light exposure using the same light source and conditions as described in the previous section. Following gelation and cell encapsulation, cell-laden hydrogels were cultured in the same culture media as described above. Cell viability and metabolic activity were determined by Live/Dead staining and AlamarBlue assay as previously described.[16, 25]
Generation of YAP reporter cells
Plasmid pEGFP-C3-hYAP1 was a gift from Marius Sudol (Addgene. Plasmid #17843).[26] To generate cells overexpressing GFP-YAP1, Huh7 cells were transfected by pEGFP-C3-hYAP1 plasmids using LTX1 plus (Mirus) following manufacturer’s protocol. One day post-transfection, 0.5 mg/mL of G415 (Santa Cruz Biotechnology) was added in culture media for selecting GFP-YAP1 overexpressing cells. Flow cytometry was performed to sort GFP (+) cells 3 weeks post-transfection and G415 selection. Sorted cells were maintained in media containing 0.5 mg/mL of G415.
RNA isolation, reverse transcription, and analysis of gene expression
Cell-laden hydrogels were collected in DNase/RNase-free microtubes on the indicated days of experiments, flash frozen with liquid nitrogen, and stored at −80°C until use. To extract RNA, frozen gels were homogenized in 600 µL of RA1 lysis buffer (NucleoSpin RNA II kit, Clontech) and freeze-thaw two more times to lyse cells. Lysates were purified by NucleoSpin Filters (NucleoSpin RNA II kit, Clontech), followed by mixing thoroughly with 600 µL of RNase-free 70% ethanol. Consequently, the mixtures were transferred to NucleoSpin RNA columns (NucleoSpin RNA II kit, Clontech) for RNA extraction following the manufacturer’s instruction. The isolated RNAs were eluted in 30µL of DNase/RNase-free water and quantified by UV spectrometry (NanoDrop 2000, Thermo Scientific). Aliquots of RNA samples were stored at −80°C until use.
Isolated total RNA (100–500 ng) were converted into single-stranded cDNA using the PrimeScript RT reagent kit (Clontech). Gene expression level was assessed by quantitative real-time PCR. In short, 2 µL of the reverse-transcribed cDNA were mixed with specific primers and SYBR Premix Ex Taq II kit (Clontech) following manufacturer’s protocol. Quantitative real-time PCR was performed on an Applied Biosystems 7500 fast real-time PCR machine. The reactions were run at 95°C for 30 sec, followed by 45 cycles of 95°C for 5 sec and 72°C for 30 sec. Gene expression levels were analyzed using 2−ΔΔCT method that, within the same experimental group, the detected levels of each gene were standardized to the level of GAPDH internal control (ΔCT), followed by normalizing the standardized gene levels to those from 2D culture (ΔΔCT, where gene levels in 2D culture were set as 1-fold for the comparison of gene expression (fold changes) under different culture conditions). Real-time PCR primer sequences for GAPDH [27] are 5'-GAAGGTGAAGGTCGGAGTC-3' (forward) and 5'-GAAGATGGTGATGGGATTTC-3' (reverse); primer sequences for CTGF[28] are 5'-AGGAGTGGGTGTGTGACGA-3' (forward) and 5'-CCAGGCAGTTGGCTCTAATC-3' (reverse).
Statistics
All statistical analyses and curve fittings were conducted using GraphPad Prism 5 software. Gel moduli (with more than three experimental groups), cell metabolic activity, and urea secretion were analyzed by Two-Way ANOVA followed by Bonferroni’s post-hoc test with control group specified in the respective figure captions. Gel points and swelling ratio with single experimental group were analyzed by One-Way ANOVA followed by Tukey’s post-hoc test. All experiments were conducted independently for at least three times. Data presented were Mean ± SEM. Single, double, and triple asterisks represent p<0.05, 0.001, and 0.0001, respectively. p <0.05 was considered statistically significant.
Results & Discussion
Effect of mono-functional thiols on moduli of visible light cured thiol-acrylate hydrogels
We have previously reported that PEG-peptide hydrogels can be formed by visible light initiated thiol-acrylate mixed mode photopolymerization and that these hydrogels were susceptible to hydrolytic and cell-mediated proteolytic degradation.[16, 17] In this report, we further explored the crosslinking of this type of hydrogels and evaluated the effect of incorporating pendant peptide, a means of enhancing bioactivity of the otherwise inert hydrogels, on the crosslinking of the resulting hydrogels. The crosslinking of thiol-acrylate hydrogels using PEGDA and bi-functional thiol linkers (with co-monomer NVP) yields a mixed-mode network with both poly(acrylate-co-VP) chains and thiol-ether ester bonds (Fig. 1A). When mono-thiol pendant peptide was added, the network possessed more thiol-ether ester bonds and as such had decreased gel crosslinking density (Fig. 1B). To examine this effect, we prepared thiol-acrylate mixed-mode hydrogels incorporating macromer PEGDA (10 wt%), crosslinker DTT (7.5 mM), photosensitizer eosin-Y (0.1 mM), co-monomer NVP (0.1 %), and different concentrations of either L-cysteine or CRGDS peptide. The shear moduli of the hydrogels were negatively and dose-dependently affected when mono-functional ligand such as L-cysteine (Fig. 1C) or CRGDS (Fig. 1D) was co-polymerized in the hydrogel network, suggesting a need to improve the crosslinking density of this mixed-mode thiol-acrylate hydrogel system. The simplest way is through increasing the concentration of co-monomer NVP (Fig. 1E). NVP is a small monomer that increases both the gel point and the final modulus of a mixed-mode hydrogel.[29] As shown in Fig. 1E, the shear moduli of the CRGDS-immobilized mixed-mode hydrogels were significantly and dose-dependently increased. Adjusting gel stiffness by tuning NVP content provides a facile way of controlling mechanical properties of the peptide-immobilized hydrogels without altering the compositions of other macromer components.
Figure 1. Effect of mono-functional thiol and co-monomer NVP on shear modulus (G’) of visible light-cured mixed-mode PEGDA hydrogel.
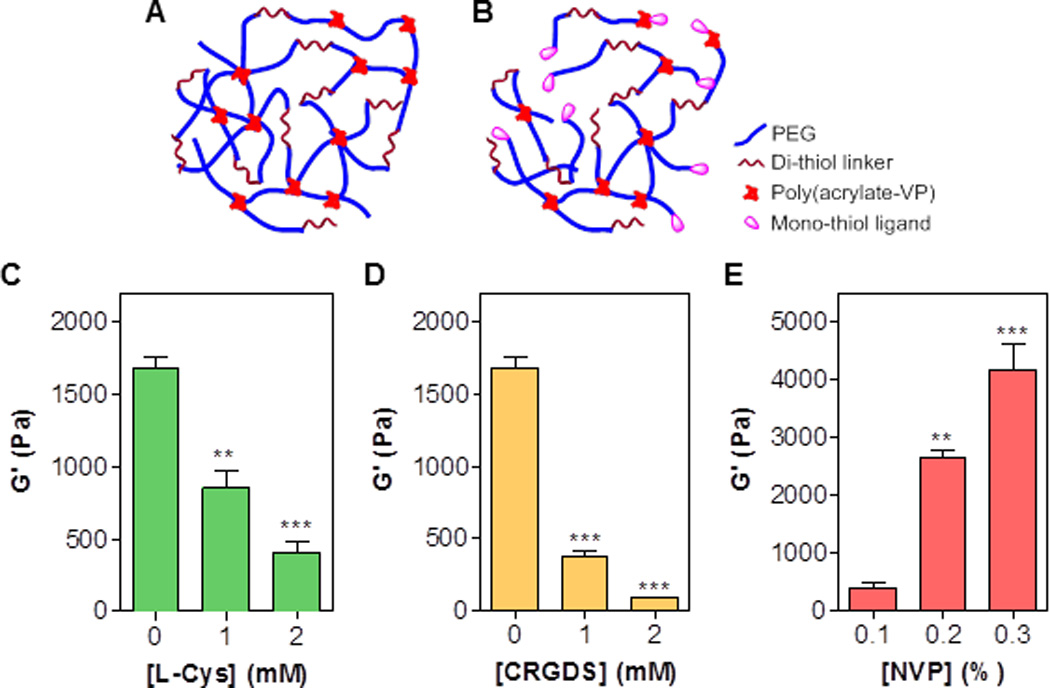
(A) Schematic of a visible light cured thiol-acrylate hydrogel without mono-cysteine peptide immobilization. (B) Schematic of a visible light cured thiol-acrylate hydrogel with immobilized pendant ligands. (C) Effect of [L-Cysteine] on shear modulus of thiol-acrylate hydrogels formed with 0.1 vol% NVP. (D) Effect of [CRGDS] on shear modulus of thiol-acrylate hydrogels formed with 0.1 vol% NVP. (E) Effect of [NVP] on shear modulus of thiol-acrylate hydrogels formed with 2mM L-Cysteine. Other conditions in the gels: 10wt% 3.4 kDa PEGDA, 7.5 mM DTT, 0.1 mM eosin-Y, and 5-min visible light exposure. Shear moduli of all hydrogels were measured after incubating in PBS for 2 hours (**p<0.001, *** p<0.0001).
Effect of bi-functional thiols on moduli of visible light cured thiol-acrylate hydrogels
Mono-thiol pendant peptide reduces the crosslinking density of a thiol-acrylate hydrogel network by means of thiol-mediated chain-transfer reactions. To explore whether it is possible to maintain hydrogel crosslinking density without adding additional co-monomer (e.g., NVP), we designed and synthesized a peptide that contains terminal RGDS and a functional peptide sequence sandwiched by two cysteines that provide two linkages in the hydrogel network (e.g., CX1X2X3C, or CX1X2X3CRGDS. X represents any amino acid residue. Fig. 2A). This multifunctional peptide provides not only thiol moieties for initiating thiol-acrylate gelation, but also sequences critical for protease-mediated cleavage and integrin-mediated receptor binding. We first evaluated the effect of bis-cysteine containing peptide on the crosslinking of thiol-acrylate PEGDA hydrogels using a model peptide CGGYC. This short peptide was selected because it can be readily cleaved by chymotrypsin and thus serves as a model for protease sensitive peptide. As shown in Fig. 2B, the incorporation of 2mM of CGGYC peptide (group B) insignificantly decreased the shear modulus of the PEGDA hydrogel. This result could be attributed to the increase in total thiol concentration (i.e., 15mM in group A and 19mM in group B) that increased chain-transfer events during gel crosslinking. In an attempt to improve this, we prepared another group of hydrogels using a fixed total concentration of thiol (i.e., total 15mM with 2mM of DTT replaced by CGGYC peptide, group C). Unfortunately, the gel shear modulus decreased to a statistically significant level, suggesting that the de-protonation efficiency of cysteines might be lower than that of the thiol groups in DTT, hence resulting in lower thiyl radicals necessary for efficient gel crosslinking.
Figure 2. Effect of bi-functional pendant peptides on shear moduli of visible light-cured mixed-mode PEG hydrogels.
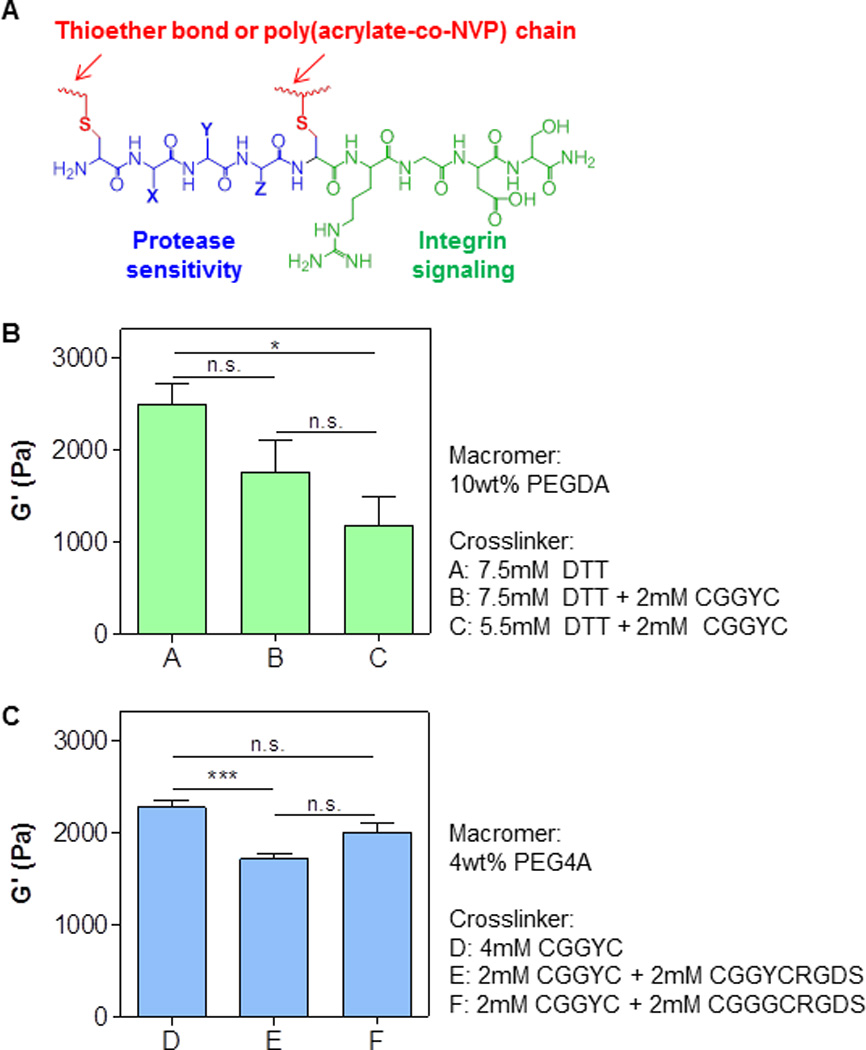
(A) Schematic of a bi-functional pendant peptide (X, Y, Z represent any amino acid residue). Bioactive motif (e.g., RGDS) was tethered at the C-terminal of the bi-functional peptide to afford cell-responsiveness in the otherwise inert PEG-based hydrogels. (B) Shear moduli of PEGDA (10wt%, 3.4 kDa) hydrogels crosslinked by DTT or DTT and CGGYC at various concentrations. (C) Shear moduli of PEG4A (4wt%, 20 kDa) hydrogels crosslinked by CGGYC or CGGYC and CGGYCRGDS. Shear moduli were measured at 2-hr post-gelation (*p<0.05, *** p<0.0001).
In addition to the linear macromer PEGDA, we also evaluated the crosslinking efficiency using 4-arm PEG acrylate (PEG4A). Multi-arm PEGs provide additional linkages inherent in their molecular structure and should enhance gel crosslinking. Indeed, the concentration of macromer and bi-functional thiol crosslinker needed to achieve a similar degree of shear modulus were higher in the PEGDA system when compared with PEG4A (i.e., 10wt% PEGDA 15mM thiol in group A; 4wt% PEG4A and 8mM thiol in group D, Fig. 2). Furthermore, when half of the peptide was replaced by CGGYCRGDS (group E), the shear moduli of the thiol-acrylate PEG4A hydrogels only decreased slightly when compared with gels crosslinked by CGGYC (group D). We also tested gel crosslinking using another bio-functional peptide – CGGGCRGDS (group F) – and found that the gel modulus was in between that of CGGYC (group D) and CGGYCRGDS (group E), suggesting that peptide sequences played a role in the efficiency of thiol-acrylate photopolymerization. Previously, Lutolf and Hubbell used Michael-type addition gel crosslinking to demonstrate the profound impact of neighboring amino acid residues on deprotonation of cysteines and subsequent thiol-addition to vinyl groups.[30] We have also shown that bis-cysteine containing amino acid sequences influence crosslinking and degradation of step-growth thiol-norbornene photoclick hydrogels.[24] Future studies may be focused on systematic alteration of peptide crosslinker sequences to gain understanding in the role of amino acid residues on the efficiency of visible light induced thiol-acrylate photopolymerization. Nonetheless, while the shear moduli of thiol-acrylate hydrogels with multifunctional peptide crosslinkers were still lower than controls (groups A and D in Fig. 2), the degree of reduction was not as drastic as that when using mono-thiol pendant peptide (Figs. 1C and 1D).
Effect of bi-functional thiol linker on hydrolytic degradation of visible light cured thiol-acrylate hydrogels
We have previously showen that, owing to the formation of thioether ester bonds, thiol-acrylate hydrogels are susceptible to hydrolytic degradation (Fig. 3A). This phenomenon was examined here using the multifunctional peptide crosslinked thiol-acrylate hydrogels. As shown in Fig. 3B, PEGDA hydrogels crosslinked by 7.5mM DTT (group A), 7.5mM DTT and 2 mM CGGYC (group B), and 5.5mM DTT and 2mM CGGYC (group C) all degraded slowly as a function of time. Because these hydrogels had different initial moduli (Fig. 2B), we evaulated the change of gel moduli as a function of time using the pseudo-first order degradation kinetics developed previously for this class of hydrogels.[24, 31] Briefly, the hydrolytic degradation of thiol-acrylate hydrogels causes a reduction in gel shear moduli, which can be expressed by the following equation:
Figure 3. Effect of gel formulations on hydrolytic degradation of visible light-cured mixed-mode PEG hydrogels.
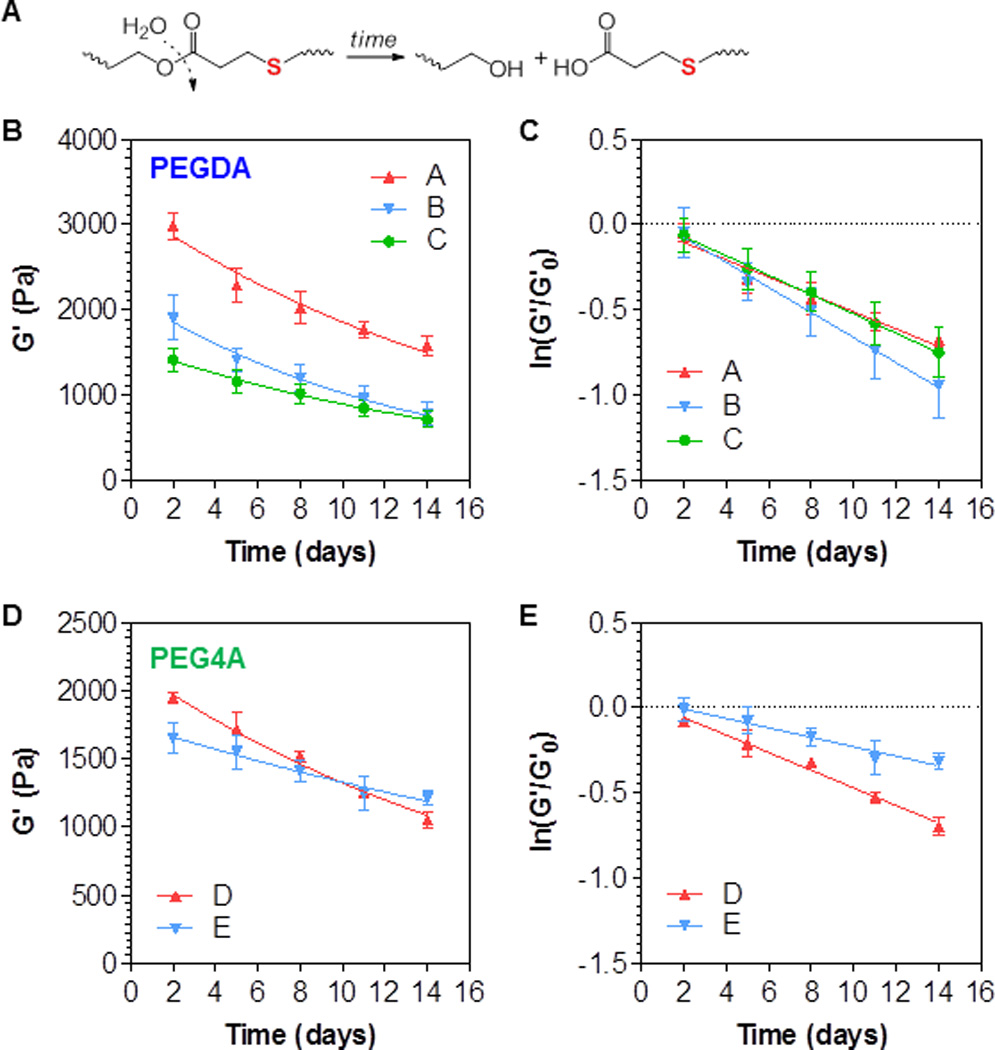
Shear moduli of the hydrogels were monitored as a function of time. (A) Schematic of hydrolytic degradation of thiol-ether ester bond formed in thiol-acrylate hydrogels. (B) Degradation of PEGDA (10 wt%, 3.4 kDa) hydrogels with different crosslinkers: A: 7.5mM DTT; B: 7.5mM DTT and 2 mM CGGYC; C: 5.5mM DTT and 2mM CGGYC. (C) Replotting the degradation data in B with ln(G’/G’0). (D) Degradation of PEG4A (4 wt%, 20 kDa) hydrogels with different crosslinkers: D: 4 mM CGGYC; E: 2 mM CGGYC and 2 mM CGGYCRGDS. (E) Replotting the degradation data in D with ln(G’/G’0).
Here G’ is the gel shear modulus at any time during degradation, G’0 is gel moduli before significant degradation has occurred (day 2 at this experimental set-up), t is the degradation time, while k is the pseudo-first order hydrolysis constant. After plotting ln(G’/G’0) as a function of time, it can be seen that all degradation kinetics followed the pseudo-first order degradation kinetics (Table 1). Although no statistically significant difference was found between any two of the three groups (Table 1), group B degraded slightly faster than the other two groups (k = 0.051, 0.074, and 0.057 day−1 for group A, B, and C, respectively), a trend similar to the results we have reported in an earlier publication.[16, 17] This was reasonable since gels in group B contained more thioether ester bonds (total thiol concentraiton = 19mM) than gels in gropus A and C (total thiol concentration = 17mM).
Table 1.
Parameters of linear regression results shown in Figures 3C and 3E.
| Group | A (control) | B | C | D (control) | E |
|---|---|---|---|---|---|
| k (day−1) | 0.051 ± 0.002 | 0.074 ± 0.075 | 0.057 ± 0.045 | 0.052 ± 0.047 | 0.028 ± 0.007 |
| R2 | 0.96 | 0.99 | 0.99 | 0.99 | 0.97 |
| p# | - | 0.10 | 0.95 | - | 0.04 |
We next evaluated hydrolytic degradation of thiol-acrylate PEG4A hydrogels crosslinked by either purely CGGYC (4mM peptide) or a mixture of CGGYC and CGGYCRGDS (2mM peptide each). As shown in Figs. 3D and 3E, the degradation of both groups of gels still followed the pseudo-first order hydrolysis kinetics (Table 1). However, gels crosslinked by purely CGGYC (group D) degraded almost two times faster (k = 0.052 vs. 0.028 day−1 for group D and E, respectively. Table 1) than the gels crosslinked by a mixture of both multifunctional peptides (group E). This was unexpected as both groups contained equal concentraiton of thioether ester bonds (i.e., 8mM). Although the exact mechanism by which the RGDS motif decreases the hydrolysis rate of the hydrogel is yet to be determined, the difference of the gel properties within the examined time frame (i.e., 2 weeks) was less likely to affect cell behaviors due to the small differences in the absolute moduli (Fig. 3D).
Proteolytic degradation of visible light cured thiol-acrylate hydrogels
Protease sensitivity is an important aspect of a biomimetic hydrogel design and labile peptide substrates are commonly used to impart proteolytic degradability in the otherwise inert synthetic hydrogels. To demonstrate the proteolytic degradability of the thiol-acrylate PEG-peptide hydrogel system, we used bis-cysteine peptide (i.e., CGGYC)[23] sensitive to α-chymotrypsin, a protease that cleaves the C-terminal peptide bond of phenylalanine, tryptophan, and tyrosine (Fig. 4A). When thiol-acrylate PEG hydrogel was crosslinked by 100% of inert linker DTT (i.e., 0% CGGYC), no significant change in gel mass was observed during the 300 minute incubation in the protease solution (Fig. 4B). When the gels were crosslinked by 100% chymotrypsin labile peptide, the hydrogels network was cleaved by infiltrated chymotrypsin, leading to increased gel swelling and higher gel mass (~10% increase, Fig. 4B). The increase in gel swelling was due to additional water uptake in the degrading network. The effect of co-monomer NVP on proteolytic degradation of PEGDA hydrogel was also investigated (Fig. 4C). Increasing NVP concentration results in higher content of poly(acrylate-co-NVP) kinetic chain in the gels. Consequently, the gels with higher NVP content (i.e., 0.3%) degraded slightly slower by chymotrypsin (Fig. 4C). Next, the effect of α-chymotrypsin concentration (1, 2, or 4 mg/mL) on degradation rate was examined. Interestingly, there was no significant difference in their degradation profiles (Fig. 4D). The insignificant difference in proteolytic gel degradation was likely caused by the high protease concentration used in the study. It was also likely that the rate of proteolytic gel degradation is limited by the peptide substrate availability. Therefore, increasing the enzyme concentration only has limited effect on gel degradation rate. Further extended the protease incubation time did not increase gel mass (up to 300 min, data not shown), potentially due to the presence of none-degradable poly(PEG-co-NVP) chains. It is worth noting that, although not degraded completely by protease, these hydrogels still contain hydrolytically labile thioether ester bonds that subject them to complete degradation over an extended period of time.
Figure 4. Proteolytic degradation of visible light-cured mixed-mode PEG hydrogel properties.
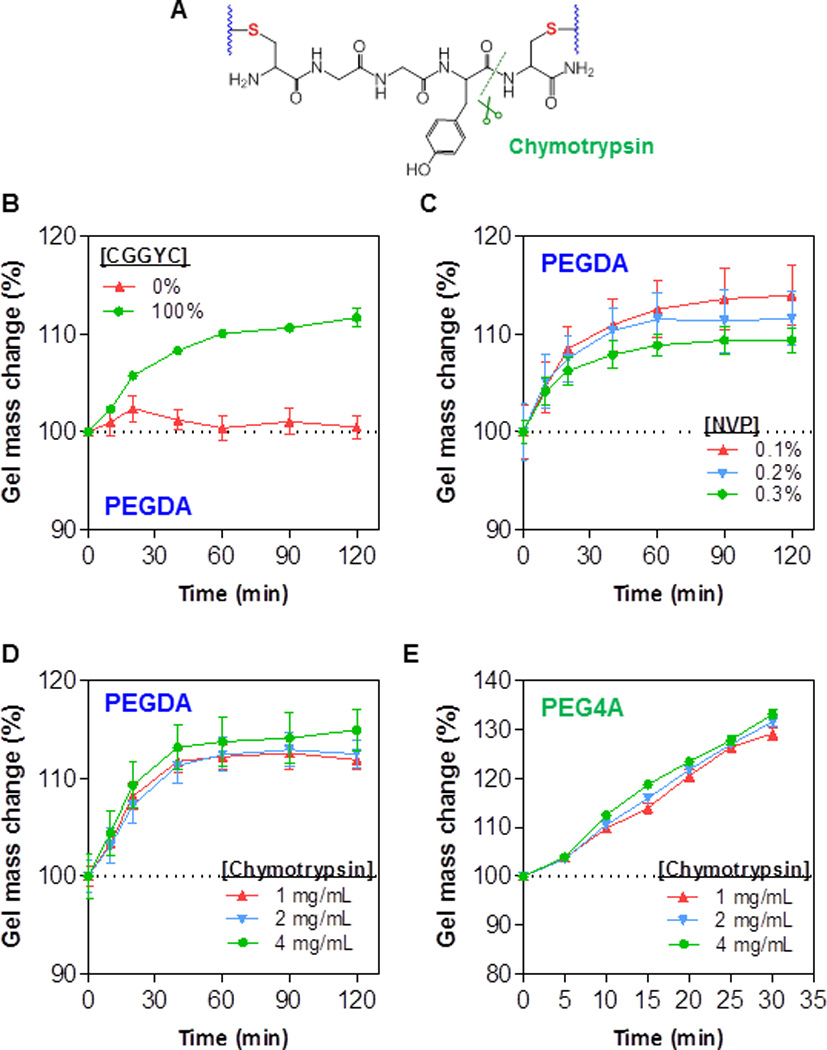
(A) Schematic of a protease sensitive peptide crosslinker (CGGYC) within a thiol-acrylate hydrogel. (B) Mass change of 10wt% PEGDA thiol-acrylate hydrogels induced by exogenous chymotrypsin (2mg/mL) treatment. Total di-thiol crosslinkers (CGGYC and DTT at indicated percentage) was 7.5mM. Other conditions: 0.1vol% NVP, 0.1mM eosin-Y, and 5-min visible light exposure. (C) Exogenous chymotrypsin (2mg/mL) induced mass change of 10wt% PEGDA thiol-acrylate hydrogels crosslinked with different NVP contents. Other conditions: 7.5mM CGGYC; 0.1mM eosin-Y, and 5-min visible light exposure. (D) Mass change of 10wt% PEGDA thiol-acrylate hydrogels induced by exogenous chymotrypsin treatment. Other conditions: 7.5mM CGGYC, 0.1vol% NVP, 0.1mM eosin-Y, and 5-min visible light exposure. (E) Mass change of 4wt% PEG4A thiol-acrylate hydrogels induced by exogenous chymotrypsin treatment. Other conditions: 4mM CGGYC, 0.1vol% NVP, 0.1mM eosin-Y, 5-min visible light exposure.
Degradation studies in Figs. 4B–4D were conducted using linear macromer PEGDA and the results showed that the proteolytic degradation was rather slow. The maximal increase in gel mass (caused by protease-mediated network cleavage and subsequent gel swelling) was about 15% in 120 minutes. This could be attributed to the higher amount of poly(acrylate-co-NVP) chains presented in the mixed-mode network as these gels were crosslinked by 10wt% of PEGDA3.4kDa (~59 mM acrylate). We also assessed the protease sensitivity of hydrogels crosslinked by 4wt% of 4-arm PEG-acrylate (PEG4A20kDa, Fig. 4E) and found that gel mass increased about 30% in 30 minutes, a rate much faster than that in PEGDA gels. This was presumably because of a much lower acrylate concentration in this gel formulation (i.e., 8mM). Note that even though the acrylate concentration in PEG4A was 7.4-fold lower than that in PEGDA hydrogel, the two set of gels actually had similar modulus (see Figs. 2B and 2C). These results support the notion that multi-arm PEG4A thiol-acrylate hydrogels may be a more attractive gel formulation for biomedical applications. Similar to results shown in Fig. 4D, the concentration of chymotrypsin did not affect the degradation rate in PEG4A hydrogels (Fig. 4E).
In situ encapsulation and growth of hepatocellular carcinoma cells in visible light cured thiol-acrylate hydrogels
Our previous works have revealed the high cytocompatibility of PEG hydrogels cured using visible light and NVP as the co-monomer.[16] To examine whether enhancing gelation efficiency using aforementioned adjustments (Figs. 1–4) affects cell viability, we encapsulated Huh7 cells in PEG4A hydrogels crosslinked with bis-cysteine peptides (CGGYC) using visible light initiated thiol-acrylate photopolymerization. Gel stiffness was adjusted using 0.1% or 0.2% of co-monomer NVP (Fig. 1E). Effects of integrin-binding motif were also examined by incorporating 1mM CGGYCRGDS motif in the gels. Hydrogel cytocompatibility was assessed by Live/Dead staining and confocal microscopy (Figs. 5A–C) and by AlamarBlue assay (Figs. 5D–E). As shown in Fig. 5A, the majority of encapsulated Huh7 cells were viable (stained green) on Day 1 (D1) post-encapsulation irrespective of hydrogel stiffness. Seven days (D7) post-encapsulation, Huh7 cells grew into clusters, and majority of the population were viable (Fig. 5B). Interestingly, less cells clusters were observed when integrin-binding RGDS motif was added to the gel formulation (Fig. 5C), suggesting the restriction of Huh7 cell growth by RGDS. Furthermore, more dead cells (stained red) were observed in softer hydrogels (0.1% NVP) than in stiffer hydrogels (0.2% NVP) when integrin-binding RGDS was present (Fig. 5C), indicating the combination of RGDS and softer environment inhibits Huh7 cell proliferation.
Figure 5. Effect of gel stiffness and integrin ligand on proliferation of encapsulated Huh7 cells.
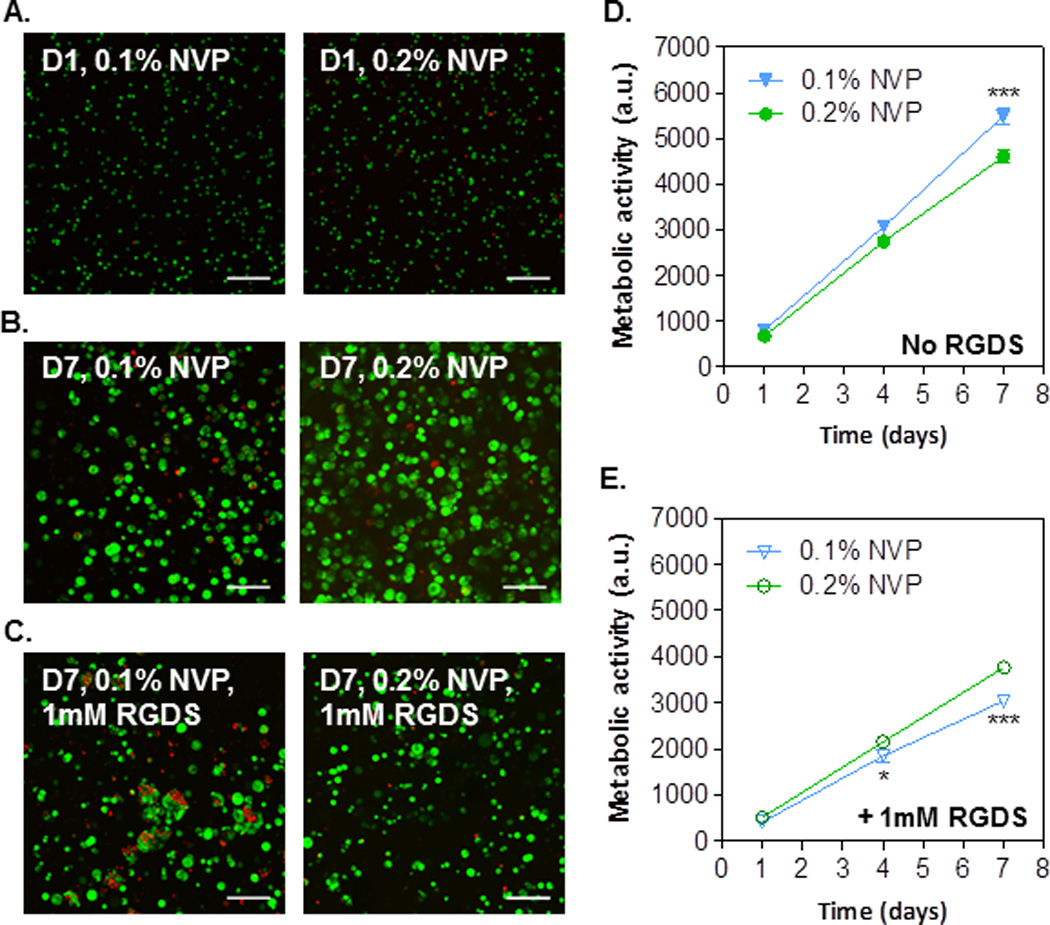
(A–C) Confocal z-stack images of Live/Dead stained Huh7 cells encapsulated in thiol-acrylate hydrogels with conditions labeled in the images. (D) Effect of gel stiffness (by controlling NVP content) on cell metabolic activity. (E) Effect of gel stiffness (by controlling NVP content) and RGDS (1mM) on cell metabolic activity. Gelation conditions: Without RGDS: 4wt% PEG4A and 4mM CGGYC. With RGDS: 4wt% PEG4A, 3mM CGGYC, and 1mM CGGYCRGDS. All gels were formed with 0.1mM eosin-Y and 5 min visible light exposure.
AlamarBlue assay quantitatively measures intracellular reducing state. The results also revealed Huh7 cell growth in the thiol-acrylate PEG4A hydrogels (Figs. 5D–E). Interestingly, Huh7 cells encapsulated in gels immobilized with RGDS exhibited significantly lower metabolic activity than without the presence of RGDS motif. This result coincides with our previous work on Huh7 cells encapsulated in UV light crosslinked thiol-norbornene hydrogels.[32] In gels without RGDS motif (Fig. 5D), Huh7 cell metabolic activity was significantly higher in softer hydrogels (i.e., 0.1% NVP) than in stiffer hydrogels (i.e., 0.2% NVP) on day 7 post-encapsulation, although no major difference was observed in Live/dead staining images between the two groups (Fig. 5B). Conversely, in gels with 1mM of RGDS motif, cells encapsulated in softer hydrogels showed significantly lower metabolic activities than in stiffer gels on day-4 and day-7 post-encapsulation (Fig. 5E). The result coincides with the live/dead staining where more dead cells were found in softer gel with 1mM of RGDS motif, suggesting that the presence of RGDS provides stimuli that inhibit Huh7 cell growth when in softer environment. While the exact roles of RGDS in molecular and cellular signaling in Huh7 cells are not the focus of the current study, the results presented in Fig. 5 provide an interesting angle for future investigation in the synergistic influence of matrix stiffness and integrin signaling on liver cancer cell fate.
YAP localization in encapsulated Huh7 cells
The HIPPO pathway is a key signaling cascade that controls organ size, cell proliferation and differentiation.[28, 33, 34] It was reported that YAP, the HIPPO pathway effector, localized in the cytoplasm when cells are cultured on top of softer environmental matrices; whereas when cells are seeded on stiffer surfaces, YAP trans-localize into the nucleus.[35] To examine whether 3D encapsulation by our hydrogels also affect YAP localization, we encapsulated GFP-YAP-overexpressing Huh7 cells in visible light-initiated PEG4A hydrogels, and the localization of GFP-YAP was visualized using confocal microscopy (Figs. 6A–B). As shown in the figures, no visible GFP-YAP1 or GFP-YAP2 proteins were observed in the nuclei. When cells were cultured on 2D glass surfaces, both GFP-YAP1 and GFP-YAP2 localized in the cytoplasm; conversely, cytoplasmic GFP-YAP1 and GFP-YAP2 were not detected by confocal microscopy when cells were encapsulated in 3D hydrogels. Taken together, these results revealed that cytoplasmic YAP proteins are down-regulated when cells are cultured in our 3D hydrogels, suggesting the activation of the HIPPO signaling pathway in inhibiting YAP activities in 3D culture environments.
Figure 6. YAP localization and inactivation in encapsulated hepatocellular carcinoma-derived Huh7 cells.
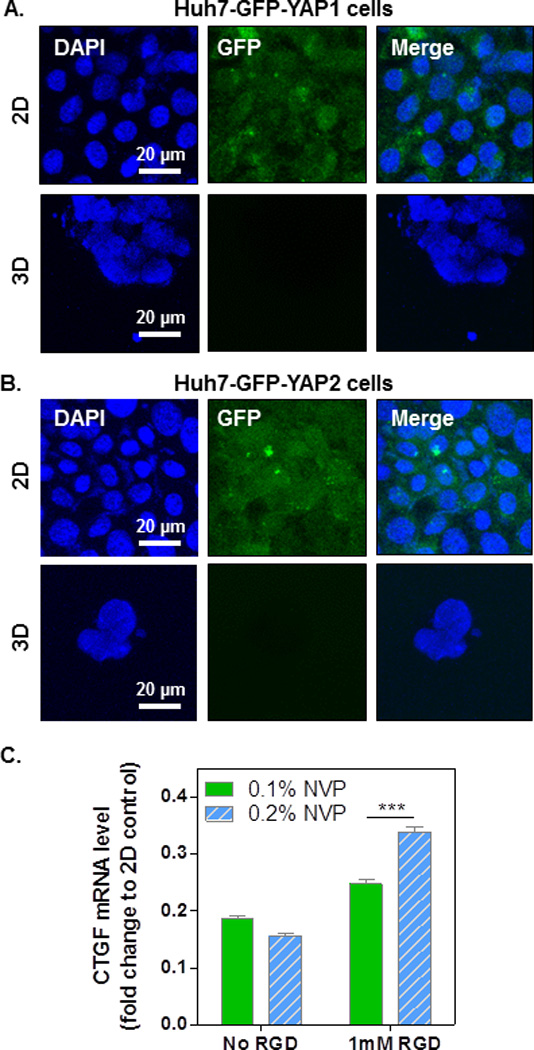
Huh7 overexpressing GFP-YAP1 (A) and GFP-YAP2 (B) were cultured in 2D or encapsulated in 4wt% PEG4A hydrogels (3D) crosslinked by 4mM of DTT with 0.2% of NVP. Cell-laden hydrogels or 2D cultured cells were collected on day 7 for visualizing the localization of GFP-YAP. Cell nuclei were counterstained with DAPI (blue). (C) Relative CTGF mRNA level in encapsulated Huh7 cells. The CTGF level in 2D counterpart was set as 1 for comparison.
To further investigate whether gene expression in Huh7 cells encapsulated in 3D hydrogels is affected by the down-regulation of cytoplasmic YAP, we isolated RNA from 2D-cultured and 3D-encapsulated cells and performed reverse transcription PCR and real-time PCR to analyze the expression of CTGF, a downstream gene up-regulated by the activation (nuclear localization) of YAP (Fig. 6C). When compared with the CTGF mRNA level (set as relative level 1) in 2D-cultured Huh7 cells, CTGF mRNA levels in 3D-encapsulated cells were significantly lower. When increasing gel stiffness using NVP, no significant difference was found between 0.1% NVP and 0.2% NVP (Fig. 6C, no RGDS). Although still significantly lower than the 2D counterparts, the presence of integrin-binging motif RGDS in 3D hydrogels enhanced the CTGF mRNA level in both softer (0.1% NVP, 1mM RGD) and stiffer (0.2% NVP, 1mM RGD) cell-laden hydrogels, suggesting a role of cell-matrix interaction in regulating YAP activation. The HIPPO pathway controls organ size and cell proliferation.[33, 34] Activated HIPPO pathway phosphorylates YAP, causing its cytoplasmic retention and degradation. It is possible that when Huh7 cells are encapsulated in 3D hydrogels, the HIPPO pathway is activated by the 3D micro-environmental stimuli. Consequently, the activated HIPPO signaling cascade leads to the phosphorylation, degradation and inhibition of cytoplasmic YAP. Although the role of integrin-binding motif RGD in the HIPPO pathway is not conclusive, our results showed that the presence of RGDS in stiffer hydrogels (i.e., 0.2% NVP) promoted CTGF expression (Fig. 6C), suggesting a positive influence of RGD-cell (integrin) interactions on YAP activation. As a result, higher degrees of cell survival and metabolic activity were observed (comparing soft and stiff hydrogel with RGDS, Fig. 5E). Future studies are required to further elucidate the delicate influence of matrix stiffness and integrin signaling on the HIPPO-dependent or independent pathways in controlling the effector YAP and subsequent cellular fate.
Conclusions
In summary, we have prepared visible light cured thiol-acrylate hydrogels with tunable degree of crosslinking and degradability. At fixed PEG macromer content, photoinitiator concentration, and light irradiation conditions, the crosslinking of thiol-acrylate PEG-peptide hydrogels can be adjusted by several parameters, including concentrations of co-monomer NVP and di-thiol peptide linkers. Gel crosslinking density is negatively affected by the inclusion of mono-thiol pendant ligand, but it can be increased by using higher co-monomer NVP content or by using multi-thiol peptide linker. The use of multi-arm PEG-acrylate (e.g., PEG4A) yields hydrogels with higher degree of crosslinking even at a lower PEG macromer concentration. Furthermore, these hydrogels were susceptible to both hydrolytic and proteolytic degradations. The tunable gel properties were explored for studying the influence of matrix stiffness and integrin activation (e.g., RGD ligand) on Huh7 cell proliferation and the activation of the HIPPO pathway. The inclusion of RGD ligand suppresses metabolic activity of Huh7 cells grown in 3D. Huh7 cells cultured in thiol-acrylate hydrogels exhibit lower YAP activation (i.e., lower CTGF mRNA expression). The inclusion of RGD motif partially rescues YAP activation only in stiffer hydrogels (i.e., gels crosslinked by 0.2% NVP), suggesting a synergistic effect of matrix stiffness and integrin activation on the activation of HIPPO pathway in hepatocellular carcinoma cells. This hydrogel crosslinking scheme may be useful in future study of HIPPO pathway activation in other tumor cells.
Acknowledgments
This work was supported in part by the National Cancer Institute of the NIH (R21CA188911) and a Research Support Funds Grant (RSFG) from the Office of the Vice Chancellor for Research (OVCR) at IUPUI. The authors acknowledge the Flow Cytometry Resource Facility of the Indiana University Simon Cancer Center which is partially funded by National Cancer Institute grant (P30CA082709).
Footnotes
The authors declare no conflict of interest.
References
- 1.DeForest CA, Anseth KS. Annu Rev Chem Biomol Eng. 2012;3:421–444. doi: 10.1146/annurev-chembioeng-062011-080945. [DOI] [PubMed] [Google Scholar]
- 2.Jabbari E. Curr Opin Biotechnol. 2011;22:655–660. doi: 10.1016/j.copbio.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 4.Lin CC. RSC Adv. 2015;5:39844–398583. doi: 10.1039/C5RA05734E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sokic S, Papavasiliou G. Tissue Eng Part A. 2012;18:2477–2486. doi: 10.1089/ten.tea.2012.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Kao WJ. Biomacromolecules. 2003;4:1055–1067. doi: 10.1021/bm034069l. [DOI] [PubMed] [Google Scholar]
- 7.Salinas CN, Anseth KS. Macromolecules. 2008;41:6019–6026. [Google Scholar]
- 8.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant SJ, Nuttelman CR, Anseth KS. J Biomater Sci Polym Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 10.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 11.Kizilel S, Scavone A, Liu XA, Nothias JM, Ostrega D, Witkowski P, Millis M. Tissue Eng A. 2010;16:2217–2228. doi: 10.1089/ten.TEA.2009.0640. [DOI] [PubMed] [Google Scholar]
- 12.Hern DL, Hubbell JA. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 13.Turturro MV, Sokic S, Larson JC, Papavasiliou G. Biomed Mater. 2013;8:025001. doi: 10.1088/1748-6041/8/2/025001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bahney CS, Lujan TJ, Hsu CW, Bottlang M, West JL, Johnstone B. Eur Cell Mater. 2011;22:43–55. doi: 10.22203/ecm.v022a04. discussion 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SH, Chu CC. Fibers Polym. 2009;10:14–20. [Google Scholar]
- 16.Hao Y, Lin CC. J Biomed Mater Res A. 2014;102:3813–3827. doi: 10.1002/jbm.a.35044. [DOI] [PubMed] [Google Scholar]
- 17.Hao Y, Shih H, Munoz Z, Kemp A, Lin CC. Acta Biomater. 2014;10:104–114. doi: 10.1016/j.actbio.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bragg JC, KHY, JYY, LKG, Lin CC. Journal of Applied Polymer Science. 2015 In press. [Google Scholar]
- 19.Reddy SK, Okay O, Bowman CN. Macromolecules. 2006;39:8832–8843. [Google Scholar]
- 20.Rydholm AE, Bowman CN, Anseth KS. Biomaterials. 2005;26:4495–4506. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Zustiak SP, Durbal R, Leach JB. Acta Biomater. 2010;6:3404–3414. doi: 10.1016/j.actbio.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zustiak SP, Leach JB. Biomacromolecules. 2010;11:1348–1357. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CC, Raza A, Shih H. Biomaterials. 2011;32:9685–9695. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shih H, Lin CC. Biomacromolecules. 2012;13:2003–2012. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shih H, Lin CC. Macromol Rapid Comm. 2013;34:269–273. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 26.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Mol Cell. 2003;11:11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 27.Ling H, Sylvestre JR, Jolicoeur P. Oncogene. 2010;29:4543–4554. doi: 10.1038/onc.2010.186. [DOI] [PubMed] [Google Scholar]
- 28.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 29.Elbert DL, Hubbell JA. Biomacromolecules. 2001;2:430–441. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- 30.Lutolf MP, Tirelli N, Cerritelli S, Cavalli L, Hubbell JA. Bioconjug Chem. 2001;12:1051–1056. doi: 10.1021/bc015519e. [DOI] [PubMed] [Google Scholar]
- 31.Metters A, Hubbell J. Biomacromolecules. 2005;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 32.Lin TY, Ki CS, Lin CC. Biomaterials. 2014;35:6898–6906. doi: 10.1016/j.biomaterials.2014.04.118. [DOI] [PubMed] [Google Scholar]
- 33.Zhao B, Tumaneng K, Guan KL. Nat Cell Biol. 2011;13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong D, Zhao Y, Men T, Teng CB. J Drug Target. 2015;23:125–133. doi: 10.3109/1061186X.2014.983522. [DOI] [PubMed] [Google Scholar]
- 35.Yang C, Tibbitt MW, Basta L, Anseth KS. Nat Mater. 2014;13:645–652. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]