

Abstract
Circadian variations in the corrected QT (QTc) interval have been documented in clinical trials. Animal models show circadian variations in expression of the cardiac ion channels that are necessary to maintain the heart's electrophysiological properties. Can these diurnal rhythms in QTc affect the ability of a drug to delay cardiac repolarization?
In the article “Levofloxacin‐Induced QTc Prolongation Depends on the Time of Drug Administration,” the authors find that drug‐induced QTc prolongation displays diurnal variation, with maximal drug effects occurring after a 2 pm dose administration and no drug effects occurring after a 6 am dose.1 This observation comes from a pharmacokinetic/pharmacodynamic (PK/PD) model using concentration and QT data collected from an uncontrolled, six‐period crossover study in 12 healthy subjects administered 1,000 mg of oral levofloxacin at different times of the day. This implies that thorough QT studies underestimate or completely miss drug‐induced QT prolongation because these studies typically dose subjects between 6 am and 10 am, when the sensitivity to drug‐induced QTc prolongation is at its lowest during the day. The authors hypothesize that diurnal fluctuations in serum potassium or cardiac ion channel expression could explain the time‐dependent slope estimate.1 The authors evaluated the influence of physiological fluctuations in serum potassium in their PK/PD model and found that it could not explain the difference in the sensitivity to levofloxacin over the course of the day. Thus, they are suggesting that the differences could be due to circadian variations in cardiac ion channel expression.
Circadian rhythms are 24‐hour cycles in biological systems that are critical for normal function of organisms, organs, tissues, or individual cells. In the cardiovascular system, changes over the day in blood pressure and heart rate are the results of circadian variations in the autonomic activity. The timing of cardiovascular adverse events shows a diurnal rhythm: for example, myocardial infarctions, sudden cardiac death, and ventricular tachyarrhythmias commonly occur in the early morning.2 The QT interval also displays diurnal variations, even when corrected for diurnal variations in heart rate, with lengthening of the QTc interval during the night.3 Some of these changes in QTc could be secondary to diurnal variations in cardiac gene expression, which has been documented in animal models. The expression of cardiac K+ and Na+ channels follows circadian variations and is important to preserve the heart's electrophysiological properties.4, 5, 6, 7 Circadian variations in the expression of genes that encode the transient outward current (Ito) and ultrarapid K+ (IKur) currents resulted in differences in the electrophysiological characteristics between light and dark periods in single myocytes and isolated‐perfused whole hearts.6 The role of clock‐dependent transcriptional factors such as Klf15 (Krüppel‐like factor 15) and BMAL1 (brain muscle arn‐like1) and their relation to the circadian variation of cardiac genes was further assessed in transgenic mice. In Klf15 and BMAL1 transgenic mice changes in circadian variation, or loss thereof, was observed for multiple genes responsible for encoding cardiac ion channels such as SCN5a, hERG, and Kcdn2, which are responsible for the INa, IKr, and Ito currents, respectively. In addition to causing a loss of circadian variation, an increased susceptibility to arrhythmias was also observed in Klf15 and BMAL1 transgenic mice.
Circadian variation in K+ channel density, however, seems unlikely to explain a greater than 10‐fold increase in the slope estimate of the levofloxacin‐QTc relationship over the day (from −0.04 ms per mg/L at 6 am to 1.7 ms per mg/L at 2 pm; table 3 in Kervezee et al.1). If normal physiological variations in cardiac ion channel density profoundly affect the sensitivity to drug‐induced QTc prolongation, then these effects would be expected with other drugs that selectively inhibit the hERG K+ channel. With drugs that directly inhibit the hERG K+ channel, the extent of the PD response varies according to the proportion of channels that are blocked, until saturation. Levofloxacin inhibits the hERG K+ channel, with an IC50 of 915 µM8 and is not thought to affect other cardiac ion channels. Dofetilide, on the other hand, is a much more potent inhibitor of hERG, with an IC50 of ∼1.5 nM,9 prolonging the QTc interval by as much as 80 ms, and without showing any evidence of saturation.10 In two recent double‐blind clinical studies, dofetilide or placebo was administered in the morning10 or after lunch9; there was no significant difference in the slope estimate of the studies (Figure 1). One would have expected a steeper slope following afternoon dosing compared to morning dosing if diurnal hERG K+ density affects drug‐induced QTc prolongation. A possible limitation of this counterexample is that the dosing and sampling timepoints were not optimal for observing potential diurnal differences in the slope estimate in either study. However, given the large variation in levofloxacin response at 6 am and 2 pm reported by the authors, one would expect dofetilide, an inhibitor that is over 4 orders of magnitude more potent than levofloxacin, to show enough variation to be evident even in these studies of morning vs. after lunch dosing.
Figure 1.
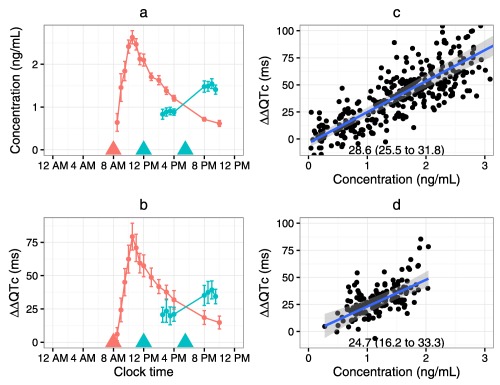
Comparison of the observed dofetilide plasma concentrations (a) and ΔΔQTc (b) in two clinical studies where dofetilide was either administered at 8 am in the morning (Ref. 10, red) or at noon and at dinner (Ref. 9, blue). The concentration‐QTc relationships for the morning administration (c) and noon/dinner administration (d) show no difference in the slope of the concentration‐QTc relationship (slope (95% confidence interval) in ms per ng/mL in each panel).
We suspect that the diurnal variation in the slope of the levofloxacin concentration‐QT relationship is an artifact of not collecting time‐matched drug‐free QT/RR pairs to inform the circadian rhythm model for QTc. These data could have been obtained by incorporating a placebo control arm. The diurnal model for QTc is based on QT/RR pairs collected predose at each dosing event (e.g., t = 2:00, 6:00, 10:00, 14:00, 18:00, and 22:00) over six separate occasions, each separated by 1 week. In Smetana et al., full 24‐hour ECG recordings were used to collect dense data on circadian variations in QTc, showing fairly flat QTc variability between approximately 9 am and 4 pm.3 This observed pattern in the QTc interval does not correspond to the authors' diurnal model using their more sparsely collected data, where the QTc appears to be still decreasing between 9 am and 3 pm. Thus, the authors' diurnal model shows more variability in QTc during daytime hours when the estimated slope of the concentration‐QTc relationship is at its largest. Any model misspecification of the diurnal model at unobserved timepoints could impact the slope estimate of the drug‐effect model. Further studies evaluating circadian variations in drug‐induced QTc prolongation should include a placebo‐control group to account for physiological variations in QTc.
The findings presented by the authors suggest a circadian pattern in the drug‐induced effects of levofloxacin, which they hypothesize could occur from circadian variations in cardiac ion channel expression. It is important to note that these interesting findings are based on a small study of one drug and without a placebo control. While studies in animal models do describe a circadian variation in ion channel expression that would certainly support the authors' hypothesis, only small changes in QTc have been observed in the absence of a disturbance of the circadian clock. Moreover, in a study with administration of a hERG K+ blocker around noon, the concentration‐QTc relationship was similar to that of a study with administration in the morning (Figure 1). It is possible, however, that if multiple cardiac ion channels are simultaneously and similarly downregulated, one might not expect to see a major change in the QTc interval, unless one channel is selectively blocked, such as with a potent hERG K+ inhibitor. Thus, further study is needed with appropriate controls and data collection to enhance our understanding of the impact of circadian variation in ion channel density on drug‐induced QTc prolongation.
DISCLAIMER
This article reflects the views of the authors and should not be construed to represent the FDA's views or policies.
References
- 1. Kervezee, L. et al Levofloxacin‐induced QTc prolongation depends on the time of drug administration. Clin. Pharmacol. Ther. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martino, T.A. & Sole, M.J. Molecular time: An often overlooked dimension to cardiovascular disease. Circ. Res. 105, 1047–1061 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Smetana, P. , Batchvarov, V.N. , Hnatkova, K. , Camm, A.J. & Malik, M. Sex differences in repolarization homogeneity and its circadian pattern. Am. J. Physiol. Heart Circ. Physiol. 282, H1889–1897 (2002). [DOI] [PubMed] [Google Scholar]
- 4. Jeyaraj, D. et al Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 483, 96–99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schroder, E.A. et al The cardiomyocyte molecular clock regulates the circadian expression of Kcnh2 and contributes to ventricular repolarization. Heart Rhythm 12, 1306–1314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamashita, T. et al Circadian variation of cardiac K+ channel gene expression. Circulation 107, 1917–1922 (2003). [DOI] [PubMed] [Google Scholar]
- 7. Schroder, E.A. et al The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am . J. Physiol. Cell. Phys. 304, C954–C965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang, J.S. , Wang, L. , Chen, X.L. , Triggle, D.J. & Rampe D. Interactions of a series of fluoroquinolone antibacterial drugs with the human cardiac K+ channel HERG. Mol. Pharmacol. 59, 122–126 (2001). [DOI] [PubMed] [Google Scholar]
- 9. Johannesen, L. et al Late sodium current block for drug‐induced long QT syndrome: results from a prospective clinical trial. Clin. Pharmacol. Ther. 99, 214–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johannesen, L. et al Differentiating drug‐induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Pharmacol. Ther. 96, 549–558 (2014). [DOI] [PubMed] [Google Scholar]