

Abstract
Protein crystallization is a major bottleneck of structure determination by X-ray crystallography, hampering the process by years in some cases. Numerous matrix screening trials using significant amounts of protein are often applied, while a systematic approach with phase diagram determination is prohibited for many proteins that can only be expressed in small amounts. Here, we demonstrate a microfluidic nanowell device implementing protein crystallization and phase diagram screening using nanoscale volumes of protein solution per trial. The device is made with cost-effective materials and is completely automated for efficient and economical experimentation. In the developed device, 170 trials can be realized with unique concentrations of protein and precipitant established by gradient generation and isolated by elastomeric valving for crystallization incubation. Moreover, this device can be further downscaled to smaller nanowell volumes and larger scale integration. The device was calibrated using a fluorescent dye and compared to a numerical model where concentrations of each trial can be quantified to establish crystallization phase diagrams. Using this device, we successfully crystallized lysozyme and C-phycocyanin, as visualized by compatible crystal imaging techniques such as bright-field microscopy, UV fluorescence, and second-order nonlinear imaging of chiral crystals. Concentrations yielding observed crystal formation were quantified and used to determine regions of the crystallization phase space for both proteins. Low sample consumption and compatibility with a variety of proteins and imaging techniques make this device a powerful tool for systematic crystallization studies.
Graphical abstract
INTRODUCTION
X-ray crystallography is the leading method to determine the structure of proteins and contributes to the vast majority of entries submitted to the Protein Data Bank. Protein crystals are needed because X-ray sources do not yet provide enough brilliance to produce detectable diffraction from a single protein molecule, and the success of X-ray crystallographic studies depends on the quality of the crystals. Traditional X-ray sources (e.g., synchrotrons) have made great progress in beam flux, where data can be collected on crystals as small as ~20 μm at microfocus beamlines under cryogenic conditions for protein crystals with small unit cells. However, X-ray damage is severe and limits the minimal size of crystals from which useful X-ray diffraction data can be collected.1 Advancements in X-ray accelerator technology and the advent of X-ray free-electron lasers (XFELs) are circumventing the X-ray damage problem with ultrashort pulse durations, allowing the study of complex protein structures that were previously unable to be determined by X-ray crystallography.2–5 XFELs produce extremely short (femtosecond) and highly brilliant X-ray pulses capable of outrunning X-ray damage processes. Consequently, diffraction can be attained from crystals as small as 200 nm.3 Small crystals (<5 μm) can more readily grow from difficult-to-crystallize proteins like membrane proteins or large complexes, which has opened new doors in structural biology.6–11
However, in all types of crystallography, crystals are still required and are made via a crystallization process where many conditions in a multiple-phase space are screened in order to determine favorable conditions where crystals grow with desired characteristics.12–15 In many cases, this can take years to optimize, making crystallization the major bottleneck of structural biology. Improved methodologies to expedite the process using limited amounts of precious sample are necessary, for example, to create large crystals for traditional X-ray sources or, more recently, to select for nano- and microcrystal growth for XFELs where very few reliable and widely applicable techniques have been developed.16 In crystallization screening, a large variety of parameters can be altered, including, but not limited to, concentration, temperature, pH, and the composition of a precipitant that is introduced to the protein suspension. The latter encumbers an even further complexity, as there are a plethora of precipitant buffer types and additives (e.g., salts, polyethylene glycols, detergents, etc.) that can be screened at different concentrations and then added to a wide range of protein concentrations to “hit” a favorable condition for crystallization to occur.
Screening can be performed with a variety of techniques, the most common for protein crystallization being vapor diffusion, while other methods like free interface diffusion, batch, and dialysis are less frequently used. Each method provides a unique pathway through the crystallization phase diagram toward crystal-forming conditions. For vapor diffusion, a “hanging” or “sitting” drop approach is generally employed in which a precipitant solution is placed in a large well and a droplet of protein suspension is placed inverted above or in an adjacent well, respectively, within the same chamber sealed air-tight.17,18 Over time, the system equilibrates due to solvent vaporization and diffusion, in which the concentrations of precipitant and protein in the droplet increase until the molar concentration in the droplet reaches the concentration in the large well. The phase diagram is ideally traversed as solute concentrations change until the nucleation zone is reached. In free interface diffusion, the highest number of nuclei (or often precipitate) are formed directly at the liquid–liquid interface where the protein and precipitant meet in a capillary. With the progression of time, the solutions mix by diffusion, leading to a concentration gradient and, after full equilibrium, reach a final stage where the protein and precipitant concentrations are equalized.19–22 Both of these diffusion-based methods have been successfully used for many proteins, yet the exact conditions under which initial nucleation and crystal growth occur cannot be determined. Additionally, vapor diffusion methods cannot be scaled up easily, as the nucleation rate depends on the high surface-to-volume ratio. Furthermore, collecting crystals from vapor diffusion experiments is very time-consuming for high-yield crystal production, which is required for serial crystallography methods such as serial femtosecond crystallography (SFX) at an XFEL where a continuous stream of concentrated protein nano-/microcrystals is delivered to the beam at room temperature in a liquid injector.23,24
The batch crystallization method, where protein and precipitant solution are directly mixed together in a desired ratio with well-defined initial concentrations, is one of the more scalable crystallization methods. However, it ideally requires some knowledge of the phase diagram so that the desired rates of nucleation and crystal growth are achieved. Originally, batch methods were employed on the macroscale using common laboratory glassware, but as technology has progressed, microbatch has become a critical method to reduce precious protein sample consumption and more rapidly parallelize crystallization screens by mixing microliter droplets of various protein samples and precipitants in large arrays of wells.25–27 The microbatch setup process has also been automated, and sample consumption has been reduced further to the sub-microliter regime using advanced robot delivery systems.27,28 However, these systems are generally very expensive, thereby limiting their use for a large number of laboratories.
Microfluidic platforms, which can be quickly fabricated with low-cost materials and accessible instrumentation, have emerged as an economical and versatile way to reduce sample consumption. Microfluidic devices have also been employed for numerous biological applications, such as diagnostic sensing and assays,29–33 separations,34–36 and protein crystallization,37–39 using the free interface diffusion,40–42 vapor diffusion,43,44 and microbatch43,45,46 approaches. The diffusion-based approaches are generally hampered by limited crystal production and quantitative analysis of crystallization conditions, while the microbatch approaches rely on the generation of droplets or the need for moving parts and complex fabrication procedures. Here, we propose an easy-to-fabricate microfluidic device implementing a batch-type crystallization approach in an automated fashion without sample interference or the need for oils, using only ~25 nL of protein sample per crystallization “well”, which is orders of magnitude below the microliter volumes required for traditional microbatch-under-oil and on par with advanced robot systems. Our method is based on a gradient generator system developed previously47–49 to create many protein and precipitant screening conditions by splitting and recombining input solutions through an array of channels. The device is fabricated in polydimethylsiloxane (PDMS), in which a previously developed “doormat” valve50–52 can be implemented, allowing each crystallization nanowell to be isolated from other nanowells. Furthermore, PDMS is compatible with common protein crystallization precipitants and organic solvents used for screening. Using this device, we demonstrate the capability of crystallizing proteins in these nanowells with the ability to image crystals using bright-field microscopy, UV fluorescence, and second-order nonlinear imaging of chiral crystals (SONICC). These regions are then analyzed quantitatively to determine conditions where crystal formation was observed to develop portions of a phase diagram. Combined, this device serves as a high-throughput, nanoscale platform, eventually allowing complex multi-dimensional protein crystallization screens in a cost-effective and adaptable package to serve a broad range of crystallography applications.
EXPERIMENTAL SECTION
Materials and Chemicals
Fluorescein sodium salt, lysozyme from chicken egg white, sodium acetate, sodium chloride, polyethylene glycol (PEG 3350), and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were all purchased from Sigma-Aldrich, USA. Magnesium chloride was purchased from Alfa Aesar, USA. PDMS and its curing agent (Sylgard 184 elastomer kit) were purchased from Dow Corning, USA. Tygon tubing, 1/8 in. o.d., was purchased from Cole Parmer, USA. PEEK tubing, 1/32 in. o.d., and NanoPort Assemblies were purchased from IDEX Health and Science, USA.
Microfluidic Chip Fabrication
The fluid (gradient generator) and control (valve) layer channels were designed using AutoCAD 2015 (Autodesk, USA) and sent for film mask printing (CAD/Art Services Inc., USA). The film mask was used to perform standard photolithography53 to pattern both designs with SU-8 photoresist onto a silicon wafer. Standard soft lithography54 was then performed to impress the negative relief of the structures into PDMS.34 To fabricate the three-layer “doormat” valve system, a thin PDMS membrane (~25 μm thickness) was spin-coated onto a silicon wafer and heated to 70 °C for 1 h to cure. An access reservoir was punched into the control channel PDMS layer, which was then oxygen plasma treated together with the thin layer. These two layers were then brought into contact with each other and heated at 70 °C for 30 min to bond them irreversibly. The control layer and thin membrane were peeled off the wafer together, and the fluid layer (with reservoirs punched into fluid inlet/outlet channels and at the location of the control layer access port) was aligned to the control layer using a stereo microscope (SZ51, Olympus, USA). NanoPort Assemblies were then adhered to the fluid inlets, and a barbed tube fitting was adhered to the control layer reservoir for connection to respective pressure pumps. After an initial investment to fabricate the master wafer and purchase reusable tubing and fittings (~50–100 USD), each chip can be manufactured for ~2 USD.
Crystallization Experiments
The assembled chip was connected to two pressure pumps: (i) a positive pressure pump (MCFS-EZ, Fluigent, France) connected to a sample reservoir, which was connected to the fluid layer inlets via PEEK tubing to the NanoPort Assemblies and (ii) a negative pressure pump (MCFS-EZ, Fluigent, France) connected to the control layer reservoir fitting directly via Tygon tubing. First, negative pressure (−400 mbar and below) was applied to the control layer to deflect the membranes and open the valves. Once all valves were opened, crystallization precipitant solution (for lysozyme: 0.1 M sodium acetate at pH 4.6 containing 2.5 M NaCl; for C-phycocyanin: 75 mM HEPES at pH 7.0 containing 17.5% w/v PEG 3350 and 20 mM MgCl2) and water were pumped into separate fluid layer inlets using positive pressure, generally between 15 and 30 mbar. This wash step was administered with ~3 times the total volume until all channels were completely filled, and then water was switched with protein sample (lysozyme, 50 mg/mL; C-phycocyanin, 25 mg/mL) and positive pressure was reapplied. In the case of the fluorescein dye calibration, water was added to both inlets for washing, and then one inlet was pumped with 1 mM fluorescein solution. The filling steps were monitored using a CCD camera (iXon, Andor, USA) controlled by MicroManager software (ver. 2.46, UCSF, USA) connected to a bright-field/fluorescence microscope (IX71, Olympus, USA) equipped with appropriate filter sets.
After the device was filled with sample and water or precipitant, all pressure sources were removed from the chip, and a slight positive pressure (100 mbar) was applied to the control layer to ensure the valve membrane rapidly returned to its normally closed state. For crystallization incubation, pressure on the control layer was maintained with a slightly pressurized syringe, and the entire system was placed in a humidity chamber set at 100% humidity to prevent solvent evaporation out of the PDMS. Crystallization was allowed to occur for 2 days, followed by imaging of the entire chip using bright-field microscopy, UV fluorescence, and second-order nonlinear imaging of chiral crystals (SONICC; Formulatrix, USA). For the calibration experiments with fluorescein, the chip was allowed to rest for 1 h after filling to ensure each well had mixed completely, and fluorescence intensities of each well on the entire chip were recorded. This was repeated four times with the same chip, and the images were analyzed for background-corrected intensities using ImageJ software (ver. 1.49, NIH, USA).
Theoretical Modeling
COMSOL Multiphysics 5.1 finite element analysis software was used for numerical modeling of the microfluidic gradient mixer to additionally quantify the concentration distribution throughout the device. The AutoCAD .dxf file for the fluid layer channels was imported into COMSOL to create the model geometry with boundaries between each of the crystallization wells. These boundaries were set to either open (for filling conditions) or closed (during crystallization), depending on the COMSOL model study being performed. Two studies were performed, first to model filling and second to model the concentration distribution during crystallization.
In the first, the Creeping Flow module was used to solve for the convective velocity field for pressure-driven flow set at the inlet boundaries representing experimental pressures of ~15 mbar. The Navier–Stokes equation was solved in a simplified form for incompressible fluids and neglecting inertial terms:
| (1) |
where μ is the dynamic viscosity of the medium and p is the pressure. The channel walls were set to a no-slip (u = 0) boundary condition, and the outlet was set as an open boundary. Fluid properties (viscosity and density) were set to those of water (for fluorescein and lysozyme/NaCl modeling) and PEG solutions at room temperature (details on the adjustments made for C-phycocyanin/PEG modeling can be found in the Supporting Information). With these conditions, eq 1 was solved at steady state to determine the fluid velocity field within the channels during the filling process.
Next, the Transport of Diluted Species module was used to solve for analyte transport by diffusion and convection generated by the solved velocity field:
| (2) |
where J is the total particle flux, D is the diffusion coefficient, c is the concentration, and u is the convective velocity field. Literature values of 5 × 10−10 m2/s for fluorescein,48 1.4 × 10−10 m2/s for lysozyme,55 and 2 × 10−9 m2/s for NaCl56 were used for D. These literature values are in agreement with values we calculated using the Stokes–Einstein equation considering hydrodynamic radii of 0.54 nm for fluorescein57,58 and 1.9 nm for lysozyme.59,60 Values of D used for C-phycocyanin and PEG are listed in the Supporting Information. Boundary properties within the microfluidic device were also established, whereby the channel walls were set to have “no flux” (J = 0). The inlet channel entrances were set to have a normalized particle concentration of 1, and the end boundary of the outlet channel was set to an outflow condition (−D∇c = 0). Combining these particle and channel conditions, eq 2 was solved at steady state to obtain the concentration profile in each of the wells.
Using the steady-state solution of all combined parameters from the first study as the initial condition, a second study was set up to model the incubation process after filling. Creeping flow was disabled such that diffusion was the only transport process occurring (J = −D∇c). For the case where no valves were present, a time-dependent study spanning 10 days was performed (see Supporting Information). For the case with valves present, a “thin impermeable membrane” boundary condition (J = 0) was set at each well entrance and exit to mimic the valve function, and the same time-dependent study was performed. For all studies, a custom ultrafine virtual mesh (minimum feature size of 1 μm) was built. The COMSOL solver was set to a relative tolerance of 0.001 using default parameters. Surface plots showing concentration distribution were generated, and normalized concentration values from the solution of the second study were extracted from each well.
RESULTS AND DISCUSSION
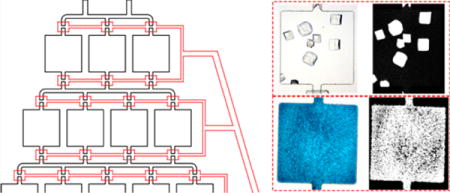
The microfluidic device developed for this work consists of a multi-layer design combining a gradient generator with a valve system. An overview top-down schematic picture of the design with integrated “doormat” valves is shown in Figure 1a with the fluid layer in black and the control layer in red (see the Experimental Section and Figure 2 for more details on the “doormat” valve). A series of “split and recombine” pathways forming a branching structure establish a concentration gradient throughout the device. However, unlike typical gradient generators which serve to form a continuous gradient in an outlet channel solution by using the same channel width throughout the device,47–49 our design contains boxed regions in between each split and recombine point which serve as crystallization nanowells with unique concentrations created during gradient generation. This is more clearly illustrated in the zoomed-in schematic in Figure 1b, which corresponds to the highlighted area marked in Figure 1a. Two inlets were used to allow protein and precipitant to be added for mixing (this can be expanded for a multi-dimensional screening platform, e.g., multiple precipitant inlets whereby gradients for each are created simultaneously and are adjustable by independently applied pressures and initial solution conditions48).
Figure 1.
(a) Overview schematic of the nanowell gradient generator (black) with incorporated “doormat” valve system (red dashes) showing 207 wells. (b) Zoom-in of the highlighted region in (a) detailing the “split and recombine” gradient generation design with incorporated valve system. Each well is isolated in the fluid layer and becomes connected by the overlapping valve region during the filling step.
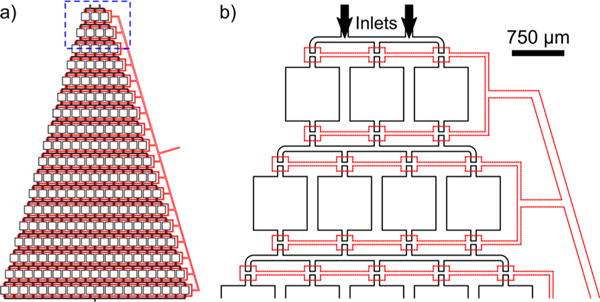
Figure 2.
“Doormat” valve schematic applied to PDMS channels and the filling process. (a) The three-layer device is shown in the normally closed state with no pressure applied, blocking the channels. (b) During filling, a vacuum is applied to the control layer which allows the thin membrane to deflect downward (due to not being strongly bonded to the fluid layer), opening a pathway between the channels for fluid to flow (designated by the red arrow). (c) Once the device is filled, the vacuum is removed, causing the thin membrane to return to its original state, isolating the wells from one another for crystallization. (d) Bright-field image of a PDMS replica when the valve is closed, showing the blocked channels, marked with corresponding locations on the schematic in (a). (e) Bright-field image of the PDMS replica of the device when the valve is actuated, showing the protruded PDMS membrane and red arrow marking the filling pathway. The speckles in the wells relate to defects due to the photolithography process.
The developed device exhibits >200 wells and generates 170 unique crystallization conditions (due to the outermost wells serving as a control containing only one of the components). The device can be easily expanded with more wells or scaled down with fewer wells (limited by the maximum size and number of wells allowing filling without flow/pressure-drop problems). We used relatively large wells of 750 × 750 × 100 μm to demonstrate that large crystals can be formed, yet each well still contains a volume of ~25 nL. Additionally, the individual well dimensions can also be increased or reduced to use even less protein (the lower limit approaches ~5 × 5 × 5 μm using the employed fabrication process).
We further integrated a developed “doormat” valve system to seal each crystallization nanowell. The necessity of these valves is described in more detail below. As outlined in red in Figure 1, the fluid layer wells were isolated from one another and the control layer was positioned to create a pathway between wells when the valve is open. Figure 2a shows a corresponding cross section schematic of the employed valve system whereby three layers of PDMS are sandwiched together. The top fluid layer is discontinuous, with a wall between entrance and exit channel regions where the valves are placed overlapping. The middle layer is a thin membrane of PDMS irreversibly sealed to a bottom control layer (and not strongly bonded to the fluid layer) containing channels where a negative pressure is applied, causing the thin membrane to deflect downward. This in turn creates a flow pathway between fluid layer channels when the valve is actuated during filling (Figure 2b). The valve is then returned to the normally closed state when the vacuum is removed to seal each crystallization well for incubation (Figure 2c). A realization of the valve in the closed state within an actual PDMS device is shown in Figure 2d, where the membrane is flat with the fluid layer, with arrows indicating corresponding locations on the cross section schematic. When the valve is actuated, the thin membrane deflects as seen by the protrusions in the PDMS in Figure 2e, making the fluid layer continuous and allowing filling to proceed along the pathway marked by the red arrow.
To determine how a concentration gradient forms within the device, we performed numerical modeling mimicking the experimental setup. A first modeling study considered species transport due to convection from a pressure-driven inlet flow and diffusion to calculate the steady-state filling concentration profile (modeling details can be found in the Experimental Section). Using this profile as the initial condition of a second modeling study, fluid flow was disabled and a closed boundary condition was established at the entrance and exit of each well to represent the closed valve during incubation. A time-dependent solution considering only diffusion was then obtained to quantify the final concentration profile of each well.
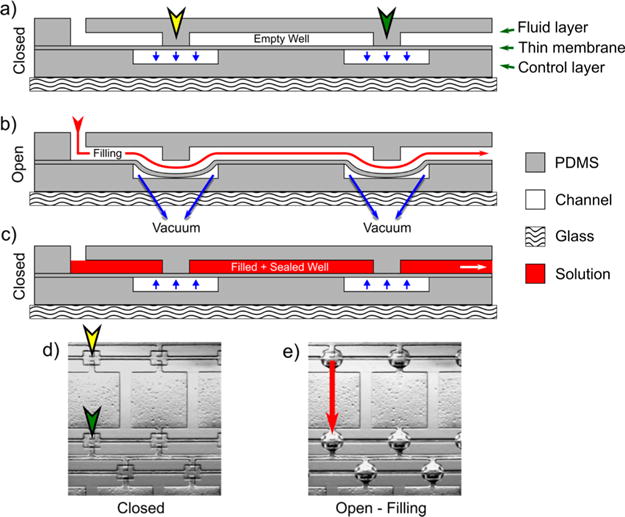
Figure 3a illustrates the overall normalized concentration distribution throughout the device, with the broad concentration gradient apparent on a green-scale. To confirm the modeling results, we tested whether a similar concentration gradient would establish experimentally by filling the device with fluorescein dye in the right inlet and water in the left inlet. Figure 3b shows fluorescence microscopy images of each well in the device, demonstrating the fluorescein concentration gradient established (concentration increases with brightness), very similar in appearance to the modeled concentration profile shown in Figure 3a. Resulting concentrations were quantified using background-corrected, normalized fluorescence intensities averaged across the entire well for five trials, where a normalized concentration of 1 represents the initial fluorescein concentration (i.e., the control wells to the far right) and 0 is background.
Figure 3.
(a) Modeled concentration profile of fluorescein illustrating the gradient formed. Concentration increases from black to green. (b) Fluorescence microscopy images showing the experimental gradient of fluorescein. Brighter areas are more concentrated. (c) Quantified normalized concentrations in each well from the fluorescein dye calibration (black dots) and model (red crosses) indicating good agreement between the two. Error bars represent standard deviation for five trials and are smaller than the data point in places they are not seen. (d) Plot of quantified concentrations from the numerical model comparing the 170 unique concentrations obtained from the dye (black squares), lysozyme (red circles), and NaCl precipitant (blue triangles). The outermost control wells with no variation are omitted.
The obtained values for all 207 wells, including the outermost control wells, were plotted (black dots) with the modeled normalized concentrations (red crosses) in Figure 3c, organized by rows corresponding to the device geometry. While the crosses correlate to their nearest neighboring black dot, some discrepancies can be seen, which is likely due to flow rate fluctuations in the middle and lower portions downstream of the inlets, where flow rates are smallest. This is also apparent by the larger experimental error in this region; however, the errors were generally small, indicating the filling procedure is robust and reproducible. Furthermore, the modeled and experimentally determined concentrations were in good agreement overall. Additionally, we note that the observed concentrations remained consistent over several days (data not shown) due to the integrated valve system sealing the chambers.
We further investigated the necessity of the integrated valves quantitatively. Numerical modeling with open valves indicated that the concentrations fully equilibrated after a period of 5–10 days, ruining the unique concentrations established by the nanowell approach (see Supporting Information, Figure S-1). With the integration of the valves, 170 unique conditions (i.e., ratios between precipitant and protein) were created as shown in Figure 3d. The plot shows correlated concentration pairs in ascending/descending order with arbitrary well numbers to illustrate the inverse concentration distribution relationship between the protein (red circles) and precipitant (blue triangles) inlet solutions (the outermost control wells were omitted for clarity as they have no concentration variation due to being at the outside boundaries of gradient generation where no mixing occurs). Furthermore, the final concentrations after mixing did not vary significantly between lysozyme and fluorescein (black squares), indicating that the establishment of various concentrations in each well is mainly dependent on the convective filling process. This allows the concentrations obtained with the fluorescein calibration to be used for determining protein concentrations in crystallization trials where fluid properties do not vary significantly. We also investigated the potential influence of an initial concentration gradient within individual wells immediately after the filling procedure at the start of the crystallization incubation phase. With the well dimensions presented in Figure 1, the diffusion times of a typical protein and precipitant across the well at the start of the incubation phase are 20–40 min and 5–10 min, respectively, which is not significant in the scheme of a multi-day crystallization experiment.
Having determined experimental success in establishing a concentration gradient throughout the device, we applied protein solutions with their respective precipitants to attempt to crystallize them and determine nucleation conditions in the crystallization phase space. We first investigated lysozyme, as it is a commonly used model protein for proof-of-concept studies with NaCl as the precipitant. The device was loaded with each solution via the separate inlets as described previously. Since both solutions were clear, qualitative confirmation by imaging that the concentration gradient established was not possible as in the case of fluorescein; thus, the results from the dye experiments were used as a reference to determine the concentrations in the device (see above). It should be noted that dyes can be incorporated into tolerant crystallization systems to aid visualization; however, they were not used in this study to demonstrate that they are not required. Once filled, the device was incubated for 2 days for the crystallization process to proceed.
Figure 4a shows a bright-field microscopy image of the wells after incubation whereby crystals are shown to have formed in a distinct region where nucleation conditions were established (highlighted by multi-colored boxes), also indicating that the concentrations throughout the device were different and did not equilibrate due to leakage or lack of gradient generation. To further clarify the varying regions of the phase space polled in the device, Figure 4b–d shows images of zoomed-in wells with unique characteristics. Figure 4b represents a well on the top left expected to contain only precipitant and has no apparent crystals in the bright-field image and no UV signal, confirming no protein is present (this would constitute the extreme oversaturated region of the phase diagram). Figure 4c highlights a well with favorable nucleation conditions for crystallization where large lysozyme crystals (P43212 space group61) are clearly visible and confirmed to be protein crystals by the UV signal. Furthermore, crystals grew throughout the well, indicating the two solutions were well-mixed. Lastly, Figure 4d provides a zoom-in of a well located at the bottom right of the device, expected to contain only protein (constituting the extreme undersaturated region of the phase diagram). The bright-field image is indistinguishable from the first scenario; however, the UV image shows a bulk signal from the noncrystallized lysozyme molecules in solution.
Figure 4.
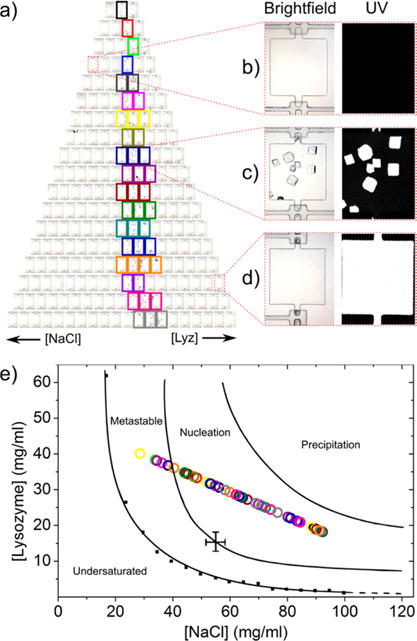
Results of lysozyme crystallization. (a) Bright-field image of a lysozyme crystallization trial after 2 days, showing a distinct region where macroscopic crystals grew, as highlighted by multi-colored boxes. (b–d) Zoomed-in images of several unique conditions showing (b) a precipitant-only case where a clear solution in the well is observed under bright-field and UV fluorescence, (c) a nucleation case where crystals are observed and confirmed to be proteinaceous from the UV signal, and (d) a protein-only case where a clear solution in the chamber is observed under bright-field, yet a UV signal is emitted from the protein molecules. (e) Experimental nucleation conditions where crystals were observed, plotted within a lysozyme phase diagram previously reported by Hekmat et al.62 (reproduced with permission, copyright 2007 Elsevier Ltd.), indicating good agreement of the nucleation region. The points are color-coded to match the colored boxes in each row in (a) and individual wells can be pinpointed using the concentration trends of lysozyme and NaCl.
To determine the nucleation concentrations of the protein and precipitant, we used the experimental calibration plot from Figure 3c. The corresponding modeled concentration profiles for lysozyme and NaCl are shown in the Supporting Information, Figure S-2. Wells where crystals were observed were marked, and, using the calibrated well concentrations, the nucleation concentrations were calculated. This equated to a lysozyme concentration range of 18–40 mg/mL and a NaCl concentration range of 29–92 mg/mL. Because this is an established system, we were able to check whether the results of our screening procedure were in agreement with previous studies. The obtained nucleation conditions are plotted as multi-colored circles in Figure 4e and overlaid onto a phase diagram reported by Hekmat et al.62 for lysozyme crystallized with NaCl in a sitting drop. Each point is color-coded to match the rows in Figure 4a, and individual wells can be pinpointed using the concentration trends of lysozyme and NaCl. As shown, our nucleation zone is in excellent agreement with this prior study, with a few points extending into the reported metastable zone that is still beyond the protein solubility limit, and none extending into the reported undersaturated or precipitation zones. We remark that the errors reported by Hekmat et al. for the supersolubility curve (as indicated in Figure 4e) are likely the reason for the overlap of observed protein crystallization into the metastable phase.
A second test of the crystallization device was performed with the pigment protein complex C-phycocyanin (C-PC), which is a study target at emerging XFEL sources due to its implications in photosynthesis. Furthermore, C-PC can crystallize with a different precipitant, PEG, which is viscous and commonly used for protein crystallization. When used with C-PC at optimal concentrations, microcrystals can be formed for SFX at XFELs.63 However, to our knowledge, quantitative phase diagrams for C-PC/PEG have not yet been reported.
The procedure to fill and incubate was performed as described for the other experiments but a different imaging technique was employed. C-PC crystals are second-harmonic generation microscopy active (H32 space group64), which allowed us to perform second-order nonlinear imaging of chiral crystals (SONICC)65,66 to confirm the presence of protein crystals as small as 100 nm with high confidence. SONICC is a powerful imaging technique that utilizes the frequency doubling of light that occurs in crystals of chiral molecules such as proteins and does not occur in most salt crystals and protein molecules in solution or in the form of amorphous aggregates. The signal-to-noise ratio depends on the space group, where the signal strength of high-symmetry space groups is very weak. (This is the reason why lysozyme crystals feature an extremely weak signal and cannot be imaged by SONICC.)
The microfluidic device is compatible with SONICC imaging39 as seen in Figure 5a, which shows the entire array of wells after 2 days of C-PC crystallization. As before, a distinct zone where crystals formed is present (highlighted by multi-color boxes), as indicated by the bright SONICC signal. The crystals that formed were much smaller than those observed for lysozyme (likely showers of microcrystals and nanocrystals) due to the precipitant type and optimal concentration range screened, as we aimed to optimize conditions for nano-/microcrystallization of C-PC for SFX experiments. For a detailed view, Figure 5b–d shows zoomed-in images of several example wells similar to those shown for lysozyme. Figure 5b exhibits a precipitant solution-only case with a bright-field image void of the visibly blue C-PC protein and no SONICC signal, as expected. In Figure 5c, a well where nucleation conditions are present and crystals were observed shows a clearer view of the blue nano-/microcrystal shower in the bright-field image, with a bright corresponding SONICC signal confirming small nano- and micro-sized protein crystals as opposed to salt or amorphous protein precipitate. Lastly, Figure 5d represents a protein-only case showing a solid blue bright-field image with no SONICC signal from the noncrystallized protein molecules in solution.
Figure 5.
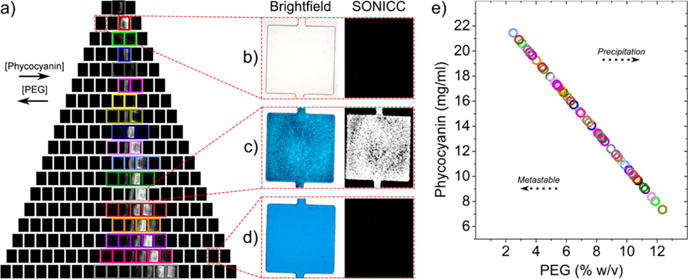
Results of C-PC crystallization. (a) SONICC image of a C-PC crystallization trial after 2 days showing a distinct region where crystals grew as highlighted by multi-colored boxes. (b–d) Zoomed-in images of several unique conditions showing (b) a precipitant-only case where nothing is observed under bright-field and SONICC, (c) a nucleation case where crystals are observed and confirmed to be proteinaceous from the SONICC signal, and (d) a protein-only case where solid blue is observed under bright-field and no SONICC signal is apparent. (e) A region of phase space showing conditions where crystals were observed, color-coded to match the rows in (a). Individual wells can be pinpointed using the concentration trends of C-PC and PEG.
Because the PEG solution has a significantly higher viscosity than the aqueous protein solution (which affects the fluid dynamics during the filling step and thus the established concentration gradient), we developed a model to more accurately quantify the conditions present in the wells where C-PC crystals were observed. When solution properties are known, a crystallization system (protein and precipitants) can be readily calibrated using a model adapted to changing experimental inputs (e.g., applied inlet pressures, channel designs/dimensions, etc.), allowing tedious, incremental experimental calibration to be avoided. In the case of C-PC/PEG, a viscosity blending function was incorporated into the model as a component of solving the flow profile throughout the device to determine the concentration profile of each crystallization well (details of changes to the model and a figure of the viscosity profile implemented can be found in the Supporting Information). When considering viscosity, the less viscous protein solution yields a >0.5 normalized concentration in more wells compared to the constant viscosity case, which is expected due to a lower viscous flow resistance. From the model, nucleation conditions were determined at C-PC concentrations of 8–22 mg/mL and PEG concentrations of 2–12% (w/v). To illustrate the relationship between the protein and precipitant and the nucleation regime uncovered, a region of phase space was constructed in Figure 5e with dotted arrows pointing toward the likely direction of neighboring phases. Additionally, each point is color-coded to match the rows in Figure 5a, and individual wells can be pinpointed using the concentration trends of C-PC and PEG. Since crystallization of C-PC with PEG has not been systematically studied previously, this nucleation region could be used as a foundation for further systematic studies and crystal optimization with different protein and precipitant concentration ranges.
CONCLUSIONS
We demonstrated a novel proof-of-concept microfluidic device to systematically crystallize proteins with nanoscale volumes of sample per trial comparable with current generation high-throughput screening platforms. The device utilizes a gradient generator to establish many unique concentrations of protein and precipitant and is easily scalable in the number of conditions, sample volume requirement, and number of inlet solutions for multi-dimensional precipitant screening. A “doormat” valve was incorporated to isolate each crystallization chamber to maintain a global concentration gradient over several days. Calibration with fluorescein dye exhibited successful concentration gradient generation quantitatively in agreement with a numerical model of the device. Two proteins were tested in the device and were successfully crystallized as observed by bright-field, UV, and SONICC imaging. Quantitative analysis of the conditions where crystals were observed provided nucleation conditions in agreement with those previously reported for lysozyme and uncovered a portion of the C-PC/PEG phase space. Large crystals of lysozyme formed with the addition of a NaCl precipitant and C-PC produced micro-/nanocrystal showers using a viscous PEG precipitant, which also demonstrated the versatility of the device with various common precipitants. To adequately model crystallization screens with varying viscosity, we also demonstrated the adaptation of a numerical model for the C-PC/PEG system. From this initial proof-of-concept study, the device can be readily used for protein crystallization screening in a wide range of laboratory settings and with a variety of proteins due to cheap and rapid fabrication, simple operation, and low sample volume requirements. Compatibility with most protein crystal imaging technologies also allows for straightforward visualization of crystal screening trials to determine crystal characteristics for X-ray crystallography. Furthermore, this device can enable studying the relationships between crystallization conditions and crystal growth characteristics (e.g., number and size), which is important in selecting optimal protein crystals for both traditional crystallography and SFX.
Supplementary Material
Acknowledgments
This work was supported by the STC Program of the National Science Foundation through BioXFEL under Agreement No. 1231306 and the National Institutes of Health Award No. R01GM095583.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.cgd.5b01748.
Modeled concentration profiles of fluorescein over a 10 day time series, modeled concentration profiles of lysozyme and NaCl, and model details and viscosity profile for C-PC/PEG crystallization (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Rupp B. Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. Garland Science; New York: 2009. [Google Scholar]
- 2.Chapman HN. Nat Mater. 2009;8:299–301. doi: 10.1038/nmat2402. [DOI] [PubMed] [Google Scholar]
- 3.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FR, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Homke A, Reich C, Pietschner D, Struder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kuhnel KU, Messerschmidt M, Bozek JD, Hau-Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jonsson O, Svenda M, Stern S, Nass K, Andritschke R, Schroter CD, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TR, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B, Spence JCH. Nature. 2011;470:73–77. [Google Scholar]
- 4.McSweeney S, Fromme P. Nature. 2014;505:620–621. doi: 10.1038/505620a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gruner SM, Lattman EE. Annu Rev Biophys. 2015;44:33–51. doi: 10.1146/annurev-biophys-060414-033813. [DOI] [PubMed] [Google Scholar]
- 6.Johansson LC, Arnlund D, Katona G, White TA, Barty A, DePonte DP, Shoeman RL, Wickstrand C, Sharma A, Williams GJ, Aquila A, Bogan MJ, Caleman C, Davidsson J, Doak RB, Frank M, Fromme R, Galli L, Grotjohann I, Hunter MS, Kassemeyer S, Kirian RA, Kupitz C, Liang M, Lomb L, Malmerberg E, Martin AV, Messerschmidt M, Nass K, Redecke L, Seibert MM, Sjohamn J, Steinbrener J, Stellato F, Wang D, Wahlgren WY, Weierstall U, Westenhoff S, Zatsepin NA, Boutet S, Spence JCH, Schlichting I, Chapman HN, Fromme P, Neutze R. Nat Commun. 2013;4:2911. doi: 10.1038/ncomms3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, Zatsepin NA, Li D, Messerschmidt M, Boutet S, Williams GJ, Koglin JE, Seibert MM, Wang C, Shah ST, Basu S, Fromme R, Kupitz C, Rendek KN, Grotjohann I, Fromme P, Kirian RA, Beyerlein KR, White TA, Chapman HN, Caffrey M, Spence JCH, Stevens RC, Cherezov V. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kupitz C, Basu S, Grotjohann I, Fromme R, Zatsepin NA, Rendek KN, Hunter MS, Shoeman RL, White TA, Wang D, James D, Yang JH, Cobb DE, Reeder B, Sierra RG, Liu H, Barty A, Aquila AL, Deponte D, Kirian RA, Bari S, Bergkamp JJ, Beyerlein KR, Bogan MJ, Caleman C, Chao T-C, Conrad CE, Davis KM, Fleckenstein H, Galli L, Hau-Riege SP, Kassemeyer S, Laksmono H, Liang M, Lomb L, Marchesini S, Martin AV, Messerschmidt M, Milathianaki D, Nass K, Ros A, Roy-Chowdhury S, Schmidt K, Seibert M, Steinbrener J, Stellato F, Yan L, Yoon C, Moore TA, Moore AL, Pushkar Y, Williams GJ, Boutet S, Doak RB, Weierstall U, Frank M, Chapman HN, Spence JCH, Fromme P. Nature. 2014;513:261–265. doi: 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, Oberthuer D, Heymann M, Kupitz C, Conrad C, Coe J, Roy-Chowdhury S, Weierstall U, James D, Wang D, Grant T, Barty A, Yefanov O, Scales J, Gati C, Seuring C, Srajer V, Henning R, Schwander P, Fromme R, Ourmazd A, Moffat K, Van Thor JJ, Spence JCH, Fromme P, Chapman HN, Schmidt M. Science. 2014;346:1242–1246. doi: 10.1126/science.1259357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JCH, Bruce Doak R, Nelson G, Fromme P, Fromme R, Grotjohann I, Kupitz C, Zatsepin NA, Liu H, Basu S, Wacker D, Han GW, Katritch V, Boutet S, Messerschmidt M, Williams GJ, Koglin JE, Marvin Seibert M, Klinker M, Gati C, Shoeman RL, Barty A, Chapman HN, Kirian RA, Beyerlein KR, Stevens RC, Li D, Shah ST, Howe N, Caffrey M, Cherezov V. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, Sawaya MR, Xu Q, Messerschmidt M, Williams GJ, Boutet S, Yefanov OM, White TA, Wang C, Ishchenko A, Tirupula KC, Desnoyer R, Coe J, Conrad CE, Fromme P, Stevens RC, Katritch V, Karnik SS, Cherezov V. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cudney R, Patel S, Weisgraber K, Newhouse Y, McPherson A. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:414–423. doi: 10.1107/S0907444994002660. [DOI] [PubMed] [Google Scholar]
- 13.Durbin SD, Feher G. Annu Rev Phys Chem. 1996;47:171–204. doi: 10.1146/annurev.physchem.47.1.171. [DOI] [PubMed] [Google Scholar]
- 14.Chayen NE, Saridakis E. Nat Methods. 2008;5:147–153. doi: 10.1038/nmeth.f.203. [DOI] [PubMed] [Google Scholar]
- 15.McPherson A, Gavira JA. Acta Crystallogr Sect F: Struct Biol Commun. 2014;70:2–20. doi: 10.1107/S2053230X13033141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kupitz C, Grotjohann I, Conrad CE, Roy-Chowdhury S, Fromme R, Fromme P. Philos Trans R Soc B. 2014;369:20130316. doi: 10.1098/rstb.2013.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luft JR, Albright DT, Baird JK, DeTitta GT. Acta Crystallogr Sect D: Biol Crystallogr. 1996;52:1098–1106. doi: 10.1107/S0907444996006932. [DOI] [PubMed] [Google Scholar]
- 18.Benvenuti M, Mangani S. Nat Protoc. 2007;2:1633–1651. doi: 10.1038/nprot.2007.198. [DOI] [PubMed] [Google Scholar]
- 19.Salemme FR. Arch Biochem Biophys. 1972;151:533–539. doi: 10.1016/0003-9861(72)90530-9. [DOI] [PubMed] [Google Scholar]
- 20.Salemme FR. Methods Enzymol. 1985;114:140–141. doi: 10.1016/0076-6879(85)14013-9. [DOI] [PubMed] [Google Scholar]
- 21.Ng JD, Gavira JA, Garcia-Ruiz JM. J Struct Biol. 2003;142:218–231. doi: 10.1016/s1047-8477(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 22.Otalora F, Gavira JA, Ng JD, Garcia-Ruiz JM. Prog Biophys Mol Biol. 2009;101:26–37. doi: 10.1016/j.pbiomolbio.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 23.DePonte DP, Weierstall U, Schmidt K, Warner J, Starodub D, Spence JCH, Doak RB. J Phys D: Appl Phys. 2008;41:195505. [Google Scholar]
- 24.Weierstall U. Philos Trans R Soc B. 2014;369:20130337. doi: 10.1098/rstb.2013.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chayen NE, Shaw Stewart PD, Blow DM. J Cryst Growth. 1992;122:176–180. [Google Scholar]
- 26.D’Arcy A, MacSweeney A, Stihle M, Haber A. Acta Crystallogr Sect D: Biol Crystallogr. 2003;59:396–399. doi: 10.1107/s0907444902022011. [DOI] [PubMed] [Google Scholar]
- 27.D’Arcy A, Sweeney AM, Haber A. Methods. 2004;34:323–328. doi: 10.1016/j.ymeth.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 28.Luft JR, Wolfley J, Jurisica I, Glasgow J, Fortier S, DeTitta GT. J Cryst Growth. 2001;232:591–595. [Google Scholar]
- 29.Kuswandi B, Nuriman, Huskens J, Verboom W. Anal Chim Acta. 2007;601:141–155. doi: 10.1016/j.aca.2007.08.046. [DOI] [PubMed] [Google Scholar]
- 30.Darain F, Gan KL, Tjin SC. Biomed Microdevices. 2009;11:653–661. doi: 10.1007/s10544-008-9275-3. [DOI] [PubMed] [Google Scholar]
- 31.Sun S, Yang M, Kostov Y, Rasooly A. Lab Chip. 2010;10:2093–2100. doi: 10.1039/c003994b. [DOI] [PubMed] [Google Scholar]
- 32.Wu J, Gu M. J Biomed Opt. 2011;16:080901. doi: 10.1117/1.3607430. [DOI] [PubMed] [Google Scholar]
- 33.Chin CD, Linder V, Sia SK. Lab Chip. 2012;12:2118–2134. doi: 10.1039/c2lc21204h. [DOI] [PubMed] [Google Scholar]
- 34.Abdallah BG, Chao T-C, Kupitz C, Fromme P, Ros A. ACS Nano. 2013;7:9129–9137. doi: 10.1021/nn403760q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharya S, Chao T-C, Ariyasinghe N, Ruiz Y, Lake D, Ros R, Ros A. Anal Bioanal Chem. 2014;406:1855–1865. doi: 10.1007/s00216-013-7598-2. [DOI] [PubMed] [Google Scholar]
- 36.Nakano A, Camacho-Alanis F, Ros A. Analyst. 2015;140:860–868. doi: 10.1039/c4an01503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen C, Quake SR. Curr Opin Struct Biol. 2003;13:538–544. doi: 10.1016/j.sbi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Ismagilov RF. Annu Rev Biophys. 2010;39:139–158. doi: 10.1146/annurev.biophys.050708.133630. [DOI] [PubMed] [Google Scholar]
- 39.Abdallah BG, Kupitz C, Fromme P, Ros A. ACS Nano. 2013;7:10534–10543. doi: 10.1021/nn402515q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen CL, Skordalakes E, Berger JM, Quake SR. Proc Natl Acad Sci USA. 2002;99:16531–16536. doi: 10.1073/pnas.262485199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segelke B. Expert Rev Proteomics. 2005;2:165–172. doi: 10.1586/14789450.2.2.165. [DOI] [PubMed] [Google Scholar]
- 42.Du W, Li L, Nichols KP, Ismagilov RF. Lab Chip. 2009;9:2286–2292. doi: 10.1039/b908978k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng B, Tice JD, Roach LS, Ismagilov RF. Angew Chem Int Ed. 2004;43:2508–2511. doi: 10.1002/anie.200453974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Sun K, Hu X, Li G, Jin Q, Zhao J. Sens Actuators B. 2015;219:105–111. [Google Scholar]
- 45.Chen DL, Gerdts CJ, Ismagilov RF. J Am Chem Soc. 2005;127:9672–9673. doi: 10.1021/ja052279v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Du W, Ismagilov RF. J Am Chem Soc. 2010;132:112–119. doi: 10.1021/ja908558m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeon NL, Dertinger SKW, Chiu DT, Choi IS, Stroock AD, Whitesides GM. Langmuir. 2000;16:8311–8316. [Google Scholar]
- 48.Dertinger SKW, Chiu DT, Jeon NL, Whitesides GM. Anal Chem. 2001;73:1240–1246. [Google Scholar]
- 49.Dertinger SK, Jiang X, Li Z, Murthy VN, Whitesides GM. Proc Natl Acad Sci USA. 2002;99:12542–12547. doi: 10.1073/pnas.192457199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hosokawa K, Maeda R. J Micromech Microeng. 2000;10:415. [Google Scholar]
- 51.Li N, Hsu CH, Folch A. Electrophoresis. 2005;26:3758–3764. doi: 10.1002/elps.200500171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frevert CW, Boggy G, Keenan TM, Folch A. Lab Chip. 2006;6:849–856. doi: 10.1039/b515560f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin D, Xia Y, Whitesides GM. Adv Mater. 1996;8:917–919. [Google Scholar]
- 54.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 55.Karlsson D, Zacchi G, Axelsson A. Biotechnol Prog. 2002;18:1423–1430. doi: 10.1021/bp0255659. [DOI] [PubMed] [Google Scholar]
- 56.Haynes WM, editor. CRC Handbook of Chemistry and Physics. CRC Press; Boca Raton, FL: 2015. [Google Scholar]
- 57.Suhling K, Siegel J, Lanigan PM, Leveque-Fort S, Webb SE, Phillips D, Davis DM, French PM. Opt Lett. 2004;29:584–586. doi: 10.1364/ol.29.000584. [DOI] [PubMed] [Google Scholar]
- 58.Devauges V, Marquer C, Lecart S, Cossec JC, Potier MC, Fort E, Suhling K, Leveque-Fort S. PLoS One. 2012;7:e44434. doi: 10.1371/journal.pone.0044434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parmar AS, Muschol M. Biophys J. 2009;97:590–598. doi: 10.1016/j.bpj.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nemzer LR, Flanders BN, Schmit JD, Chakrabarti A, Sorensen CM. Soft Matter. 2013;9:2187–2196. [Google Scholar]
- 61.Judge RA, Jacobs RS, Frazier T, Snell EH, Pusey ML. Biophys J. 1999;77:1585–1593. doi: 10.1016/S0006-3495(99)77006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hekmat D, Hebel D, Schmid H, Weuster-Botz D. Process Biochem. 2007;42:1649–1654. [Google Scholar]
- 63.Roy-Chowdhury S. School of Molecular Sciences. Arizona State University; 2015. personal communication. [Google Scholar]
- 64.Fromme R, Ishchenko A, Metz M, Chowdhury SR, Basu S, Boutet S, Fromme P, White TA, Barty A, Spence JCH, Weierstall U, Liu W, Cherezov V. IUCrJ. 2015;2:545–551. doi: 10.1107/S2052252515013160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wampler RD, Kissick DJ, Dehen CJ, Gualtieri EJ, Grey JL, Wang HF, Thompson DH, Cheng JX, Simpson GJ. J Am Chem Soc. 2008;130:14076–14077. doi: 10.1021/ja805983b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kissick DJ, Wanapun D, Simpson GJ. Annu Rev Anal Chem. 2011;4:419–437. doi: 10.1146/annurev.anchem.111808.073722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.