

Abstract
Inosine 5′-monophosphate dehydrogenase (IMPDH) catalyzes the first committed step of guanosine 5′-monophosphate (GMP) biosynthesis, and thus regulates the guanine nucleotide pool, which in turn governs proliferation. Human IMPDHs are validated targets for immunosuppressive, antiviral and anticancer drugs, but as yet microbial IMPDHs have not been exploited in antimicrobial chemotherapy. Selective inhibitors of IMPDH from Cryptosporidium parvum have recently been discovered that display anti-parasitic activity in cell culture models of infection. X-ray crystal structure and mutagenesis experiments identified the structural features that determine inhibitor susceptibility. These features are found in IMPDHs from a wide variety of pathogenic bacteria, including select agents and multiply drug resistant strains. A second generation inhibitor displays antibacterial activity against Helicobacter pylori, demonstrating the antibiotic potential of IMPDH inhibitors.
Keywords: antibacterial, Cryptosporidium parvum, guanine nucleotide biosynthesis, Helicobacter pylori, IMPDH, inosine 5′-monophosphate dehydrogenase, select agents, Mycobacterium tuberculosis
Introduction
Multiply drug resistant microbes are on the rise and poised to breach the last line of antibiotic defense [1, 2]. Mycobacterium tuberculosis strains resistant to both isoniazid and rifampicin, the front-line anti-tuberculosis drugs, are found in ~4% of new tuberculosis cases worldwide and are quickly spreading [3]. A significant fraction of these strains are also resistant to fluoroquinolines and at least one second line drug; some strains are also resistant to all second line drugs [4]. Drug resistant strains of Staphylococcus aureus are similarly threatening. In the United States, approximately 50% of hospital-acquired S. aureus infections are methicillin resistant (MRSA) and MRSA is now resident in the community as well. Vancomycin-resistant S. aureus (VRSA) has also appeared, suggesting that this second line drug may soon be obsolete. Drug resistant Streptococcus pneumoniae, Enterococcus faecilis, Clostridium difficile, Klebsiella pneumonia and Helicobacter pylori are increasingly prevalent. No effective antibiotic treatment exists for Acinetobacter baumannii. The threat of biowarfare agents also looms large; no drugs are available to deal with the catastrophic effects of accidental or intentional release of Coxiella burnetii, Francisella tularensis, Burkholderia mallei/pseudomallei or drug resistant Bacillus anthracis. These challenges demand a new arsenal of antibiotics and create an urgent need for novel scaffolds and targets [5-7].
Inosine 5′-monophosphate dehydrogenase (IMPDH) presents an intriguing, but as yet unexploited, target for antimicrobial drug discovery. This enzyme catalyzes the penultimate and rate-limiting step in guanine nucleotide biosynthesis, the oxidation of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) with the concomitant reduction of NAD+, Fig. (1A). Guanosine 5′-monophosphate synthetase (GMPS) catalyzes the conversion of XMP to guanosine 5′-monophosphate (GMP) in a reaction that consumes ATP and glutamine. Proliferation requires an expansion of the guanine nucleotide pool, so rapidly growing cells depend on IMPDH. Thus IMPDH is a target for immunosuppressive, cancer and antiviral chemotherapy [8]. IMPDH-targeted drugs include mycophenolic acid (MPA), mizoribine, tiazofurin, merimepodib and ribavirin, Fig. (1B). Rapid proliferation is also a feature of most microbial infections, which suggests that the inhibition of IMPDH may also be successful strategy for the design of antimicrobial chemotherapy.
Fig. (1).

Purine nucleotide biosynthesis. (A) The commonly occurring guanine nucleotide biosynthetic and salvage reactions are shown, as is the adenine nucleotide biosynthetic pathway. The IMPDH reaction is boxed. R5P, ribose 5′-monophosphate; NK, nucleoside kinase; HPRT, hypoxanthine phosphoribosyl transferase; XPRT, xanthine phosphoribosyl transferase; GPRT, guanine phosphoribosyl transferase; GMPR, guanosine 5′-monophosphate reductase; ADSS, adenylosuccinate synthetase; ADSL, adenylosuccinate lyase. (B) Structures of drugs that target IMPDH.
Here we review recent advances toward IMPDH-targeted antibiotics. The structure, mechanism and inhibition of IMPDH have recently been reviewed in [8, 9]. Though several more IMPDHs have been characterized in the last decade, the general properties of eukaryotic and prokaryotic IMPDHs noted in reference [10] still hold.
Overview of the structure and mechanism of IMPDH
IMPDH is a tetramer; each monomer has a catalytic domain with the classic β8/α8 barrel structure (also known as a TIM barrel, Fig. (2A)) [8]. Most IMPDHs also contain a subdomain containing two cystathionine beta synthase domains (CBS or Bateman domains). CBS domains bind adenine nucleotides in other proteins [11], but their function in IMPDH is currently under debate [8]. Deletion of these domains has no effect on enzymatic activity [8]Gan, 2002 #767}.
Fig. (2).

Structure of IMPDH. (A) The tetramer of S. pyogenes IMPDH is shown (PDB accession 1zfj). Each monomer is colored differently, IMP is depicted in spheres, the catalytic β8/α8 domain and subdomain are circled, and arrows mark the proposed bacterial signature segments, residues 12-21 and 453-462 (T. foetus IMPDH numbering is used for consistency; [78]). (B) The active site of T. foetus IMPDH. IMP and tiazofurin adenine dinucleotide (TAD) (PDB 1lrt) are shown as ball-and-stick with transparent surface. The protein is colored by conservation (alignment from [79]). Main chain within 5 Å of IMP or TAD is shown in ribbon, as are the bacterial signature segments. Side chains within 5 Å of IMP or TAD are shown in stick. (C) The CpIMPDH inhibitor binding site. The structure of CpIMPDH (PDB accession number = 3khj; salmon) with IMP (spheres, colored by element) and C64 (spheres, carbon atoms are magenta) is superimposed on structures of mammalian IMPDHs are superimposed (PDB accesssion 1jr1 and 1nfb; blue). Residues within 4 Å of C64 are shown. CpIMPDH residues Ala165 and Tyr358 are labeled. The surface of NAD+ bound to mammalian IMPDH is shown, as are residues that stack against the adenine ring.
The IMPDH reaction involves two chemical transformations, a dehydrogenase reaction where the catalytic Cys319 attacks C2 of IMP to form the covalent intermediate E-XMP* and NADH, and a subsequent hydrolase reaction where E-XMP* is released as XMP, Fig. (3A). During the dehydrogenase reaction, IMPDH assumes an open conformation that allows NAD+ to bind. However, a mobile flap binds in the NAD+ site during the hydrolase reaction, bringing Arg418 into the active site where it can act as a general base catalyst, Fig. (3B) [8]. The affinity of NAD+ and the flap are precisely balanced- if the flap binds too tightly, NAD+ will not be able to compete effectively and the dehydrogenase reaction cannot occur. If NAD+ binds too tightly, then the flap will not close and E-XMP* cannot hydrolyze. The flap also competes with many inhibitors, and this competition can be a major determinant of inhibitor potency [8]. The open conformation comprises a significant fraction (≥10%) of most IMPDHs, so this competition does not profoundly influence inhibitor design.
Fig. (3).

Mechanism of selective IMPDH inhibition. (A) Mechanism of the IMPDH reaction. T. foetus IMPDH numbering is shown. (B) A simplified kinetic mechanism is shown. Under the condition of the high throughput screen for CpIMPDH inhibitors, IMP adds first followed by NAD+, hydride transfer occurs, NADH is released, the flap folds into the NAD+ site and E-XMP* is hydrolyzed. The accumulation of each complex under the screening conditions is shown as a percentage; note that E•IMP and E-XMP* are the most abundant forms, so the screen is most likely to yield inhibitors that bind in the divergent NAD+ site. Panel B is reproduced from [67] with permission (copyright Elsevier Press).
Validation of IMPDH as a bacterial target
Although IMPDH catalyzes the pivotal step in guanine nucleotide biosynthesis, it is not generally considered an essential enzyme. Many microorganisms can salvage guanine and/or xanthine via phosphoribosyltranferase reactions, Fig. (1A). Although less well documented, the salvage of xanthosine and/or guanosine via kinase/phosphotransferase reactions is also possible. Such salvage pathways may provide resistance to IMPDH inhibitors. Of course, the efficiency of a salvage pathways depends on the availability of xanthine/guanine in the particular environmental niche and the idiosyncrasies of a given bacteria’s purine transporters and enzymes. The mere presence of a salvage enzyme in a microbial genome does not guarantee that the organism can establish an infection in the absence of IMPDH activity.
To illustrate the complexities of the purine pathways, the protozoan parasite Tritrichomonas foetus can salvage xanthine and guanine, yet is susceptible to IMPDH inhibitors when cultured in rich media. This parasite contains a single phosphoribosyltransferase that has a strong preference for hypoxanthine over xanthine and guanine [12, 13]. Therefore hypoxanthine salvage drives purine biosynthesis, so that T. foetus relies on IMPDH. This parasite becomes resistant to IMPDH inhibitors via two loss-of-function mutations that prevent hypoxanthine uptake and accumulation. These mutations reconfigure the salvage pathways to enable xanthine to supply the purine nucleotide pools [13].
T. foetus is not an exception- many microorganisms are susceptible to IMPDH inhibition when cultured in rich media despite the apparent presence of xanthine/guanine salvage pathways. In fact, the IMDPH-targeted immunosuppressive drug mycophenolic acid was originally discovered by virtue of its inhibition of Bacillus anthracis growth in Loffler's broth [14]. Mycophenolic acid also blocks proliferation of Staphylococcus aureus [15, 16] as well as the eukaryotic pathogens Candida albicans [17], Pneumocystis carinii ; [18], Leishmania donovani [19], Trypanosoma brucei gambienese [20], Eimeria tenella [21] and Cryptosporidium parvum [22]. Mizoribine, another natural product IMPDH inhibitor, blocks the growth of C. albicans [17] and Plasmodium falciparum [23]. Unfortunately, both MPA and mizoribine are potent inhibitors of mammalian IMPDHs, and so can only serve to provide proof of concept. A prokaryotic IMPDH-specific inhibitor C91 does display antibacterial activity against Helicobacter pylori cultured on rich medium (described in more detail below)[24]. These observations suggest that the salvage pathways are not sufficient to support proliferation in most microorganisms. Similar observations have been made in mammalian cells. Intriguingly, MPA and other IMPDH inhibitors induce differentiation in mammalian cells [25-28], and it is possible that such inhibitors will also disrupt the developmental program of bacteria.
Mutations in the IMPDH gene (guaB) profoundly decrease the virulence of many, but not all, bacteria, providing further target validation. Intriguingly, ablation of the adenine nucleotide biosynthetic pathways usually render bacteria avirulent, but mutations that block the biosynthesis of IMP often have no effect on virulence (see discussion in [29]). This observation suggests that the balance, rather than the supply, of the adenine and guanine nucleotide pools is critical. The loss of IMPDH renders B. burgdorferi [30, 31] and Yersinia pestis noninfectious [32]; guanine-requiring strains of K. pneumoniae are also avirulent [33]. The ability of Shigella flexneri to invade Hela cells and proliferate is also dramatically reduced by the loss of both IMPDH and GMPS (guaA); almost 100 fewer ΔguaBA bacteria are found intracellularly than wild-type [34]. The ability of both Salmonella typhimurium and F. tularensis to invade and proliferate in mouse macrophages is severely impaired when guaB is disrupted [29, 35, 36]; these strains are also ~104 less virulent in mouse peritoneal infections. In addition, a decrease in IMPDH activity appears to underlie loss of virulence in stk1 mutants of Group B Streptococcus [37]. Lastly, although M. tuberculosis contains two IMPDH genes, deletion of guaB2 alone is sufficient to inhibit growth [38].
In contrast, guaB mutations have comparatively little effect on the virulence of Salmonella dublin (only by 10-100-fold [29]) and Streptococcus suis (2.5-fold; [39]). Similarly, while the growth of guanine-dependent B. anthracis was impaired in the mouse bloodstream and peritoneal cavity, virulence was not significantly decreased [40]. Some bacteria, e.g., M. tuberculosis, contain multiple genes encoding IMPDH [41], further complicating the design of IMPDH-targeted antibiotics (although, as noted above, guaB2 is required for growth) . These observations suggest that IMPDH-targeted antibiotics are unlikely to have the broad spectrum of betalactams. Nonetheless, such drugs could be valuable addition to the antibiotic arsenal.
IMPDH has additional moonlighting functions
IMPDH is found in a surprisingly diverse array of cellular contexts and several observations suggest that this protein has additional cellular functions beyond its enzymatic activity. In E. coli, IMPDH plays a role in maintaining balance between the adenine and guanine nucleotide pools that is unrelated to its enzymatic function [9, 42]. In yeast, IMPDH associates with actively transcribed promoters [43]. Whole genome screens have identified >100 potentially interacting proteins, including proteins involved in transcription regulation, splicing and rRNA processing [44-49]. In eukaryotic cells, IMPDH associates with polyribosomes [50], lipid vesicles [51] protein kinase B [52] and actively transcribing promoters [43]. Though the physiological consequences of these interactions have not yet been elucidated and a cohesive regulatory model has not yet emerged, the clear conclusion is that the cellular functions of IMPDH extend well beyond the provision of the guanine nucleotide pool. These observations argue that IMPDH may be “mission critical” for infection.
How IMPDH-targeted drugs will affect these 'moonlighting' functions is an open question. Tyrosine kinase inhibitors provide one instructive example. Simply blocking the kinase activity of the Bcr-Abl tyrosine kinase is not sufficient to block activation of all its downstream signaling pathways [53]. Another useful lesson is found in the selective antimalarial action of dihydrofolate reductase (DHFR) inhibitors [54]. DHFR binds to its own cognate mRNA and blocks translation. In mammalian cells, inhibitors release DHFR from its mRNA and translation is restored. Thus mammalian DHFR is over-expressed, protecting the host cell. In contrast, the malaria parasite DHFR remains bound to its cognate mRNA in the presence inhibitors; no new protein is made, and the malaria parasite cannot proliferate. Given the undefined nature of the 'moonlighting' functions of IMPDH, it is impossible to predict how they may be perturbed by the presence of IMPDH inhibitors. Nonetheless, it is not unreasonable to believe that IMPDH-targeted chemotherapy may have more profound effects on proliferation and virulence than predicted by an IMPDH knockout strain that removes all traces of the protein.
The prospects for the development of resistance to IMPDH-targeted antibiotics
The propensity to develop drug resistance is an important consideration in antibiotic chemotherapy. Though it is difficult to predict the ability of a microorganism to develop drug resistance a priori, there are several reasons to be cautiously optimistic with respect to IMPDH-targeted therapy. The conformational gymnastics of the IMPDH catalytic cycle place additional functional constraints on the active site that should deter the development of drug resistance- a mutation would have to decrease the affinity of the inhibitor without disrupting the delicate balance between the binding of the flap and NAD+. The experience with the development of resistance to MPA, an eukaryotic-specific IMPDH inhibitor, is illustrative. In vitro, many organisms develop resistance to MPA by amplifying the IMPDH gene [19, 20, 55]. As noted above, the observation that IMPDH may be involved in transcription and translation suggests that over-production of IMPDH may have other consequences for a cell that may limit the amount of IMPDH that can be tolerated. MPA-resistant mutations in IMPDH are also observed, but the resulting changes in MPA affinity are generally small in magnitude and/or accompanied by a decrease in enzymatic activity [55-57]. As noted above, two loss-of-function mutations were required to create a MPA-resistant strain of T. foetus [13]. The requirement of IMPDH for virulence but not survival also bodes well- virulence factor targeted therapy is proposed to be less prone to develop resistance [58-60]. These arguments suggest that there are some reasons to believe that bacteria should not develop resistance to IMPDH inhibitors readily. However, even if resistance does occur, it will simply mean that these inhibitors will have to be used in combination with other drugs. Such therapy is already standard for many pathogens.
A comparison of prokayotic and eukaryotic IMPDHs
Prokaryotic and eukaryotic IMPDHs have distinct structural features and kinetic properties, which suggest that it should be possible to identify selective inhibitors [8, 10]. In general, the values of kcat and Km of the human IMPDHs are lower than those of the prokaryotic enzymes (Table 1). Most importantly for the prospects of selective inhibitor design, a eukaryotic IMPDH-selective inhibitor already exists: MPA. This compound binds 1000 times more tightly to the human enzymes than to prokaryotic IMPDHs (Table 1). MPA binds in the nictotinamide half of the NAD+ site, trapping the covalent E-XMP* intermediate. Only two residues are variable in the MPA/nicotinamide binding site: residues 310/431 are Arg/Gln and Lys/Glu in eukaryotic and prokaryotic enzymes, respectively (T. foetus IMPDH numbering). These substitutions account for part of the resistance to MPA, while the remaining resistance derives from the competition between MPA and the flap [61].
Table 1.
Steady state kinetic parameters of IMPDH from various organisms.
| IMPDH |
kcat (s−1) |
Km IMP (μM) |
Km NAD+ (μM) |
Kii NAD+ (mM) |
Ki MPA (nM) |
|---|---|---|---|---|---|
| Human type I | 1.2-1.8a | 14-18a | 42-70a | 2.0b | 11-33c |
| Human type II | 0.4-1.4d | 4-9d | 6-32d | 0.59e | 7-14f |
| Aerobacter aerogenes t | n.d. | 60 | 800 | n.d. | n.d. |
| Borellia burgdorferi g | 2.6 | 30 | 1100 | 2.3 | 8,000 |
| Cryptosporidium parvum h | 3.3 | 29 | 150 | 2.9 | 9,300 |
| Escherichia coli | 13i | 61i | 2000i | 2.8i | ~20,000j |
| Helicobacter pylori k | 3.0 | 18 | 73 | 1.7 | n.d. |
| Mycobacterium tuberculosis l | 0.53 | 78 | 1000 | 5.0 | 62,000 |
| Streptococcus pyogenes m | 24 | 62 | 1180 | n.d. | >10,000 |
| Tritrichomonas foetus n | 1.9g | 1.7g | 150g | 6.8g | 9,000g |
n.d., no data. Assay buffers are generally similar, but temperature varies between 25-37 degrees C. Values of kcat are reported per active site, not per tetramer. Note that while C. parvum and T. foetus are protozoan parasites, their IMPDHs are most similar bacterial enzymes, suggesting that the genes where obtained by horizontal transfer.
[82];
[83];
[85];
[66];
[86];
N. Benfield and L. Hedstrom, unpublished data;
[24];
[62];
[78];
[87].
With the exception of the two residues noted above, the IMP and nicotinamide sites are highly conserved, and therefore do not appear to be good candidates for the design of selective inhibitors, Fig. (2B). In contrast, the adenosine and pyrophosphate portions of the NAD+ site are highly diverged, Fig. (2B), which suggests that prokaryotic IMPDH-specific inhibitors can be developed by interacting with these regions. A recently reported MPA-adenine dinucleotide (MAD) provides some support for this strategy [62]. As expected, MPA is a very poor inhibitor of M. tuberculosis IMPDH (MtIMPDH) (Ki = 62 μM, Table 1), and eleven MAD derivatives failed to inhibit this enzyme. However, MAD9 does inhibit MtIMPDH with good potency (Ki = 1.5 μM, Fig. (4)). Although MAD9 is still a more better inhibitor of the human enzymes, the selectivity is only ~20, in contrast to 2000 for MPA. Modeling studies suggest that this change in selectivity originates from interactions of the triazole linker with the pyrophosphate binding portion of the NAD site of MtIMPDH, which is considerably more hydrophobic than the analogous region in the human IMPDHs due to the substitution of Thr and Ala for two Ser residues [62]. Perhaps the combination of MPA and triazole linker with a moiety selective for the adenosine subsite will produce MtIMPDH-selective inhibitors.
Fig. (4).

Structure of MAD9. This compound inhibits MtIMPDH with Ki = 1.5 μM.
Though the active site is contained within a single subunit, it is near the subunit interface. Two regions of the neighboring subunit, residues 12-21 and 453-462, display distinct sequence motifs in eukaryotic and prokaryotic IMPDHs and may be good targets for selective inhibitors (T. foetus IMPDH numbering; [10]). As described below, prokaryotic IMPDH-selective inhibitors have been discovered that span the nicotinamide subsite and 453-462 region.
Prokaryotic-specific IMPDH inhibitors
The protozoan parasite C. parvum is a major cause of diarrhea and malnutrition and a potential bioterrorism agent [63]. The parasite has a streamlined purine salvage pathway that depends on IMPDH for the production of guanine nucleotides [22, 64]. Surprisingly, C. parvum IMPDH (CpIMPDH) is closely related to prokaryotic IMPDHs, Fig. (5), suggesting that C. parvum obtained its IMPDH gene by horizontal transfer [22, 65]. As expected, the recombinant CpIMPDH has the high values of Km for IMP and NAD+ and resistance to MPA typical of prokaryotic IMPDHs (Table 1; [66]).
Fig. (5).

Phylogeny of IMPDHs from selected organisms. CpIMPDH is groups with bacterial enzymes, suggesting that C. parvum obtained its IMPDH gene by horizontal gene transfer. The dotted line demarcates IMPDHs that are predicted to be sensitive to CpIMPDH inhibitors [24]. IMPDHs from H. pylori, B. burgdorferi and S. pyogenes have been shown to be sensitive to CpIMPDH inhibitors, while human, T. foetus and E. coli IMPDHs are resistant.
CpIMPDH-selective inhibitors were identified in a high throughput screen [67]. To target the highly diverged NAD site, the screen utilized saturating concentrations of IMP but low concentrations of NAD+, Fig. (3B). Ten compounds were obtained with values of IC50 ranging from 200 nM to 20 μM that did not inhibit the human IMPDHs (the structures of 8 of these compounds, A-H, are shown in Table 2). Intriguingly, with one exception, all the CpIMPDH inhibitors contained two aromatic systems linked via an amide or similar functional group. Kinetic characterization confirmed that the inhibitors bind in the NAD+ site as predicted, and further localized binding to the nicotinamide subsite [67]. Several of these inhibitors display antiparasitic activity in a tissue culture model of C. parvum infection [67, 68].
Table 2.
The values of IC50 for inhibition of bacterial IMPDHs.
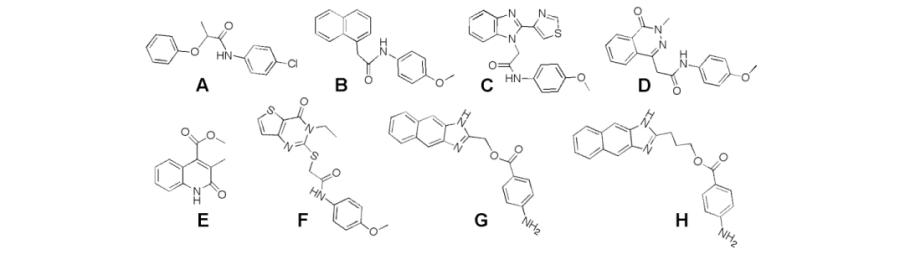
Medicinal Chemistry Optimization
Inhibitors A and C were chosen for further optimization based on preliminary structure activity relatinships (SAR), potency, antiparasitic activity and synthetic accessibility [69, 70]. Optimization initially focused on potency in the enzyme inhibition assay, while subsequence efforts addressed nonspecific protein binding, antiparasitic activity and metabolic stability.
For the A series, small alkyl groups in the (S) configuration are necessary at R1, Fig. (5A) [68, 69]; isopropyl and cyclopropyl are tolerated. Modest increases in potency are observed when R2 is p-fluorine, p-hydroxyl, and p-methoxymethyl ether. The best substitutions on the phenoxy ring are lipophilic (+π) and electron withdrawing (+σ) substituents such as 2,3-dichloro and 1-naphthyl. However, even with these improvements, the potency of these amide compounds remained >100 nM.
Potency increased 10-fold when the amide group was replaced with the 1,2,3-triazole. Although the SAR of the aniline and phenoxy groups was similar to the earlier amide compounds, this substitution changed the stereoisomer preference to (R). The naphthyl group further increased potency, but also bound to bovine serum albumin with high affinity. N-oxide derivatives decreased this non-specific protein binding. The selectivity for CpIMPDH was maintained throughout optimization; the values of IC50 for human IMPDH remained >10 μM. The resulting compound A110 displays good selectivity in a Toxoplasma gondii model of C. parvum infection [68].
The optimization of the C series displayed similar, though not identical, SAR, Fig. (5B). Potency increased 10-fold when the 4-OMe group was replaced with chlorine [70]. Replacement of the aniline with naphthyl increased potency by another factor of 10. Changing the connection between the benzimidazole and thiazole ring also increased potency, as did substitution of the thiazole with pyridine. The resulting compound, C91, is 1000 more potent than the initial hit in the enzyme assay. As described below, C91 displays antibacterial activity against H. pylori. Unfortunately, issues with metabolic stability may preclude further advancement of this series.
The structural basis of inhibitor selectivity
X-ray crystal structures show that IMPDH-targeted drugs such as MPA and merimepodib bind within a single subunit of an IMPDH homotetramer, stacking against the purine ring of the substrate in a manner similar to the nicotinamide portion of NAD+ [71, 72]. Surprisingly, the structure of CpIMPDH in complex with IMP and compound C64 revealed a new binding mode: the benzimidazole-thiazole portion stacks against the purine ring of IMP as expected, but the rest of the inhibitor bends across Ala165 and extends out of the active site and across a dimer interface into the bacterial signature segment (residues 453-462 in T. foetus IMPDH numbering; these residues are 351-360 in CpIMPDH [70]. The aniline group binds in a pocket in the adjacent subunit, where it stacks against Tyr358, Fig. (2C). Mammalian IMPDHs contain substitutions at both Ala165 and Tyr358 that account for selectivity, Fig. (2C).
Spectrum of IMPDH inhibition
The prokaryotic origins of CpIMPDH suggested that compounds A-H might also inhibit bacterial IMPDHs. Indeed, IMPDHs from H. pylori (HpIMPDH), Borrelia burgdorferi (BbIMPDH) and Streptococcus pyogenes (SpIMPDH) are inhibited by these compounds. As shown in Table 2, HpIMPDH and BbIMPDH display very similar SAR to CpIMPDH, while the SAR for SpIMPDH is significantly different. No inhibition is observed for E. coli IMPDH (EcIMPDH). However, the installation of both Ala165 and Tyr358 into EcIMPDH is sufficient to make this enzyme susceptible to compounds A-H (these residues are 250 and 444 in EcIMPDH numbering; Table 2). Thus Ala165 and Tyr358 comprise a structural motif that defines enzymes susceptible to CpIMPDH inhibitors. Importantly, the optimized compound C91 is a potent inhibitor (IC50<10 nM) of all five IMPDHs that contain Ala165 and Tyr358.
Inhibition of H. pylori growth
The availability of the potent HpIMPDH inhibitor C91 presented an opportunity to test the antibacterial potential of the CpIMPDH inhibitors. The susceptibility of H. pylori to C91 was assessed in Brucella broth, a nutrient rich medium that contains purines. This bacteria does contain a gene encoding a likely hypoxanthine-guanine phosphoribosyltransferase [73, 74], although the literature is conflicting with respect to the ability of H. pylori to salvage guanine [75, 76]. H. pylori will be resistant to IMPDH inhibitors if this salvage pathway can provide sufficient guanine nucleotides to support proliferation, so these assay conditions provide a demanding test for the antibiotic potential of IMPDH-targeted inhibitors. C91 blocked the growth of both stationary phase and exponentially growing H. pylori and was bacteriocidal at high concentrations, Fig. (6A). As expected, little inhibition of E. coli growth is observed at similar concentrations of C91, suggesting that the antibacterial activity results from the inhibition of HpIMPDH, Fig. (6B). H. pylori was cultured on defined media to further confirm the target of C91, (Ham's F12, Fig. (6C)). No proliferation is observed in the absence of fetal bovine serum; C91 is bacteriocidal under these conditions. Addition of fetal bovine serum induced proliferation. Again C91 blocked growth; addition of 10 μg/ml guanine protected against C91, as expected. Curiously, higher concentrations of guanine and xanthine inhibit H. pylori growth [75]. These results demonstrate the antibiotic potential of IMPDH-targeted antibiotics, as well as difficulty in predicting the contribution of salvage pathways to microbial metabolism.
Fig. (6).

Medicinal chemistry optimization. Initial hits A and C, structure activity relationships are summarized and example inhibitors are shown [67, 69, 70]. (A) The A series. (B) The C series.
CpIMPDH-like enzymes are present in many pathogenic bacteria
A BLAST search reveals that Ala165 and Tyr358 are present in IMPDHs from a wide variety of pathogenic bacteria (a partial list, with disease in parentheses): A. baumannii (wound infection), Arcobacter butzleri (food poisoning), B. anthracis (anthrax), Bacteroides capillosis (abscesses), Bac. fragilis (infection), Borrelia burgdorferi (Lyme disease), Brucella spp (brucellosis), Burkholderia cepacia (opportunistic infections), Bu. mallei/pseudomallei (glanders/melioidosis), Campylobacter jejuni (food poisoning), C. lari (food poisoning), Clostridia botulinum (botulism), Coxiella burnetii (Q fever), F. tularensis (tularemia), H. pylori (stomach ulcers), L. monocytogenes (listeriosis), Mycobacterium leprae (leprosy), M. tuberculosis (tuberculosis), Neisseria gonorrhoeae (gonorrhea), N. meningitides (bacterial meningitis), Pseudomonas aeruginosa (opportunistic infections), S. aureus (major cause of nosocomial infection), and Str. pneumoniae (pneumonia) and Str. pyogenes (erysipelas, pharyngitis, impetigo, cellulitis). To quote Payne and colleagues, "Gram-positive …and Gram-negative..bacteria share less in common genetically than do humans and paramecia [77]”, yet the IMPDHs predicted to be sensitive to the CpIMPDH inhibitors span the bacterial world. No effective therapy exists for many of these pathogens, while others have developed multi-drug resistant strains, underscoring the urgent need for new antibiotics. These observations suggest that IMPDH inhibition provides a promising strategy for the development of a new broad spectrum antibiotic. Importantly, E. coli IMPDH lacks the crucial Ala165 and Tyr358, and are therefore resistant to the CpIMPDH inhibitors, indicating that CpIMPDH-targeted therapy will spare some commensal bacteria.
Outlook
The many obstacles inherent in the development of broad-spectrum antibiotics were recently articulated in reference [77]. To encapsulate the main points: (1) the genomic divergence of Gram-positive and Gram-negative bacteria make common targets difficult to find; (2) Gram-positive and Gram-negative organisms pose distinct challenges in drug permeability that are difficult to negotiate with a single compound; and (3) the propensity to develop resistance is difficult to predict a priori. IMPDH is a rare target common to many Gram-negative and Gram-positive pathogens. While it would take some luck to overcome the remaining hurdles and develop IMPDH-targeted inhibitors into broad spectrum antibiotics, narrow spectrum antibiotics seem well within reach, and, given the current onslaught of multiple drug resistance, well worth pursuing.
Fig. (7).

Antibacterial activity of C91. (A) Compound C91 in DMSO was added to freshly diluted stationary cultures of H. pylori strain G27 in Brucella broth. Samples were removed at the indicated time points, diluted, and plated to determine bacterial proliferation/survival. Each point is the average of duplicate determinations; a representative of three experiments is shown. C91 concentrations are listed in μM. (B) Compound C91 was added to freshly diluted cultures of E. coli MG1655 in Luria broth. Each point is the average of three determinations; the standard deviations are smaller than the point. C91 concentrations are listed in μM. Panels A and B are reproduced from [24] with permission from Elsevier Press. (C) Guanine protects against C91. H. pylori was cultured in defined media (Ham's F12) in the presence and absence of C91 and 10 μg/ml guanine. FBS is fetal bovine serum.
Acknowledgements
This work was supported by NIH-NIAID U01 AI075466 (L.H.). G.W.L. was supported in part by the University of Virginia Cancer Training Grant (5T32 CA 009109).
Abbreviations
- ADSL
adenylosuccinate lyase
- ADSS
adenylosuccinate synthetase
- BbIMPDH
B. burgdorferi IMPDH
- CpIMPDH
C. parvum IMPDH
- DHFR
dihydrofolate reductase
- EcIMPDH-S250A/L444Y
E. coli IMPDH engineered to resemble CpIMPDH.
- GMP
guanosine 5′-monophosphate
- GMPR
guanosine 5′-monophosphate reductase
- GPRT
guanine phosphoribosyl transferase
- HpIMPDH
H. pylori IMPDH
- HPRT
hypoxanthine phosphoribosyl transferase
- IMP
inosine 5′-monophosphate
- MAD
mycophenolic acid adenine dinucleotide
- MPA
mycophenolic acid
- MtIMPDH
M. tuberculosis IMPDH
- NAD+
nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide, reduced form
- NK
nucleoside kinase
- R5P
ribose 5′-monophosphate
- SpIMPDH
S. pyogenes IMPDH
- TAD
tiazofurin adenine dinucleotide
- XMP
xanthosine 5′-monophosphate
- XPRT
xanthine phosphoribosyl transferase
References
- [1].Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10(12 Suppl):S122–129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- [2].Woodford N, Livermore DM. Infections caused by Gram-positive bacteria: a review of the global challenge. J Infect. 2009;59(Suppl 1):S4–16. doi: 10.1016/S0163-4453(09)60003-7. [DOI] [PubMed] [Google Scholar]
- [3].Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 Report on surveillance and response. World Health Organization; 2010. [Google Scholar]
- [4].Jain A, Mondal R. Extensively drug-resistant tuberculosis: current challenges and threats. FEMS Immunol Med Microbiol. 2008;53(2):145–150. doi: 10.1111/j.1574-695X.2008.00400.x. [DOI] [PubMed] [Google Scholar]
- [5].Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325(5944):1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Walsh CT, Fischbach MA. Repurposing libraries of eukaryotic protein kinase inhibitors for antibiotic discovery. Proc Natl Acad Sci U S A. 2009;106(6):1689–1690. doi: 10.1073/pnas.0813405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, Karnovsky A, Kuhn M, Limberakis C, Liu JY, Mehrens S, Mueller WT, Narasimhan L, Ogden A, Ohren J, Prasad JV, Shelly JA, Skerlos L, Sulavik M, Thomas VH, VanderRoest S, Wang L, Wang Z, Whitton A, Zhu T, Stover CK. A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc Natl Acad Sci U S A. 2009;106(6):1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hedstrom L. IMP Dehydrogenase: structure, mechanism and inhibition. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pimkin M, Pimkina J, Markham GD. A regulatory role of the Bateman domain of IMP dehydrogenase in adenylate nucleotide biosynthesis. J Biol Chem. 2009 doi: 10.1074/jbc.M808541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang R, G. E, Rotella F, Westbrook E, Huberman E, Joachimiak A, Collart FR. Differential signatures of bacterial and mammalian IMP dehydrogenase enzymes. Curr. Med. Chem. 1999;6:537–543. [PubMed] [Google Scholar]
- [11].Kemp BE. Bateman domains and adenosine derivatives form a binding contract. J Clin Invest. 2004;113(2):182–184. doi: 10.1172/JCI20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hedstrom L, Wang CC. Purine base transport in wild type and mycophenolic acid resistant Tritrichomonas foetus. Mol. Biochem. Parasit. 1989;35:816–820. doi: 10.1016/0166-6851(89)90208-9. [DOI] [PubMed] [Google Scholar]
- [13].Hedstrom L, Cheung K, Wang CC. A novel mechanism of mycophenolic acid resistance in the protozoan parasite Tritrichomonas foetus. Biochem. Pharm. 1990;39:151–160. doi: 10.1016/0006-2952(90)90659-9. [DOI] [PubMed] [Google Scholar]
- [14].Bentley R. Mycophenolic Acid: A One Hundred Year Odyssey from Antibiotic to Immunosuppressant. Chem. Rev. 2000;100:3801–3826. doi: 10.1021/cr990097b. [DOI] [PubMed] [Google Scholar]
- [15].Florey HW, Gilliver K, Jennings MA, Sanders AG. Mycophenolic acid, an antibiotic from Penicillium brevicompactum Dierckx. Lancet. 1946;i:46–49. doi: 10.1016/s0140-6736(46)90242-5. [DOI] [PubMed] [Google Scholar]
- [16].Abraham EP. The effect of mycophenolic acid on the growth of Staphylococcus aureus in heart broth. Biochem. J. 1945;39:398–408. doi: 10.1042/bj0390398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Quinn C, Bugeja V, Gallagher J, Whittaker P. The effect of mycophenolic acid on the cell cycle of Candida albicans. Mycopathologia. 1990;111:165–168. doi: 10.1007/BF02282799. [DOI] [PubMed] [Google Scholar]
- [18].O'Gara MJ, Lee C-H, Weinberg GA, Nott JM, Queener SF. IMP Dehydrogenase from Pneomocystis carinii as a potential drug target. Antimicrob. Agents Chemother. 1997;41:40–48. doi: 10.1128/aac.41.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wilson K, Collart F, Huberman E, Stringer J, Ullman B. Amplification and molecular cloning of the IMP dehydrogenase gene of Leishmania donovani. J. Biol. Chem. 1991;266:1665–1671. [PubMed] [Google Scholar]
- [20].Wilson K, Berens RL, Sifri CD, Ullman B. Amplification of the inosinate dehydrogenase gene in Trypanosoma brucei gambienese due to an increase in chromosome copy number. J. Biol. Chem. 1994;269:28979–28987. [PubMed] [Google Scholar]
- [21].Hupe D, Azzolina B, Behrens N. IMP dehydrogenase from the intracellular parasitic protozoan Eimeria tenella and its inhibition by mycophenolic acid. J. Biol. Chem. 1986;261:8363–8369. [PubMed] [Google Scholar]
- [22].Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci U S A. 2004;101(9):3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Webster HK, Whaun JM. Antimalarial properties of bredinin. Prediction based on identification of differences in human host-parasite purine metabolism. J. Clin. Invest. 1982;70:461–469. doi: 10.1172/JCI110636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gollapalli DR, Macpherson IS, Liechti G, Gorla SK, Goldberg JB, Hedstrom L. Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem Biol. 2010;17(10):1084–1091. doi: 10.1016/j.chembiol.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sokoloski JA, Blair OC, Sartorelli AC. Alterations in glycoprotein synthesis and guanosine triphosphate levels associated with the differentiation of HL-60 leukemia cells produced by inhibitors of inosine 5′-monophosphate dehydrogenase. Cancer Res. 1986;46:2314–2319. [PubMed] [Google Scholar]
- [26].Olah E, Natsumeda Y, Ikegami T, Kote Z, Horanyi M, Szelenyi J, Paulik E, Kremmer T, Hollan SR, Sugar J, et al. Induction of erythroid differentiation and modulation of gene expression by tiazofurin in K-562 leukemia cells. Proc Natl Acad Sci U S A. 1988;85(17):6533–6537. doi: 10.1073/pnas.85.17.6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Goldstein BM, Leary JF, Farley BA, Marquez VE, Levy PC, Rowley PT. Induction of HL60 cell differentiation by tiazofurin and its analogues: characterization and efficacy. Blood. 1991;78(3):593–598. [PubMed] [Google Scholar]
- [28].Floryk D, Tollaksen SL, Giometti CS, Huberman E. Differentiation of human prostate cancer PC-3 cells induced by inhibitors of inosine 5′-monophosphate dehydrogenase. Cancer Res. 2004;64(24):9049–9056. doi: 10.1158/0008-5472.CAN-04-1553. [DOI] [PubMed] [Google Scholar]
- [29].McFarland WC, Stocker BA. Effect of different purine auxotrophic strains on mouse virulence of a Vi positive strain of Salmonella dublin and of two strains of Salmonella typhimurium. Microb. Pathog. 1987;3:129–141. doi: 10.1016/0882-4010(87)90071-4. [DOI] [PubMed] [Google Scholar]
- [30].Botkin DJ, Abbott AN, Stewart PE, Rosa PA, Kawabata H, Watanabe H, Norris SJ. Identification of potential virulence determinants by Himar1 transposition of infectious Borrelia burgdorferi B31. Infect Immun. 2006;74(12):6690–6699. doi: 10.1128/IAI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jewett MW, Lawrence KA, Bestor A, Byram R, Gherardini F, Rosa PA. GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J Bacteriol. 2009;191(20):6231–6241. doi: 10.1128/JB.00450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Brubaker R. The interconversion of purine mononucleotides in Pasteurella pestis. Infect. Immun. 1970;1:446–454. doi: 10.1128/iai.1.5.446-454.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garber ED, Hacket AJ, Franklin R. The virulence of biochemical mutants of Klebsiella pneumoniae. Genetics. 1952;38:693–697. doi: 10.1073/pnas.38.8.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Noreiga FR, Losonsky G, Lauderbaugh C, Liao FM, Wang JY, Levine MM. Engineered DguaB-A DvirG Shigella flexneri 2a strain CVD 1205: construction, safety, immunogenicity and potential efficacy as a mucosal vaccine. Infection and Immunity. 1996;64:3055–3061. doi: 10.1128/iai.64.8.3055-3061.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fields PI, Swanson RV, Haidaris CG, Heffron F. Mutants of Salmonella typhimurium that can not survive within the macrophage are avirulent. Proc. Natl. Acad. Sci. USA. 1986;83:5189–5193. doi: 10.1073/pnas.83.14.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Santiago AE, Cole LE, Franco A, Vogel SN, Levine MM, Barry EM. Characterization of rationally attenuated Francisella tularensis vaccine strains that harbor deletions in the guaA and guaB genes. Vaccine. 2009;27(18):2426–2436. doi: 10.1016/j.vaccine.2009.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol Microbiol. 2005;56(5):1329–1346. doi: 10.1111/j.1365-2958.2005.04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48(1):77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- [39].Zhang XH, He KW, Duan ZT, Zhou JM, Yu ZY, Ni YX, Lu CP. Identification and characterization of inosine 5-monophosphate dehydrogenase in Streptococcus suis type 2. Microb Pathog. 2009;47(5):267–273. doi: 10.1016/j.micpath.2009.09.001. [DOI] [PubMed] [Google Scholar]
- [40].Ivanovics G, Marjai E, Dobozy A. The growth of purine mutants of Bacillus anthracis in the body of the mouse. J. Gen. Microbiol. 1968;53:147–162. doi: 10.1099/00221287-53-2-147. [DOI] [PubMed] [Google Scholar]
- [41].Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- [42].Pimkin M, Markham GD. The CBS subdomain of inosine 5′-monophosphate dehydrogenase regulates purine nucleotide turnover. Molecular Microbiology. 2008;69:342–359. doi: 10.1111/j.1365-2958.2008.06153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park JH, Ahn SH. IMP dehydrogenase is recruited to the transcription complex through serine 2 phosphorylation of RNA polymerase II. Biochem Biophys Res Commun. 2010;392(4):588–592. doi: 10.1016/j.bbrc.2010.01.079. [DOI] [PubMed] [Google Scholar]
- [44].Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- [45].Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR, 3rd, Hartzog GA. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol. 2003;23(4):1368–1378. doi: 10.1128/MCB.23.4.1368-1378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ho Y, Gruhler A, Hellbut A, Bader GD, Moore L, Adams S-L, Millar A, Taylor P, Bennett K, Boutliller K, Yang L, Wolting C, Donaldson I, Scchandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sasl H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansens LK, Jespersen H, Podtelejnikov A, Neilsen E, Crawford J, Poulsens V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CWV, Figeys D, Tyers M. Systematic identification of protein complexes in Sacccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- [47].Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, Beattie BK, Lalev A, Zhang W, Davierwala AP, Mnaimneh S, Starostine A, Tikuisis AP, Grigull J, Datta N, Bray JE, Hughes TR, Emili A, Greenblatt JF. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell. 2004;13(2):225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- [48].Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell. 2002;9(1):31–44. doi: 10.1016/s1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- [49].Collins SR, Kemmeren P, Zhao XC, Greenblatt JF, Spencer F, Holstege FC, Weissman JS, Krogan NJ. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics. 2007;6(3):439–450. doi: 10.1074/mcp.M600381-MCP200. [DOI] [PubMed] [Google Scholar]
- [50].Mortimer SE, Xu D, McGrew D, Hamaguchi N, Lim HC, Bowne SJ, Daiger SP, Hedstrom L. IMP Dehydrogenase Type 1 Associates with Polyribosomes Translating Rhodopsin mRNA. J Biol Chem. 2008;283(52):36354–36360. doi: 10.1074/jbc.M806143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Whitehead JP, Simpson F, Hill MM, Thomas EC, Connolly LM, Collart F, Simpson RJ, James DE. Insulin and oleate promote translocation of inosine-5′ monophosphate dehydrogenase to lipid bodies. Traffic. 2004;5(10):739–749. doi: 10.1111/j.1600-0854.2004.00217.x. [DOI] [PubMed] [Google Scholar]
- [52].Ingley E, Hemmings BA. PKB/Akt interacts with inosine-5′ monophosphate dehydrogenase through its plekstrin honology domain. FEBS Lett. 2000;478:253–259. doi: 10.1016/s0014-5793(00)01866-4. [DOI] [PubMed] [Google Scholar]
- [53].Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103(45):16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang K, Rathod PK. Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science. 2002;296:545–547. doi: 10.1126/science.1068274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lightfoot T, Snyder FF. Gene amplification and dual point mutations of mouse IMP dehydrogenase associated with cellular resistance to mycophenolic acid. Biochim. Biophys. Acta. 1994;1217:156–162. doi: 10.1016/0167-4781(94)90029-9. [DOI] [PubMed] [Google Scholar]
- [56].Kohler GA, White TC, Agabian N. Overexpression of a cloned IMP dehydrogenase gene of Candida albicans confers resistance to the specific inhibitor mycophenolic acid. J. Bact. 1997;179:2331–2338. doi: 10.1128/jb.179.7.2331-2338.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid resistant mutants of inosine 5′monophosphate dehydrogenase. J. Biol. Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]
- [58].Alekshun MN, Levy SB. Targeting virulence to prevent infection: to kill or not to kill? Drug Discovery Today: Therapeutic Strategies. 2004;1:483–489. [Google Scholar]
- [59].Lee YM, Almqvist F, Hultgren SJ. Targeting virulence for antimicrobial chemotherapy. Curr Opin Pharmacol. 2003;3(5):513–519. doi: 10.1016/j.coph.2003.04.001. [DOI] [PubMed] [Google Scholar]
- [60].Barczak AK, Hung DT. Productive steps toward an antimicrobial targeting virulence. Curr Opin Microbiol. 2009;12(5):490–496. doi: 10.1016/j.mib.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hedstrom L. IMP dehydrogenase: mechanism of action and inhibition. Curr. Med. Chem. 1999;6:545–560. [PubMed] [Google Scholar]
- [62].Chen L, Wilson DJ, Xu Y, Aldrich CC, Felczak K, Sham YY, Pankiewicz KW. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J Med Chem. 2010;53(12):4768–4778. doi: 10.1021/jm100424m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fayer R. Cryptosporidium: a water-borne zoonotic parasite. Veterinary Parasitology. Veterinary Parasitology. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- [64].Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304(5669):441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- [65].Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr., Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci U S A. 2002;99(9):6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- [67].Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem Biol. 2008;15(1):70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. A screening pipeline for antiparasitic agents targeting cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl Trop Dis. 2010;4(8):e794. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J Med Chem. 2009;52(15):4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D'Aquino JA, Zhang M, Lu J, Cuny GD, Hedstrom L. The Structural Basis of Cryptosporidium-Specific IMP Dehydrogenase Inhibitor Selectivity. J. Am. Chem. Soc. 2010 doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko M, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- [72].Sintchak MD, Nimmesgern E. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology. 2000;47:163–184. doi: 10.1016/s0162-3109(00)00193-4. [DOI] [PubMed] [Google Scholar]
- [73].Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388(6642):539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- [74].Duckworth M, Menard A, Megraud F, Mendz GL. Bioinformatic analysis of Helicobacter pylori XGPRTase: a potential therapeutic target. Helicobacter. 2006;11(4):287–295. doi: 10.1111/j.1523-5378.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- [75].Reynolds DJ, Penn CW. Characteristics of Helicobacter pylori growth in a defined medium and determination of its amino acid requirements. Microbiology. 1994;140(Pt 10):2649–2656. doi: 10.1099/00221287-140-10-2649. [DOI] [PubMed] [Google Scholar]
- [76].Mendz GL, Shepley AJ, Hazell SL, Smith MA. Purine metabolism and the microaerophily of Helicobacter pylori. Arch Microbiol. 1997;168(6):448–456. doi: 10.1007/s002030050521. [DOI] [PubMed] [Google Scholar]
- [77].Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- [78].Zhang R-G, Evans G, Rotella FJ, Westbrook EM, Beno D, Huberman E, Joachimiak A, Collart FR. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:4691–4700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- [79].Min D, Josephine HR, Li H, Lakner C, MacPherson IS, Naylor GJ, Swofford D, Hedstrom L, Yang W. An enzymatic atavist revealed in dual pathways for water activation. PLoS Biol. 2008;6(8):e206. doi: 10.1371/journal.pbio.0060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J. Biol. Chem. 1993;268:27286–27290. [PubMed] [Google Scholar]
- [81].Hager PW, Collart FR, Huberman E, Mitchell BS. Recombinant human inosinate dehydrogenase type I and type II proteins. Purification and characterization of inhibitor binding. Biochem. Pharm. 1995;49:1323–1329. doi: 10.1016/0006-2952(95)00026-v. [DOI] [PubMed] [Google Scholar]
- [82].Mortimer SE, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390(Pt 1):41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wang W, Hedstrom L. The kinetic mechanism of human inosine 5′-monophosphate type II: random addition of substrates, ordered release of products. Biochemistry. 1997;36:8479–8483. doi: 10.1021/bi970226n. [DOI] [PubMed] [Google Scholar]
- [84].Sintchak MD, Badia MC, Futer O, Nimmesgern E. X-ray crystal structure of the antiviral drug ribavirin monophosphate bound to IMP dehydrogenase. Antiviral Research. 1999;41:A56. [Google Scholar]
- [85].Zhou X, Cahoon M, Rosa P, Hedstrom L. Expression, purification and charcterization of inosine-5′-monophosphate dehydrogenase from Borrelia burgdorferi. J. Biol. Chem. 1997;272:21977–21981. doi: 10.1074/jbc.272.35.21977. [DOI] [PubMed] [Google Scholar]
- [86].Kerr K. The role of carboxylate residues in the mechanism of E. coli IMP dehydrogenase. Brandeis University; Waltham, MA: 1998. [Google Scholar]
- [87].Digits JA, Hedstrom L. Kinetic mechanism of Tritrichomonas foetus inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:2295–2306. doi: 10.1021/bi982305k. [DOI] [PubMed] [Google Scholar]