

Abstract
A practical and convenient procedure for the nucleophilic aromatic substitution of aryl fluorides and chlorides with dimethylamine was developed using a hydroxide assisted, thermal decomposition of N,N-dimethylforamide. These conditions are tolerant of nitro, nitrile, aldehyde, ketone, and amide groups but will undergo acyl substitution to form amides for methyl esters and acyl chlorides. Isolated yields of the products range from 44 – 98%, with the majority being greater than 70% for seventeen examples.
Keywords: Nucleophilic Aromatic Substitution, dimethylamine, aryl halides, aniline derivatives, DMF decomposition
INTRODUCTION
Aromatic compounds substituted with a dimethylamino group compose an important class of molecules that possess a range of biological activities including central nervous system stimulant,[1] antimicrobial,[2] anti-cancer,[3,4] anti-HIV,[5] AT2 receptor antagonist,[6–8] progesterone receptor modulator,[9,10] and rho kinase antagonists.[11] Additionally, numerous aromatic natural products are substituted with a dimethylamino group with some notable examples being Tigecycline,[2] Minocycline,[12] Orthoformimycin,[13] and Dendrodione.[14] While installation of the dimethylamino group can be carried out by nucleophilic aromatic substitution (SNAr), the dimethylamine nucleophile often requires high temperatures and reaction times[15–17] to install, which can be challenging when using premade solutions given dimethylamine’s volatility. Sealed tubes can be used to contain the dimethyamine gas, but the buildup of pressure can create a potentially dangerous environment and the scalability of the reaction is limited. To circumvent these issues, several procedures have been developed which involve the thermal decomposition of N,N-dimethylamine (DMF) to slowly and safely liberate dimethylamine.[18] To avoid high temperature reaction conditions, the decomposition is generally catalyzed by acidic[19,20] or basic conditions,[21–26] however, the scope of these reactions are limited, and temperatures greater than 100 °C are still required or specialized microwave reactors are utilized to obtain good yields. Herein, we report a general procedure for the SNAr of halides with dimethylamine using hydroxide assisted decomposition of DMF. This procedure can be performed using common laboratory equipment, is safe and scalable, and can be run using only DMF, and aqueous hydroxide solution.
RESULTS AND DISCUSSION
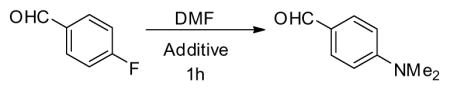
To better understand the scope of acids and bases that can facilitate DMF decomposition and the desired SNAr reaction, a range of additives were tested to estimate relative rates of conversion of 4-fluorobenzaldehyde to 4-(dimethylamino)benzaldehyde at various temperatures for a reaction time of an hour (Table 1). Trifluoroacetic acid (TFA, entry 1) did not demonstrate any detectable conversion and similarly aqueous ammonia and sodium carbonate solutions (entries 2 and 3) produced very little conversion at 95 °C. Using aqueous solutions of potassium hydroxide as an additive, however, generated considerably more substitution product (entry 4), which improved by increasing the reaction temperature (entries 5 and 6). A relatively high concentration of potassium hydroxide is required for a timely reaction as decreasing the concentration of hydroxide resulted in lower conversion (entry 7). Interestingly, no hydroxide substituted side products were detected by HPLC analysis. The conversion was optimized further by adding the potassium hydroxide solution in two aliquots (entry 8), resulting in a complete conversion. The portion-wise hydroxide addition produced two DMF decomposition cycles providing a more steady concentration of dimethylamine, which improved the overall yield.
Table 1.
Optimization of Decomposition of DMF-SNAr Conditions for 4-(dimethylamino)-benzaldehyde.
| |||
|---|---|---|---|
| Entry | Additivea | Temp (°C) | Conversion (%)b |
| 1 | TFA (0.5 M) | 95c | 0 |
| 2 | NH3 (aq., 7.3 M) | 95c | < 5 |
| 3 | Na2CO3 (aq., 1.5 M) | 95c | < 5 |
| 4 | KOH (aq., 5 M) | 65 | 59 |
| 5 | KOH (aq., 5 M) | 80 | 68 |
| 6 | KOH (aq., 5 M) | 95 | 78 |
| 7 | KOH (aq., 0.5 M) | 95 | 23 |
| 8 | KOH (aq., 5 M) | 95 | 100d |
Final concentration after mixing with equal volumes of DMF.
Conversion determined by HPLC.
Reaction temperatures were also examined at 25 °C, 45 °C, and 65 °C but a conversion was not detected.
KOH solution was added in two separate portions separated by 0.5 h.
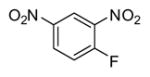
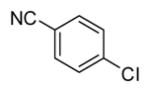
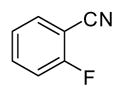
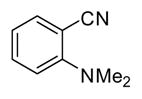
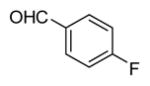
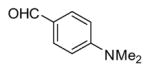
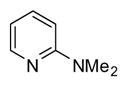
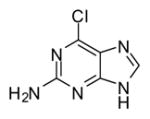
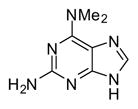
In applying the optimized reaction conditions to a range of substrates, typical SNAr trends were observed (Table 2). Fluorine served as the best leaving group and underwent conversion notably faster than chlorine. Substitution of bromine does not occur readily and is relatively inert using these reaction conditions (a conversion was not detected after a 72 h reaction time). The SNAr reaction occurred most rapidly for 1-fluoro-2,4-dinitrobenzene (entry 1), in which the reaction was completed within three minutes of introducing the substrate to provide the product as pure yellow crystals in 81% yield. Similarly, p-fluoronitrobenzene and p-chloronitrobenzene (entries 2 and 3) reacted readily in high yields generating pure crystals that were washed and filtered directly from the reaction mixture. Chlorinated and fluorinated benzonitriles (entries 4–6) possessed similar reaction times as the corresponding nitrobenzene examples, however, the yields for all of the substrates were lower. Fluorinated aldehydes (entries 7–9) reacted smoothly in moderate to excellent yields. Entry 8 illustrates a disubstitution reaction, in which dimethylamine replaces fluorine atoms situated at positions ortho and para to the nitrile electron withdrawing group. The addition of the first equivalent of dimethylamine, however, substantially decreases the reaction rate of the second addition, and therefore required 24 h for the reaction to complete. In exploiting the unreactive nature of bromine in this reaction, it is possible to replace a fluorine atom and retain a bromine atom in very good yield as demonstrated in entry 9. Ortho and para substituted acetophenones and benzamides (entries 10 – 12) generally result in good to excellent yields in 1–6 h reaction times, with the exception of para-chloroacetophenone (entry 11) which required a reaction time of 24 h to obtain a 45% yield of dimethyl-substituted acetophenone. Using these conditions, it is also possible to perform an acyl substitution in addition to the SNAr reaction, as demonstrated in entries 13 and 14. However, the acyl chloride (entry 13) reaction completed in significantly less time and in substantially higher yield than the corresponding methyl ester (entry 14). Substituted heterocyclic compounds (entries 15–17) also readily undergo SNAr using the optimized conditions to provide dimethylamino substituted products in good yields. Pyridine analogs, 2-fluoropyridine and 2-chloro-3-nitropyridine reacted in the highest yields of all the substrates tested in the study (95 and 98% respectively) with reaction times of an hour or less. Purine derivative, 2-amino-6-chloropurine, also smoothly converted the amino substituted product in an 81% yield. Interestingly, the purine product was crystallized directly from the reaction mixture also providing a simplified method of purification than previously reported for this compound.26
Table 2.
Dimethylamine SNAr of Aryl Fluorides/Chlorides Using the Optimized DMF-KOH Conditions.
| Entry | Reactant | Product | Time (h) | Yield (%)a |
|---|---|---|---|---|
| 1 |
|
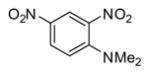
|
0.05 | 81 |
| 2 |
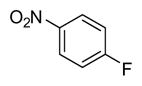
|
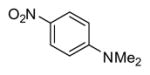
|
1 | 90 |
| 3 |
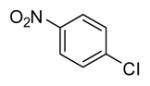
|
|
2 | 93 |
| 4 |
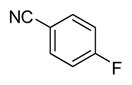
|
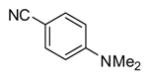
|
1 | 78 |
| 5 |
|
|
6 | 72 |
| 6 |
|
|
1 | 70 |
| 7 |
|
|
6 | 89 |
| 8 |
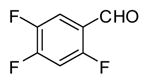
|
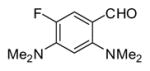
|
24 | 64 |
| 9 |
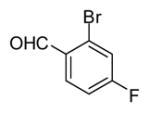
|
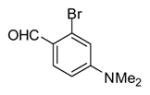
|
0.6 | 88 |
| 10 |
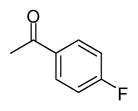
|
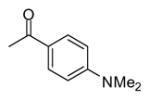
|
6 | 91 |
| 11 |
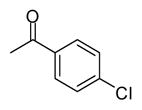
|
|
24 | 45 |
| 12 |
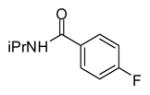
|
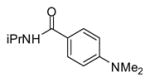
|
1 | 70 |
| 13 |
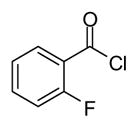
|
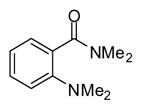
|
1 | 76 |
| 14 |
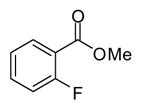
|
|
5 | 44 |
| 15 |
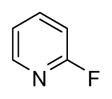
|
|
1 | 95 |
| 16 |
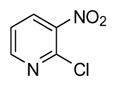
|
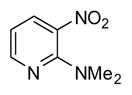
|
0.5 | 98 |
| 17 |
|
|
2 | 81 |
Isolated Yields after purification.
CONCLUSION
In summary, a method for installing dimethylamine groups via SNAr of aryl fluorides and chlorides using the thermal decomposition of DMF in the presence of KOH is discribed. This method uses relatively mild reaction temperatures (95 °C), is tolerant of nitro, nitrile, aldehyde, ketone, and amide functional groups, and the reactions can be scaled up to gram quantities without a significant change in yield. The reaction conditions can also be used in a chemoselective fashion to substitute fluorine in the presence and without substution of bromine groups. Multiple substitutions on a single molecule can also occur in the same reaction pot.
EXPERIMENTAL
All of the solvents and chemicals used were purchased from commercial suppliers and were used without any further purification. Thin layer chromatography (TLC) analysis was performed using P254 silica gel 60 aluminum backed plates and column chromatography purification was accomplished using BDH silica gel (230 – 400 mesh). Melting point ranges were acquired with a Bibby scientific SMP10 melting point apparatus and are uncorrected. FT-IR measurements were obtained using a Shimadzu GladiATR 10 Single Reflection ATR accessory and UV-Vis measurements were obtained by using a Beckman Coulter DU 800 Spectrophotometer. HPLC analysis was performed using a Beckman Coulter System Gold unit equipped with UV-Vis detection. 1H (400 MHz) NMR spectra was acquired on Varian VNMRS-400 instrument using approximately 0.3 M solutions. High resolution electrospray mass spectrum (ESI HRMS) analysis was conducted at the University of California, Irvine.
General Procedure
A mixture of DMF (2.00 mL, 25.8 mmol) and 10 M KOH (0.500 mL, 5.00 mmol) were heated at reflux for 5 min. The corresponding aryl halide (1.00 mmol) was added and the resulting mixture was heated for 0.5 h at 95 °C. The mixture was treated with additional 10 M KOH (0.500 mL, 5.00 mmol) and heated for an additional 0.5 h. The reaction was monitored by TLC and additional portions 10M KOH (0.500 mL, 5.00 mmol) were added in 0.5 h intervals until completion was evident. The reaction mixture was then cooled, diluted with water and vacuum filtered (for solid products) or extracted (for product oils) with Et2O (3x). For reaction products that were extracted, the organic fractions were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the final product.
Selected Spectral Data
2,4-bis(dimethylamino)-5-fluorobenzaldehyde (Entry 8): 1H NMR (CDCl3, 400 MHz) δ 9.93 (d, J = 2.8 Hz, 1H), 7.27 (d, J = 14.5 Hz, 1H), 6.18 (d, J = 7.7 Hz, 1H), 2.92 (d, J = 1.9 Hz, 6H), 2.76 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 188.24 (d, J = 1.8 Hz), 154.34, 148.37 (d, J = 239.7 Hz), 145.43 (d, J = 9.2 Hz), 118.26 (d, J = 5.3 Hz), 116.53 (d, J = 22.5 Hz), 104.97 (d, J = 3.7 Hz), 41.87 (d, J = 6.1 Hz); IR (thin film) 3248, 3069, 2972, 2933, 2876, 1628, 1614, 1547, 1445, 1219, 758 cm−1; HRMS (ESI) m/z 233.1066 (M+Na+, C11H15FN2ONa requires 233.1061).
2-bromo-4-(dimethylamino) benzaldehyde (Entry 9): mp = 87–90 °C; 1H NMR (CDCl3, 400 MHz) δ 10.08 (d, J = 0.8 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 6.79 (d, J = 2.5 Hz, 1H), 6.63 (ddd, J = 8.9, 2.5, 0.8 Hz, 1H), 3.07 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 190.23, 154.47, 131.05, 129.70, 122.06, 114.84, 110.58, 77.32, 77.00, 76.69, 40.08; IR (thin film) 2893, 2833, 2755, 1659, 1587, 1520, 1327, 1258, 804 cm−1; HRMS (ESI) m/z 249.9839 (M+Na+, C9H1079BrNONa requires 249.9843).
Supplementary Material
Acknowledgments
We would like to thank the National Center for Research Resources (5P20RR016464-11) and the National Institute of General Medical Sciences (8 P20 GM103440-11) from the National Institutes of Health and the National Science Foundation (IIA-1301726) for their generous funding, as well as Mr. Jungjae Koh for his help in acquiring 400 MHz 1H NMR spectra and Dr. John Greaves for aquiring ESI HRMS.
Footnotes
Full experimental details and 1H NMR, 13C NMR, and HRMS for all novel compounds can be found in the Supplementary Content section which can be accessed through the articles web page.
References
- 1.Hansch C, Björkroth JP, Leo A. J Pharm Sci. 1987;76:663–687. doi: 10.1002/jps.2600760902. [DOI] [PubMed] [Google Scholar]
- 2.Hung YP, Lee JC, Lin HJ. Antibiotics. 2015;4:216–229. doi: 10.3390/antibiotics4020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueller S, Rodriguez R. Expert Rev Clin Pharmacol. 2014;7:663–679. doi: 10.1586/17512433.2014.945909. [DOI] [PubMed] [Google Scholar]
- 4.Neidle S. The discovery of G-quadruplex telomere targeting drugs. Royal Society of Chemistry; London: 2006. [Google Scholar]
- 5.Perrone R, Butovskaya E, Daelemans D, Palu G, Pannecouque C, Richter SN. J Antimicrob Chemother. 2014;69:3248–3258. doi: 10.1093/jac/dku280. [DOI] [PubMed] [Google Scholar]
- 6.Tamura M, Takagi T, Howard EF. J Hypertens. 2000;18:1239–1246. doi: 10.1097/00004872-200018090-00010. [DOI] [PubMed] [Google Scholar]
- 7.Boulay G, Servant G, Luong TT, Escher E, Guillemette G. Mol Pharmacol. 1992;41:809–815. [PubMed] [Google Scholar]
- 8.Blankley CJ, Hodges JC, Klutchko SR. J Med Chem. 1991;34:3248–3260. doi: 10.1021/jm00115a014. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Wang J, Shao J. Med Res Rev. 2014;34:979–1000. doi: 10.1002/med.21311. [DOI] [PubMed] [Google Scholar]
- 10.Bouchard P, Chabbert-Buffet N, Fauser BCJM. Fertil Steril. 2011;96:1175–1189. doi: 10.1016/j.fertnstert.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 11.Henderson AJ, Hadden M, Guo C. Bioorg Med Chem Lett. 2010;20:1137–1140. doi: 10.1016/j.bmcl.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Sharma V. Elixir Int J. 2015:29716–29719. [Google Scholar]
- 13.Maffioli SI, Fabbretti A, Brandi L. ACS Chem Biol. 2013;8:1939–1946. doi: 10.1021/cb4004095. [DOI] [PubMed] [Google Scholar]
- 14.Heitz S, Durgeat M, Guyot M, Brassy C, Bachet B. Tetrahedron Lett. 1980;21:1457–1458. [Google Scholar]
- 15.Alihodzic S, Mutak S, Pavlovic D. Preparation of azithromycin and erythromycin macrolides substituted at the 4′-position as antibacterial agents. 2005108412. US Patent. 2005
- 16.Rewcastle GW, Atwell GJ, Zhuang L, Baguley BC, Denny WA. J Med Chem. 1991;34:217–222. doi: 10.1021/jm00105a034. [DOI] [PubMed] [Google Scholar]
- 17.Urner RS, Bergstrom FW. J Am Chem Soc. 1945;67:2108–2110. [Google Scholar]
- 18.Wan Y, Alterman M, Larhed M, Hallberg A. J Org Chem. 2002;67:6232–6235. doi: 10.1021/jo025965a. [DOI] [PubMed] [Google Scholar]
- 19.Pechkovskii VV, Cherches GK, Barannikova TI, Sorin VS, Korablina AA. Zh Neorg Khim. 1988;33:2949–2951. [Google Scholar]
- 20.Coppinger GM. J Am Chem Soc. 1954;76:1372–1373. [Google Scholar]
- 21.Seong CM, Park WK, Park CM, Kong JY, Park NS. Bioorg Med Chem Lett. 2008;18:738–743. doi: 10.1016/j.bmcl.2007.11.045. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal A, Chauhan P. Synth Commun. 2004;34:2925–2930. [Google Scholar]
- 23.Liu X, Wang Y, Gao Y. A process for preparing N,N-dimethylamines using DMF as starting material. 103224438. US patent. 2013
- 24.Cho YH, Park JC. Tetrahedron Lett. 1997;38:8331–8334. [Google Scholar]
- 25.Petersen TP, Larsen AF, Ritzén A, Ulven T. J Org Chem. 2013;78:4190–4195. doi: 10.1021/jo400390t. [DOI] [PubMed] [Google Scholar]
- 26.Cechova L, Jansa P, Sala M, Dracinsky M, Holy A, Janeba Z. Tetrahedron. 2011;67:866–871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.