

Abstract
A facile one-pot two-enzyme chemoenzymatic approach has been established for gram (Neu4,5Ac2α3Lac, 1.33 g) and preparative-scale (Neu4,5Ac2α3LNnT) synthesis of monotreme milk oligosaccharides. Other O-acetyl-5-N-acetylneuraminic acid (Neu4,5Ac2)- or 4-O-acetyl-5-N-glycolylneuraminic acid (Neu4Ac5Gc)-containing α2–3-sialosides have also been synthesized in preparative scale. Used as an effective probe, Neu4,5Ac2α3GalβpNP was found to be a suitable substrate by human influenza A viruses but not bacterial sialidases.
Graphical abstract
One-pot multienzyme (OPME) synthesis: Monotreme milk oligosaccharides and other sialosides containing a 4-O-acetyl-sialic acid were synthesized in gram (1.33 g for Neu4,5Ac2α3Lac) or preparative (10–56 mg) scales using a one-pot two-enzyme (OP2E) sialylation system containing a bacterial cytidine 5′-monophosphate (CMP)-sialic acid synthetase and Pasteurella multocida sialyltransferase 3 (PmST3).
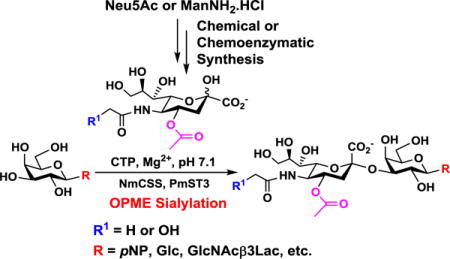
Introduction
Sialic acids commonly presented as the terminal residues on mammalian oligosaccharides or glycoconjugates are key recognition components of bacterial adhesins, viral surface proteins, selectins, Siglecs, and other sialic acid-binding proteins involved in infection, inflammation, cancer metastasis, and immunoregulation.1–4 Sialic acids present tremendous structural diversity in nature and more than 50 members have been identified so far. These include N-acetylneuraminic acid (Neu5Ac), the non-human sialic acid N-glycolylneuraminic acid (Neu5Gc), 2-keto-3-deoxy-nonulosonic acid (Kdn), their derivatives with single or multiple O-acetylation and less frequent 8-O-methylation, 8-O-sulfation, 9-O-lactylation, or 9-O-phosphorylation.1–4
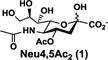
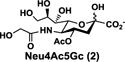
O-Acetylation is the most frequently occurring modification of sialic acids. Single or multiple O-acetylation at positions 4, 7, 8, and/or 9 of sialic acid has been identified and the presentation varies among different types of species, organs, and cells.1–4 Comparing to 9-O-acetylation which is the most common sialic acid O-acetylation, 4-O-acetylated sialic acids (Fig. 1) including 4-O-acetyl-N-acetylneuraminic acid (Neu4,5Ac2, 1) and 4-O-acetyl-N-glycolylneuraminic acid (Neu4Ac5Gc, 2) are less common.5 Neu4,5Ac2 (1) has been found in horse,6 donkey,7 Australian monotreme Echidna,5, 8 Japanese dace,9 South American pit viper,10 rabbit,11 guineapig.6 Neu4,5Ac2-containing oligosaccharides have been found to be the dominant components of the acidic milk oligosaccharides (MOSs) of monotremes echidnas and platypus.12–14 Both Neu4,5Ac2 and Neu4Ac5Gc have been found in horse glycoproteins.5 The presence of Neu4Ac5Gc-GM3 ganglioside in human colon cancer tissues was detected using purified antibodies.15 Neu4Ac5Gc (2) has also been found in α2–8-linked polysialic acids of glycoproteins from unfertilized kokanee salmon egg16 and in the serum of guinea pig in trace amounts.17
Fig. 1.
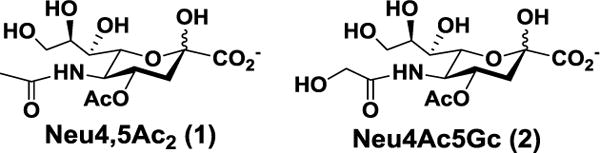
Structures of naturally occurring 4-O-acetyl-N-acetylneuraminic acid (Neu4,5Ac2, 1) and 4-O-acetyl-N-glycolylneuraminic acid (Neu4Ac5Gc, 2).
4-O-Acetylation of sialic acids provides resistance or decreased sensitivity of sialosides to various sialidases.6, 18, 19 This property is believed to contribute to the inhibitory activity of Neu4,5Ac2-rich horse and guinea pig α2-macroglobulins against hemagglutination and infectivity of several human influenza viruses.6, 18, 19 On the other hand, Neu4,5Ac2 is selectively recognized by the hemagglutinin-esterase (HE)20, 21 of mouse hepatitis virus strain S (MHV-S), a type of murine coronavirus, and infectious salmon anemia virus (ISAV).22 Nevertheless, the significance of naturally existing 4-O-acetylated sialic acids is not well understood partly due to the lack of access to sufficient amounts of the corresponding sialosides.
To our knowledge, the synthesis of sialosides containing 4-O-aceylated sialic acids by either chemical or enzymatic methods has not been reported. Here we present a facile chemoenzymatic approach for preparative and gram-scale synthesis of monotreme milk oligosaccharides including Neu4,5Ac2-containing α2–3-linked sialyl lactose (Neu4,5Ac2α3Lac) and sialyl lacto-N-neotetraose (Neu4,5Ac2α3LNnT). Other Neu4,5Ac2- or Neu4Ac5Gc-containing α2–3-linked sialosides representating common terminal sialosides in mammals are also synthesized.
Results and discussion
We envisioned that the desired 4-O-acetyl-sialic acid-containing sialosides could be chemoenzymatically synthesized using an effective one-pot two-enzyme sialylation system containing a cytidine 5′-monophosphate-sialic acid (CMP-Sia) synthetase (CSS) and a sialyltransferase (ST).23 To test the feasibility, the corresponding 4-O-acetyl-sialic acids were chemically synthesized. As shown in Scheme 1, Neu4,5Ac2 (1) was synthesized from N-acetylneuraminic acid (Neu5Ac, 3) by esterification of the C1-carboxyl group with benzyl bromide to provide benzyl ester 4 with a 90% yield. Protection of 4 at C8 and C9 by adding 2,2-dimethoxypropane in the presence of a catalytical amount of p-toluenesulfonic acid produced partially protected 5 in an excellent 92% yield. Regioselectively acetylation of 5 at O-4 using acetic anhydride in pyridine at room temperature provided the key intermediated 6 in a 81% yield. Final deprotection of the O-isopropylidene group in 6 by treating with 80% acetic acid and removal of the benzyl ester group by catalytic hydrogenolysis with H2 in the presence of Pd/C in methanol resulted in Neu4,5Ac2 (1) in a 82% yield. The obtained product has a proton nuclear magnetic resonance (1H NMR) spectrum consistent with that reported.24 The 4-O-acetyl modification caused a downfield shift of Neu5Ac H-4 signal.
Scheme 1.
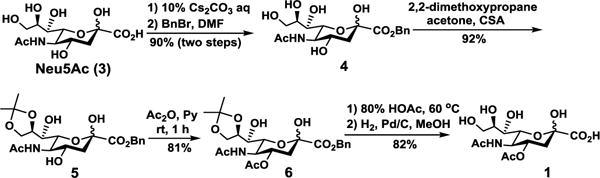
Chemical synthesis of Neu4,5Ac2 (1).
Neu4Ac5Gc (2) was chemoenzymatically synthesized from D-mannosamine hydrochloride (ManNH2·HCl, 7). As shown in Scheme 2, N-(2-Benzyloxyacetyl)-D-mannosamine 8 was obtained by treating ManNH2·HCl (7) with 2-benzyloxyacetyl chloride in the presence of NaHCO3 in CH3CN and water. P. multocida sialic acid aldolase (PmNanA)25-catalyzed aldol reaction of 8 with sodium pyruvate at 37 °C for 48 h produced the corresponding sialic acid derivative 9 in an excellent quantitative yield. The introduction of 4-O-acetyl group was carried out similarly to that described above for the synthesis of Neu4,5Ac2 (1). Briefly, benzylation of 9 with benzyl bromide formed benzyl ester 10 in a 90% yield. Treating 10 with 2,2-dimethoxypropane produced 8,9-isopropylidene-protected 11 in a 96% yield. Selective acetylation of 11 with Ac2O and pyridine produced the corresponding 4-O-acetyl derivative 12 in a good yield (81%). Subsequent removal of the isopropylidene group using HOAc/H2O followed by debenzylation using catalytic hydrogenation produced the target Neu4Ac5Gc (2) in a 78% yield.
Scheme 2.

Chemoenzymatic synthesis of Neu4Ac5Gc (2).
Sialyltransferase-catalyzed reactions use CMP-sialic acids which can be potentially accessed by a suitable CMP-Sia synthetase (CSS, EC 2.7.7.43). The tolerance of using Neu4,5Ac2 (1) as a potential substrate was tested for several bacterial CSSs including those from Neisseria meningitidis (NmCSS),26 Pasteurella multocida (PmCSS), Haemophilus ducreyi (HdCSS), as well as two NmCSS mutants (NmCSS_S81R and NmCSS_Q163A).27 Small-scale reactions carried out in Tris-HCl buffer (100 mM, pH 7.1) at 37 °C for 15 h followed by thin-layer chromatography (TLC) and mass spectrometry (MS) analyses indicated that all CSSs tested could use Neu4,5Ac2 (1) efficiently (Fig. S1, †ESI). These results were quite exciting as a previous report showed that CSSs purified from calf brain, bovine, and equine submaxillary glands could not use Neu4,5Ac2 (1) as a substrate for the synthesis of the corresponding CMP-sialic acid.28 NmCSS was chosen for further studies and for preparative-scale and gram-scale syntheses.
A coupled enzymatic assay was used to evaluate several bacterial sialyltransferases in using CMP-Neu4,5Ac2 and CMP-Neu4Ac5Gc synthesized in situ by NmCSS from Neu4,5Ac2 (1) and Neu4Ac5Gc (2), respectively, as potential donor substrates. Sialyltransferases tested include α2–3-sialyltransferases from Pasteurella multocida (PmST1,29 PmST2,30 PmST331) and PmST1_M144D mutant;32 α2–6-sialyltransferases Pd26ST33 and Psp26ST;34 as well as α2–3/8-sialyltransferase CstII.35, 36 Among these sialyltransferases, only PmST3 showed good activity as shown by TLC and MS studies. PmST3 was previously characterized as a monofunctional α2–3-sialyltransferase encoded by Pm1174 gene in Pasteurella multocida strain Pm70. It had broad acceptor substrate specificity and could use oligosaccharides, glycolipids, and glycopeptides as acceptors.31, 37 PmST3 could use oligosaccharides containing a terminal β1–4-or β1–3-llinked galactose as acceptors, but had poor activity on using galactosides containing a monosaccharide unit as acceptors. It could not use fucosylated oligosaccharides, such as Lex, as acceptor substrates. It was delightful to identify the unique efficiency of PmST3, among various bacterial sialyltransferases, in catalyzing the synthesis of sialosides containing 4-O-acetyl sialic acid.
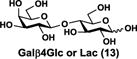
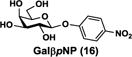
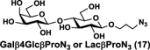
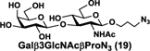
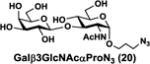
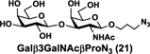
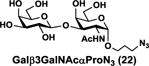
NmCSS and PmST3 were used in an efficient one-pot two-enzyme (OP2E) sialylation system23, 38 (Scheme 3) for preparative-scale synthesis of target sialosides. To minimize loss of the 4-O-acetyl group, enzymatic reactions were carried out at 37 °C in a Tris-HCl buffer (100 mM, pH 7.1) containing NmCSS, PmST3, Mg2+, cytidine 5′-triphosphate (CTP), Neu4,5Ac2 or Neu4Ac5Gc, and a galactoside acceptor. Monotreme milk oligosaccharides Neu4,5Ac2α3Lac (24) and Neu4,5Ac2α3LNnT (25) were readily produced from lactose (Galβ4Glc or Lac, 13) and previously synthesized lacto-N-neotetraose (Galβ4GlcNAcβ3Galβ4Glc or LNnT, 14)39 in 82% and 78% yields, respectively (Table 1). Neu4,5Ac2α3LNT (26) was also synthesized from commercially available lacto-N-tetraose (Galβ3GlcNAcβ3Galβ4Glc or LNT, 15) in a 65% yield. GalβpNP (16) with a monosaccharide unit was a less efficient acceptor and Neu4,5Ac2α3GalβpNP (27) was obtained in a 31% yield (the yield could not be improved further by adding more enzymes). Other α2–3-linked sialylated disaccharides containing a propyl azide aglycone (28–33) were also successfully synthesized from the corresponding disaccharides29, 40, 41 17–22 in 74–88% yields. To demonstrate the efficiency of the OP2E system, monotreme milk trisaccharide Neu4,5Ac2α3Lac (24) was further synthesized in a gram scale (1.33 g) with a 71% yield.
Scheme 3.
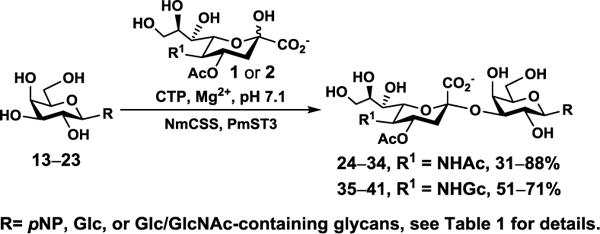
One-pot two-enzyme (OP2E) synthesis of sialosides containing Neu4,5Ac2 (1) or Neu4Ac5Gc (2). Enzymes: NmCSS, Neisseria meningitidis CMP-sialic acid synthetase; PmST3, Pasteurella multocida α2–3-sialyltransferase 3. The pH of the reaction was controlled at 7.1 to minimize de-O-acetylation during the enzymatic reaction. Yields were calculated for purified products based on the amounts of acceptors (limiting reagents) used.
Table 1.
One-pot two-enzyme (OP2E) preparative-scale synthesis of α2–3-linked 4-O-acetylated sialosides. Yields were calculated for purified products based on the amounts of acceptors (limiting reagents) used.
| Entry | Acceptor | Donor | Sialosides(Neu4,5Ac2/Neu4Ac5Gca3OR) | yield |
|---|---|---|---|---|
| a |
|
|
|
82% |
| b |
|
1 |
|
78% |
| c |
|
1 |
|
65% |
| d |
|
1 |
|
31% |
| e |
|
1 |
|
88% |
| f |
|
1 |
|
85% |
| g |
|
1 |
|
86% |
| h |
|
1 |
|
80% |
| i |
|
1 |
|
81% |
| j |
|
1 |
|
74% |
| k |
|
1 |
|
81% |
| l | 17 |
|
|
67% |
| m | 18 | 2 |
|
71% |
| n | 19 | 2 |
|
65% |
| o | 20 | 2 |
|
56% |
| p | 21 | 2 |
|
51% |
| q | 22 | 2 |
|
52% |
| r | 23 | 2 |
|
62% |
A longer propyl azide-containing oligosaccharide Galβ4GlcNAcβ3Galβ4GlcβProN3 or LNnTβProN3 (23) was synthesized from LacβProN3 (17) using a sequential two-step one-pot multienzyme (OPME)38 process (Scheme 4) similar to that reported previously for LNnT.39 Briefly, Lc3 trisaccharide GlcNAcβ3Galβ4GlcβProN342 was synthesized from LacβProN3 (17) and GlcNAc in a 94% yield using a one-pot four-enzyme (OP4E) GlcNAc activation and transfer system containing Bifidobacterium longum N-acetylhexosamine-1-kinase (BLNahK, NahK_ATCC55813),43 Pasteurella multocida N-acetylglucosamine uridyltransferase (PmGlmU),44 Pasteurella multocida inorganic pyrophosphatase (PmPpA),40 and Neisseria meningitidis β1–3-N–acetylglucosaminyltransferase (NmLgtA).45–47 LNnTβProN3 (23)42 was then produced from Lc3 and galactose in an excellent (99%) yield using a OP4E galactosylation system37, 39 containing Escherichia coli galactokinase (EcGalK),48 Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),49 PmPpA,40 and Neisseria meningitidis β1–4-galactosyltransferase (NmLgtB).40, 47 Synthesis of Neu4,5Ac2α3LNnTβProN3 (34) from LNnTβProN3 (23) and Neu4,5Ac2 (1) was successfully achieved in a good 81% yield using NmCSS and PmST3 in one-pot (Scheme 3).
Scheme 4.
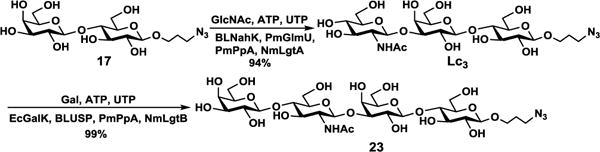
Sequential one-pot multienzyme (OPME) synthesis of LNnTβProN3 (23). Enzymes: BLNahK or NahK_ATCC55813, Bifidobacterium longum N-acetylhexosamine-1-kinase; PmGlmU, Pasteurella multocida N-acetylglucosamine uridyltransferase; PmPpA, Pasteurella multocida inorganic pyrophosphatase; NmLgtA, Neisseria meningitidis β1–3-N–acetylglucosaminyltransferase; EcGalK, Escherichia coli galactokinase; BLUSP, Bifidobacterium longum UDP-sugar pyrophosphorylase; NmLgtB, Neisseria meningitidis β1–4-galactosyltransferase.
Although less efficient compared to the synthesis of their Neu4,5Ac2-containing matching pairs, Neu4Ac5Gc-containing α2–3-linked sialosides with a propyl azide aglycone (35–41) were successfully obtained from Neu4Ac5Gc (2) and the corresponding disaccharides (17–22) or tetrasaccharide (23) in 51–71% yields using the OP2E sialylation reaction (Scheme 3). This indicated that CMP-Neu4,5Ac2 generated in situ is a better donor substrate than in situ generated CMP-Neu4Ac5Gc for PmST3. Longer reaction time needed for the synthesis of Neu4Ac5Gc-containing compounds also led to de-O-acetylation which also contributed to lower sialylaton yields.
All sialoside products synthesized in preparative scale were purified by gel filtration chromatography and semi-prep high-performance liquid chromatography (HPLC) using a reverse-phase C18 column. The structures were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy as wells as high resolution mass spectrometry (HRMS). Neu4,5Ac2α3Lac (24) synthesized in gram scale was purified (1.33 g) using a CombiFlash® Rf 200i system with a ODS-SM column (51 g, 50 μM, 120 Å, Yamazen).
The synthesized pNP-tagged sialoside Neu4,5Ac2α3GalβpNP (27) was used in a 384-well plate-based high-throughput colorimetric assay50 for substrate specificity studies of the α2–3-sialidase activity of Pasteurella multocida sialyltransferase 1 (PmST1),29 four commercially available bacterial sialidases, and four human influenza A viruses.51 While all bacterial sialidases tested including PmST1 as well as commercially available sialidases from Arthrobacter ureafaciens (Prozyme), Clostridium perfringens (Prozyme), Vibrio cholerae (Prozyme), and Streptococcus pneumoniae (Prozyme) did not show any sialidase activity towards Neu4,5Ac2α3GalβpNP (27) under the conditions used, human influenza A viruses (presumably the surface neuraminidases) could hydrolyze it (Fig. 2 black columns). As expected, all bacterial sialidases and viruses tested could cleave non-O-acetylated sialoside Neu5Acα3GalβpNP50 which was used as a positive control (Fig. 2 white columns).
Fig. 2.
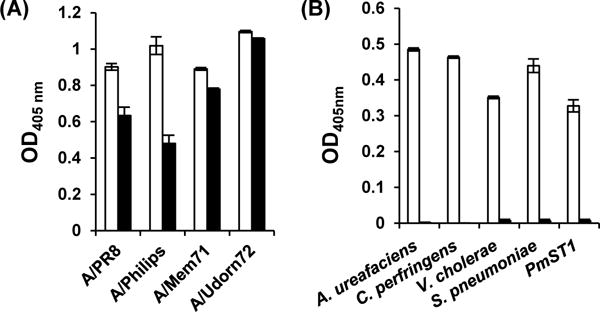
Human influenza A virus (A) and bacterial sialidase (B) substrate specificity studies using Neu4,5Ac2α3GalβpNP (27) as the substrate (black columns). Non-O-acetylated sialoside Neu5Acα3GalβpNP50 (white columns) was used as a positive control. Human influenza A viruses (purified) used: A/PR8, A/Puerto Rico/34/8 H1N1; A/Philips, A/Philippines/2/82/X-79 H3N2; A/Mem71, A/Memphis/71 H3N1; A/Udorn72, A/Udorn/307/72 H3N2.
In order to test whether the cleavage of Neu4,5Ac2 by human influenza A viruses was caused by de-O-acetylation or direct de-sialylation, Neu4,5Ac2α3Lac was incubated with purified human influenza A viruses followed by high-resolution mass spectrometry analysis. A major product peak for Neu4,5Ac2 was seen and only a trace amount of Neu5Ac was observed (Fig. S2, ESI), indicating a direct de-sialylation of Neu4,5Ac2 mechanism was used by human influenza A viruses.
Conclusions
In conclusions, we report here a convenient and efficient chemoenzymatic sialylation approach for synthesizing α2–3-linked sialosides containing a 4-O-acetylated sialic acid. PmST3, but not other bacterial sialyltransferases, was identified as a unique sialyltransferase that can catalyze the transfer of 4-O-acetylated sialic acid. Using chemically or chemoenzymatically synthesized Neu4,5Ac2 (1) or Neu4Ac5Gc (2), α2–3-linked sialosides were readily obtained by a one-pot two-enzyme (OP2E) system containing NmCSS and PmST3. Using Neu4,5Ac2α3GalβpNP as an effective probe, substrate specific studies showed that while Neu4,5Ac2 was not hydrolyzed by bacterial sialidases tested, it was effectively removed by human influenza A virus neuraminidases.
Experimental Section
Materials and methods
Chemicals were purchased and used as received. NMR spectra were recored in the NMR facility of University of California, Davis on a Varian Inova 400 (400 MHz for 1H, 100 MHz for 13C) and a Bruker Avance-800 NMR spectrometer (800 MHz for 1H, 200 MHz for 13C). Chemical shifts are reported in parts per million (ppm) on the δ scale. High resolution (HR) electrospray ionization (ESI) mass spectra were obtained using a Thermo Electron LTQ-Orbitrap Hybrid MS at the Mass Spectrometry Facility in the University of California, Davis. Silica gel 60 Å (230–400 mesh, Sorbent Technologies) was used for flash column chromatography. Thin layer chromatography was performed on silica gel plates (Sorbent Technologies) using anisaldehyde sugar stain for detection. Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with Bio-Gel P-2 Fine resins (Bio-Rad). D-Galactose (Gal) and N-acetyl-D-glucosamine (GlcNAc) were from Fisher Scientific. GalβpNP was from Sigma. N-Acetyl-D-mannosamine (ManNAc) and N-acetylneuraminic acid (Neu5Ac) was from Inalco (Italy). Adenosine 5′-triphosphate (ATP), uridine 5′-triphosphate (UTP), and cytosine 5′-triphosphate (CTP) were purchased from Hangzhou Meiya Pharmaceutical Co. Ltd. Lacto-N-tetraose (LNT) was from Elicityl (Crolles, France). Aspergillus oryzae β-galactosidase was from Sigma (St. Louis, MO). Recombinant enzymes Bifidobacterium longum strain ATCC55813 N-acetylhexosamine-1-kinase (BLNahK or NahK_ATCC55813),43 Pasteurella multocida N-acetylglucosamine uridyltransferase (PmGlmU),44 Pasteurella multocida inorganic pyrophosphatase (PmPpA),40 Neisseria meningitidis β1–3-N-acetylglucosaminyltransferase (NmLgtA),45, 46 Escherichia coli galactokinase (EcGalK),48 Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),49 Neisseria meningitidis β1–4-galactosyltransferase (NmLgtB),40 Pasteurella multocida sialic acid aldolase (PmNanA),25 Neisseria meningitidis CMP-sialic acid synthetase (NmCSS),26 Pasteurella multocida CSS (PmCSS), Haemophilus ducreyi CSS (HdCSS), and two NmCSS mutants (NmCSS_S81R and NmCSS_Q163A),27 Pasteurella multocida multifunctional α2–3-sialyltransferase 1 (PmST1)29 and Pasteurella multocida α2–3-sialyltransferase 3 (PmST3)31 were expressed and purified as described previously. Purified human influenza A viruses A/Puerto Rico/34/8 H1N1 (A/PR8), A/Philippines/2/82/X-79 H3N2 (A/Philips), A/Memphis/71 H3N1 (A/Mem71), and A/Udorn/307/72 H3N2 (A/Udorn72) were described previously.51 Sialidases from Arthrobacter ureafaciens, Clostridium perfringens, Vibrio cholerae, and Streptococcus pneumoniae were purchased from Prozyme (Hayward, CA).
LacβProN3 (17), LacNAcβProN3 (18), disaccharides 19–22, tetrasaccharide LNnT (14) were prepared as described previously.29, 40, 41 Tetrasaccharide LNnTβProN3 (23) was chemoenzymatically synthesized via sequential one-pot multienzyme (OPME) glycosylation approach from LacβProN3 (17) as described below:
Chemical synthesis of 5-N-acetyl-4-O-acetylneuraminic acid (Neu4,5Ac2, 1)
Benzyl 5-acetamido-3,5-dideoxy-D-glycero-β-D-galacto-2-nonulopyranosonate (4)
To a stirring solution of Neu5Ac (3, 5 g, 16.2 mmol) in water (20 mL), 10% Cs2CO3 was added dropwisely to adjust the pH value of solution to neutral. The reaction mixture was then evaporated and dried for overnight using a vacuum pump. The obtained solid was dissolved in N,N-dimethylformamide (20 mL), and benzyl bromide (3 mL) was added dropwisely. The mixture was stirred under argon for overnight at room temperature and then filtered. The filtrate was evaporated to produce a syrup. Crystallization using 2-propanol produced 4 (5.8 g, 90%). 1H NMR (800 MHz, D2O) δ 7.59–7.41 (m, 5H), 5.27 (dd, J = 27.2 and 10.8 Hz, 2H), 4.06–4.03 (m, 2H), 3.89 (t, J = 10.4 Hz, 1H), 3.80 (dd, J = 12.8 and 3.2 Hz, 1H), 3.67 (m, 1H), 3.58 (dd, J = 12.0 and 6.4 Hz, 1H), 3.53 (d, J =8.8 Hz, 1H), 2.29 (dd, J = 12.8 and 5.6 Hz, 1H), 2.03 (s, 3H), 1.88 (t, J = 12.8 Hz, 1H); 13C NMR (200 MHz, D2O) δ 174.71, 170.47, 134.80, 132.95, 130.65, 129.03, 128.77, 128.21, 95.20, 70.30, 69.98, 68.19, 68.02, 66.53, 63.00, 51.92, 39.90, 21.94.
Benzyl 5-acetamido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-β-D-galacto-2-nonulopyranosonate (5)
Compound 4 (3.1 g, 7.76 mmol) was dissolved in 2,2-dimethoxypropane (50 mL) and camphorsulfonic acid (50 mg) was added. The mixture was stirred for overnight at room temperature. Then trimethylamine (100 μL) was added and the mixture was stirred for 15 min before it was concentrated. The residue was purified by a silica gel column (EtOAc:MeOH = 15:1, by volume) to produce compound 5 (3.7 g, 92%). 1H NMR (800 MHz, CDCl3) δ 7.59–7.41 (m, 5H), 5.29 (d, J = 12.8 Hz, 1H), 5.22 (d, J = 12.0 Hz, 1H), 4.27–3.84(m, 7H), 2.33 (dd, J = 12.8 and 4.8 Hz, 1H), 2.08 (s, 3H), 2.01 (t, J = 12.8 Hz, 1H), 1.44 (s, 3H), 1.38 (s, 3H); 13C NMR (200 MHz, CDCl3) δ 173.97, 169.61, 134.97, 128.48 (2C), 128.38, 128.00 (2C), 109.30, 95.43, 74.15, 71.19, 69.82, 69.72, 67.36, 67.05, 53.09, 39.06, 26.70, 25.07, 22.52.
Benzyl 5-acetamido-4-O-acetyl-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-β-D-galacto-2-nonulopyranosonate (6)
To a solution of compound 5 (2.0 g, 4.55 mmol) in pyridine (4 mL), acetic anhydride (2 mL) was added. The mixture was stirred for 1 h at room temperature and then ethanol (10 mL) was added. The solution was evaporated to dryness and the residue was purified by a silica gel column (EtOAc:Hexane = 2:1, by volume) to produce compound 6 (1.7 g, 81%). 1H NMR (400 MHz, CDCl3) δ 7.36–7.29 (m, 5H), 6.22 (d, J = 8.0 Hz, 1H, NH), 5.36 (td, J = 10.8 and 5.6 Hz, 1H, H-4), 5.26 (d, J = 12.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 4.54–3.82 (m, 7H), 2.23 (dd, J = 12.8 and 4.8 Hz, 1H), 2.04 (t, J = 12.8 Hz, 1H), 2.03 (s, 3H), 1.97 (s, 3H), 1.34 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.78, 172.23, 168.76, 134.85, 128.68 (2C), 128.62, 128.16 (2C), 109.19, 94.92, 74.17, 71.80, 69.81, 68.93, 67.89, 67.44, 51.91, 35.90, 27.00, 25.22, 23.04, 21.09.
5-Acetamido-4-O-acetyl-3,5-dideoxy-D-glycero-β-D-galacto-2-nonulopyranosonate (1)
A solution of compound 6 (1.1 g, 2.28 mmol) in 90% acetic acid was stirred for 2 h at 60 °C. The reaction mixture was evaporated to produce a syrup which was dissolved in methanol (20 mL) and treated with H2 in the presence of 10% Pd/C for 2 h at room temperature. The solution was then filtered through Celite and concentrated. The residue was purified by a silica gel column (EtOAc:MeOH:H2O = 5:2:1, by volume) to produce a white powder 1 (658 mg, 82%). 1H NMR (800 MHz, D2O) δ 5.27 (td, J = 11.2 and 4.8 Hz, 1H, H-4), 4.19 (d, J = 10.4 Hz, 1H, H-8), 4.15 (t, J = 10.4 Hz, 1H, H-5), 3.82 (dd, J = 12.0 and 2.4 Hz, 1H, H-9a), 3.74 (m, 1H, H-6), 3.60 (dd, J = 12.0 and 6.4 Hz, 1H, H-9b), 3.56 (d, J = 9.6 Hz, 1H, H-7), 2.32 (dd, J = 12.8 and 5.6 Hz, 1H, H-3eq), 2.04 (s, 3H), 1.99 (t, J = 11.2 Hz, 1H, H-3ax), 1.97 (s, 3H); 13C NMR (200 MHz, D2O) δ 174.39, 173.31, 173.29, 95.16, 70.00, 69.98, 69.91, 67.95, 63.02, 49.31, 35.95, 21.76, 20.22; HRMS (ESI) calculated for C13H20NO10 (M-H) 350.1093, found 350.1093.
Multigram scale synthesis of Neu4,5Ac2 (1)
A multigram scale synthesis of Neu4,5Ac2 (1) was also achieved using the procedure described above. Briefly, Neu5Ac (10.1 g, 32.65 mmol) was dissolved in water (50 mL) and 10% Cs2CO3 was added to adjust the solution to neutral. After removal of the solvent and dried under vacuum, the obtained solid was dissolved in dry DMF (60 mL), and benzyl bromide (6 mL) was added at 0 °C. The mixture was stirred at room temperature under argon for overnight and then filtered. The filtrate was evaporated to a syrup which was crystallized using 2-propanol to produce 4 (11.1 g, 85%). Compound 4 (10.08 g, 25.23 mmol) was then dissolved in acetone (150 mL), and 2,2-dimethoxypropane (20 mL) and camphorsulfonic acid (150 mg) were added. The mixture was stirred at room temperature for 2 hours. Trimethylamine (500 μL) was added and the mixture was stirred for 15 min before it was concentrated. The obtained residue was purified using a 150 g RediSep Rf silica column (EtOAc:MeOH = 5:1, by volume) on a CombiFlash® Rf 200i system to produce 5 (10.5 g, 95%). Compound 5 (9.89 g, 22.50 mmol) was dissolved in pyridine (40 mL) and Ac2O (15 mL) was added. The mixture was stirred for 1 h at room temperature and ethanol (30 mL) was added at 0 °C. After being concentrated, the residue was purified using a 150 g RediSep Rf silica column (EtOAc:MeOH = 10:1, by volume) on a CombiFlash® Rf 200i system to produce compound 6 (9.1 g, 84%). A solution of compound 6 (8.58 g, 17.83 mmol) in 90% acetic acid (40 mL) was stirred at 60 °C for 2 h. The reaction mixture was evaporated and the obtained syrup was dissolved in methanol (50 mL) and treated with H2 in the presence of 10% Pd/C for 2 h at room temperature. The solution was filtered and concentrated. The residue was purified using a ODS-SM column (51 g, 50 μM, 120 Å, Yamazen) on a CombiFlash® Rf 200i system and was eluted with a gradient of 0–100% acetonitrile in water containing 0.05% formic acid over 30 minutes with a 30 mL/min flow rate. The fractions containing the pure product were collected and concentrated to produce the final product 1 (5.2 g, 83%) as a white powder.
Synthesis of Neu4Ac5Gc (2)
N-(2-Benzyloxyacetyl)-D-neuraminic acid (9)
Solid K2CO3 (14.4 g, 104.3 mM) was added to a stirred mixture of ManNH2·HCl (7, 4.5 g, 20.8 mM) in dry MeOH (100 mL). The mixture was stirred for 30 min and benzyloxyacetyl chloride (5.8 g, 31.3 mM) was added dropwisely. When the reaction was completed as monitored by TLC, the mixture was concentrated and purified by flash chromatography (DCM:MeOH = 10:1, by volume) to produce ManNGc analog 8 (5.8 g, 85%). Compound 8 (3.5 g, 10.7 mM) and pyruvate (4.11 g, 37.4 mM) were dissolved in water (35 mL) in a 50 mL centrifuge tube. After the addition of an appropriate amount of PmNanA (52 mg), the reaction mixture was incubated in an isotherm incubator for 48 h at 37 C with gentle shaking. The reaction progress was monitored by TLC (nPrOH:H2O:HOAc = 5:2:1, by volume) and mass spectrometer (MS). When the reaction was completed, ice-cold ethanol (35 mL) was added and the reaction mixture was incubated at 4 °C for 30 min. The mixture was centrifuged and the supernatant was concentrated. The residue was purified by silica gel column flash chromatography (EtOAc:MeOH:H2O = 6:2:1 then 4:2:1, by volume) to produce the desired Neu5GcOBn 9 (4.4 g, quant.) as a white solid. 1H NMR (800 MHz, D2O) δ 7.45–7.41 (m, 5H), 4.64 (s, 2H), 4.12 (s, 2H), 4.09 (m, 1H), 4.05 (d, J = 10.4 Hz, 1H), 3.96 (t, J = 10.4 Hz, 1H), 3.81 (dd, J = 12.0 and 3.2 Hz, 1H), 3.75 (m, 1H), 3.58 (dd, J = 12.0 and 6.4 Hz, 1H), 3.50 (d, J = 9.6 Hz, 1H), 2.21 (dd, J = 12.8 and 4.8 Hz, 1H), 1.82 (t, J = 12.8 Hz, 1H); 13C NMR (200 MHz, D2O) δ 176.65, 173.01, 136.45, 128.72(2C), 128.56(2C), 128.48, 96.33, 73.20, 70.30, 69.88, 68.38, 68.33, 66.86, 63.10, 51.88, 39.37. HRMS (ESI) m/z calcd for C18H24NO10 (M-H) 414.1400, found 414.1399.
Benzyl 5-glycolylamido-3,5-dideoxy-D-glycero-β-D-galacto-2-nonulopyranosonate (10)
To a stirring solution of Neu5GcOBn (9, 3.0 g, mmol) in water (25 mL), 10% CsCO3 was added dropwisely to adjust pH to neutral. The solution was then evaporated and the residue was dried for overnight by a vacum pump to produce the cesium salt of Neu5GcOBn. The salt was dissovled in N,N-dimethylformamide (40 mL), benzyl bromide (1.5 mL) was then added to the solution. The mixutre was strired under argon for 2 h at room temperature. After evaporation, the residue was purified by silica gel column chromatography (EtOAc:MeOH:H2O = 6:2:1, by volume) to produce compound 10. 1H NMR (400 MHz, CD3OD) δ 7.35–7.24 (m, 10H), 5.26 (m, 2H, PhCH2), 4.65 (m, 2H, PhCH2), 4.39–3.53 (m, 9H), 2.36 (dd, J = 12.8 and 4.8 Hz, 1H), 2.17 (t, J = 12.8 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 172.27, 170.95, 132.14, 131.50, 127.86 (4C), 127.78, 127.70 (4C), 126.90, 97.80, 73.37, 71.18, 70.23, 69.22, 68.92, 68.61, 67.31, 66.96, 53.05, 39.06.
Benzyl 5-glycolylamido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-β-D-galacto-2-nonulopyranosonate (11)
To a solution of compound 10 (1.5 g, 2.97 mmol) in acetone (40 mL), 2,2-dimethoxypropane (4 mL) and camphorsulfonic acid (15 mg) were added. The mixture was stirred for 3 h at room temperature. Then trimethylamine (200 μL) was added and the solution was stirred for 15 min. The reaction mixture was concentrated and the residue was purified by a silica gel column (EtOAc:Hexane = 5:1, by volume) to produce compound 11 (1.56 g, 96%) as a white solid. 1H NMR (800 MHz, D2O) δ 7.43–7.37 (m, 10H), 7.17 (d, J = 8.8 Hz, 1H), 5.27 (d, J = 12.0 Hz, 1H), 5.20 (d, J = 12.0 Hz, 1H), 4.60 (d, J = 12.0 Hz, 1H), 5.27 (d, J = 11.2 Hz, 1H), 4.49 (d, J = 6.4 Hz, 1H), 4.25–3.99 (m, 9H), 3.59 (t, J = 7.2 Hz, 1H), 2.36 (dd, J = 12.0 and 4.0 Hz, 1H), 2.15 (t, J = 12.0 Hz, 1H), 1.45 (s, 3H), 1.37 (s, 3H); 13C NMR (200 MHz, D2O) δ 172.51, 169.48, 136.54, 134.94, 128.61(2C), 128.58(2C), 128.45, 128.27, 128.01(2C), 127.99(2C), 109.15, 95.39, 74.59, 73.41, 71.23, 69.56, 68.96, 67.61, 66.93, 66.76, 52.44, 38.98, 26.97, 25.28.
5-Glycolylamido-4-O-acetyl-3,5-dideoxy-D-glycero-β-D-galacto-2-nonulopyranosonate (2)
To a solution of 8,9-O-isopropylidene-Neu5GcOBn (11, 1.1 g, 2.02 mmol) in pyridine (4 mL), acetic anhydride (2 mL) was added. The mixture was stirred for 1 h at room temperature and ethanol (10 mL) was added. The solution was evaporated to dryness and the residue was purified by a silica gel column (EtOAc:toluene = 1:2, by volume) to produce compound 12 (0.96 g, 81%). A solution of compound 12 (0.8 g, 1.36 mmol) in 90% acetic acid was stirred for 2 h at 60 °C. The reaction mixture was evaporated to give a syrup which was dissolved in methanol (20 mL) and treated with H2 in the presence of 10% Pd/C for 2 h at room temperature. The solution was filtered through Celite and concentrated. The residue was purified by a silica gel column (EtOAc:MeOH:H2O = 5:2:1, by volume) to produce a white powder 2 (0.31 g, 78%). 1H NMR (800 MHz, D2O) δ 5.36 (td, J = 11.2 and 4.8 Hz, 1H, H-4), 4.32 (d, J = 10.4 Hz, 1H, H-8), 4.23 (t, J = 10.4 Hz, 1H, H-5), 4.05 (d, J = 6.4 Hz, 2H, OCH2-), 3.82 (dd, J = 12.0 and 2.4 Hz, 1H, H-9a), 3.75 (m, 1H, H-6), 3.61 (dd, J = 12.0 and 6.4 Hz, 1H, H-9b), 3.56 (d, J = 8.8 Hz, 1H, H-7), 2.35 (dd, J = 12.8 and 5.6 Hz, 1H, H-3eq), 2.04 (s, 3H), 2.01 (t, J = 12.8 Hz, 1H, H-3ax); 13C NMR (200 MHz, D2O) δ 75.34, 173.25, 172.97, 95.06, 69.99, 69.75, 69.70, 67.86, 62.98, 60.71, 48.97, 35.98, 20.22; HRMS (ESI) calculated for C13H20NO11 (M-H) 366.1042, found 366.1042.
Chemoenzymatic synthesis of GlcNAcβ3Galβ4GlcβProN3 (Lc3βProN3) and Galβ4GlcNAcβ3Galβ4GlcβProN3 (LNnTβProN3, 23)
One-pot four-enzyme preparative-scale synthesis of trisaccharide GlcNAcβ3Galβ4GlcβProN3 (Lc3βProN3)
A reaction mixture in a total volume of 40 mL containing Tris-HCl buffer (100 mM, pH 7.5), LacβProN3 (17, 0.45 g, 1.06 mmol), N-acetylglucosamine (GlcNAc, 0.304 g, 1.38 mmol), ATP (0.758 g, 1.38 mmol), UTP (0.756 g, 1.38 mmol), MgCl2 (20 mM), BLNahK (12.0 mg), PmGlmU (6.0 mg), NmLgtA (6.0 mg), and PmPpA (4.0 mg) was incubated in a shaker with agitation (100 rpm) at 37 °C for 15 h and then 72 h at room temperature. The product formation was monitored by TLC (EtOAc:MeOH:H2O:HOAc = 4:2:1:0.2 by volume and detected by p-anisaldehyde sugar stain) and mass spectrometry (MS). When the reaction was completed, the same volume (40 mL) of 95% ethanol was added to the reaction mixture and the mixture was incubated at 4 °C for 30 min. After centrifugation, the supernatant was concentrated and passed through a Bio Gel P-2 gel filtration column (water was used as an eluent). The fractions containing the product were collected, concentrated, further purified by silica gel column (EtOAc:MeOH:H2O = 5:2:1, by volume) and finally by a Bio-Gel P-2 column (eluted with H2O) to produce Lc3 trisaccharide GlcNAcβ3Galβ4GlcβProN342 (0.625 g, 94%). 1H NMR (800 MHz, D2O) δ 4.67 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz,1H), 4.43 (d, J = 8.0 Hz, 1H), 4.15 (d, J = 3.2 Hz, 1H), 4.01–3.97 (m, 2H), 3.90 (d, J = 11.2 Hz,1H), 3.76–3.55 (m, 13H), 3.47–3.44 (m, 4H), 3.30 (d, J = 8.0 Hz,1H), 2.03 (s, 3H), 1.91 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.85, 102.81, 102.74, 101.99, 81.83, 78.20, 75.52, 74.76, 74.65, 74.23, 73.43, 72.66, 69.87, 69.54, 68.21, 67.24, 60.83, 60.34, 59.92, 55.53, 47.73, 28.10, 22.03. HRMS (ESI) m/z calculated for C23H40N4O16Na (M+Na) 651.2337, found 651.2337.
One-pot four-enzyme preparative-scale synthesis of Galβ4GlcNAcβ3Galβ4GlcβProN3 (LNnTβProN3, 23)
The reaction mixture in a total volume of 30 mL containing Tris-HCl buffer (100 mM, pH 8.0), Lc3 trisaccharide (0.55 g, 0.875 mmol), galactose (0.204 g, 1.14 mmol), ATP (0.627 g, 1.14 mmol), UTP (0.626 g, 1.14 mmol), MgCl2 (20 mM), EcGalK (12.0 mg), BLUSP (10.0 mg), NmLgtB (7.0 mg), and PmPpA (8 mg) was incubated in a shaker with agitation (100 rpm) at 37 °C for 5 hrs. The product formation was monitored by TLC (n-PrOH:H2O:NH4OH = 5:2:1 and detected by p-anisaldehyde sugar stain) and mass spectrometry. When an optimal yield was achieved, ethanol (30 mL) was added and the mixture was incubated at 4 °C for 30 min. The precipitates were removed by centrifugation and the supernatant was concentrated and purified by a Bio-Gel P-2 gel column (water was used as an eluate). Further purification was achieved by silica gel chromatography (EtOAc:MeOH:H2O = 5:3:1.5) and finally Bio-Gel P-2 column (eluted with H2O) to produce Galβ4GlcNAcβ3Galβ4GlcβProN342 (23, 0.613 g, 99%). 1H NMR (800 MHz, D2O) δ 4.69 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 7.2 Hz,1H), 4.47 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 7.2 Hz, 1H), 4.15 (d, J = 3.2 Hz, 1H), 4.01–3.92 (m, 4H), 3.85–3.57 (m, 19H), 3.53 (t, J =8.8 Hz, 1H), 3.46 (t, J = 7.2 Hz, 2H), 3.30 (d, J = 8.0 Hz,1H), 2.03 (s, 3H), 1.90 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.80, 102.82, 102.73, 102.64, 101.99, 81.92, 78.20, 78.00, 75.23, 74.75, 74.65, 74.42, 74.23, 72.66, 72.37, 72.05, 70.84, 69.83, 68.42, 68.20, 67.24, 60.91, 60.83, 59.92, 59.72, 55.06, 47.73, 28.10, 22.05. HRMS (ESI) m/z calculated for C29H50N4O21Na (M+Na) 813.2865, found 813.2865.
General procedures for one-pot two-enzyme (OP2E) preparative-scale synthesis of α2–3-linked sialosides containing a 4-O-acetyl sialic acid
An acceptor (10 mM, 1.0 eq, chosen from 13–23), 4-O-acetyl-sialic acid (1.2 eq, 1 or 2), and CTP (2.0 eq), MgCl2 (20 mM) were dissolved in Tris-HCl buffer (100 mM, pH 7.1), containing NmCSS (3 mg/mL), and PmST3 (5 mg/mL). The reaction mixture was incubated at 37 °C in an incubator with agitation (100 rpm) for 14 h. The product formation was monitored by MS. The reaction was terminated by adding the same volume (10 mL) of ice-cold EtOH followed by incubation at 4 °C for 30 min. The mixture was centrifuged to remove precipitates. The supernatant was concentrated and passed through a Bio-gel P-2 gel filtration column with water as the eluate to obtain the desired product. The compound was further purified by a reverse-phase C18 column (Phenomenex, 10 μm, 21.2 × 250 mm) with a flow rate of 10 mL/min using a gradient elution of 0–100% acetonitrile in water containing 0.05% formic acid over 20 minutes [Mobile phase A: 0.05% formic acid in water (v/v); Mobile phase B: acetonitrile (v/v); Gradient: 0% B for 3 minutes, 0% to 100% B over 12 minutes, 100% B for 2 minutes, then 100% to 0% B over 3 minutes]. The fractions containing the pure product were collected and concentrated to provide the final purified product.
Neu4,5Ac2α3Galβ4Glc (24)
56 mg, yield 82%; white solid. 1H NMR (800 MHz, D2O) δ 5.20 (d, J = 4.0 Hz, 0.4H, α-isomer), 4.94 (m, 1H), 4.64 (d, J = 8.0 Hz, 0.6H, β-isomer), 4.51 (d, J = 8.0 Hz, 1H), 4.12–3.25 (m, 18H), 2.75 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 1.94 (s, 3H), 1.91 (t, J = 12.0 Hz, 1H); 13C NMR (200 MHz, D2O) δ 174.43, 173.42, 173.09, 102.49, 99.62, 95.65, 78.03, 75.41, 75.02, 74.68, 74.20, 73.67, 72.74, 71.63, 71.04, 69.23, 67.78, 67.05, 62.41, 60.91, 59.93, 49.68, 36.54, 21.75, 20.20; HRMS (ESI) m/z calcd for C25H40NO20 (M-H) 674.2144, found 674.2149.
Neu4,5Ac2α3Galβ4GlcNAcβ3Galβ4Glc (25)
34 mg, yield 78%; white solid. 1H NMR (800 MHz, D2O) δ 5.21 (d, J = 4.0 Hz, 0.4H, α-isomer), 4.97 (m, 1H), 4.69 (d, J = 8.8 Hz, 0.4H, α-isomer), 4.68 (d, J = 8.0 Hz, 0.6H, β-isomer), 4.65 (d, J = 8.0 Hz, 0.6H, β-isomer), 4.55 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.15 (d, J = 4.0 Hz, 1H), 4.13 (dd, J = 3.2 and 9.6 Hz, 1H), 4.09 (t, J = 9.6 Hz, 1H), 3.96–3.26 (m, 28H), 2.76 (dd, J = 4.8 and 12.0 Hz, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.96 (s, 3H), 1.94 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) α/β isomers: δ 174.77, 174.45, 173.08, 172.92, 102.80, 102.77, 102.67, 102.40, 95.61, 81.92, 81.90, 78.25, 78.14, 77.88, 75.42, 74.99, 74.77, 74.68, 74.42, 74.23, 73.65, 72.35, 72.03, 71.44, 71.28, 71.00, 70.86, 70.00, 69.87, 69.84, 69.25, 68.21, 67.78, 67.39, 62.50, 60.87, 60.85, 59.94, 59.72, 55.06, 49.12, 46.55, 36.41, 22.05, 21.77, 20.20. HRMS (ESI) m/z calculated for C39H63N2O30 (M-H) 1039.3466, found 1039.3442.
Neu4,5Ac2α3Galβ3GlcNAcβ3Galβ4Glc (26)
29 mg, yield 65%; white solid. 1H NMR (800 MHz, D2O) δ 5.20 (d, J = 3.2 Hz, 0.4H, α-isomer), 4.93 (m, 1H), 4.71 (d, J = 8.0 Hz, 1H), 4.65 (d, J = 8.0 Hz, 0.6H, β-isomer), 4.50 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.13 (d, J = 3.2 Hz, 1H), 4.09 (dd, J = 3.2 and 9.6 Hz, 1H), 4.07 (t, J = 9.6 Hz, 1H), 3.94–3.26 (m, 27H), 2.75 (dd, J = 4.0 and 12.0 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 3H), 1.95 (s, 3H), 1.89 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) β-isomer: δ 174.81, 174.41, 173.41, 173.09, 103.25, 102.78, 102.40, 99.45, 95,61, 81.99, 81.80, 78.16, 75.24, 75.08, 74.95, 74.78, 74.68, 74.23, 73.65, 72.19, 71.73, 71.03, 69.89, 68.95, 68.33, 67.74, 67.13, 62.30, 60.91, 60.85, 60.38, 59.93, 54.47, 49.14, 36.70, 22.17, 21.76, 20.21. HRMS (ESI) m/z calculated for C39H63N2O30 (M-H) 1039.3466, found 1039.3487.
Neu4,5Ac2α3GalβpNP (27)
13 mg, yield 31%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 5.30 (d, J = 8.0 Hz, 1H), 4.95 (m, 1H), 4.25 (dd, J = 9.6 and 4.0 Hz, 1H), 4.09 (t, J = 10.4 Hz, 1H), 4.04 (d, J = 2.4 Hz, 1H), 3.93–3.57 (m, 10H), 2.79 (dd, J = 12.0 and 4.8 Hz, 1H), 2.06 (s, 3H), 1.95 (s, 3H), 1.92 (t, J = 12.0 Hz, 1H); 13C NMR (200 MHz, D2O) δ 174.48, 173.37, 173.10, 163.03, 162.87, 161.61, 142.42, 126.03, 116.31, 99.69, 99.51, 75.43, 75.31, 72.28, 71.69, 71.02, 68.66, 67.79, 67.16, 62.43, 60.54, 49.17, 36.63, 21.82, 20.27; HRMS (ESI) calculated for C25H33N2O17 (M-H) 633.1779, found 633.1807.
Neu4,5Ac2α3Galβ4GlcβProN3 (28)
28 mg, yield 88%; white solid. 1H NMR (800 MHz, D2O) δ 4.98 (m, 1H), 4.51 (d, J = 8.0 Hz, 1H), 4.47 (d, J = 8.0 Hz, 1H), 4.11 (dd, J = 9.6 and 3.2 Hz, 1H), 4.07 (t, J = 10.4 Hz, 1H), 3.98–3.94 (m, 3H), 3.89–3.55 (m, 14H), 3.44 (t, J = 6.4 Hz, 2H), 3.29 (t, J = 8.8 Hz, 1H), 2.75 (dd, J = 12.8 and 4.8 Hz, 1H), 2.10 (s, 3H), 1.95 (s, 3H), 1.91 (t, J = 12.8 Hz, 1H), 1.89 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.44, 173.42, 173.09, 102.51, 102.02, 99.63, 78.12, 75.41, 75.02, 74.65, 74.22, 72.68, 72.26, 71.63, 71.04, 69.23, 67.78, 67.35, 67.24, 62.41, 60.90, 59.92, 49.16, 47.74, 36.54, 28.11, 21.76, 20.21; HRMS (ESI) calculated for C28H45N4O20 (M-H) 757.2627, found 757.2633.
Neu4,5Ac2α3Galβ4GlcNAcβProN3 (29)
32 mg, yield 85%; white solid. 1H NMR (800 MHz, D2O) δ 4.97 (m, 1H), 4.53 (d, J = 8.0 Hz, 1H), 4.501 (d, J = 8.8 Hz, 1H), 4.12 (dd, J = 10.4 and 3.2 Hz, 1H), 4.08 (t, J = 10.4 Hz, 1H), 3.99–3.54 (m, 18H), 3.36 (m, 2H), 2.75 (dd, J = 12.8 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.02 (s, 3H), 1.95 (s, 3H), 1.94 (t, J = 12.0 Hz, 1H), 1.82 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.44, 174.38, 173.09, 172.78, 102.42, 101.02, 99.29, 78.17, 75.39, 74.97, 74.61, 72.36, 72.22, 71.36, 70.79, 69.24, 67.78, 67.39, 67.00, 62.52, 60.85, 59.90, 54.95, 49.10, 47.63, 36.34, 27.97, 22.02, 21.75, 20.19; HRMS (ESI) calculated for C30H48N5O20 (M-H) 798.2893, found 798.2893.
Neu4,5Ac2α3Galβ3GlcNAcβProN3 (30)
24 mg, yield 86%; white solid. 1H NMR (800 MHz, D2O) δ 4.98 (m, 1H), 4.54 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.10–4.07 (m, 2H), 3.96–3.36 (m, 20H), 2.75 (dd, J = 12.0 and 4.8 Hz, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.95 (s, 3H), 1.94 (t, J = 12.0 Hz, 1H), 1.83 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.46 (2C), 173.12, 172.33, 103.23, 100.77, 98.92, 82.44, 75.53, 75.24, 74.85, 72.36, 71.28, 70.61, 68.96, 68.61, 67.77, 67.26, 67.03, 62.53, 60.80, 60.60, 54.32, 49.06, 47.66, 36.29, 27.97, 22.17, 21.78, 20.19; HRMS (ESI) calculated for C30H48N5O20 (M-H) 798.2893, found 798.2900.
Neu4,5Ac2α3Galβ3GlcNAcαProN3 (31)
21 mg, yield 80%; white solid. 1H NMR (800 MHz, D2O) δ 4.93 (m, 1H), 4.84 (d, J = 4.0 Hz, 1H), 4.50 (d, J = 8.0 Hz, 1H), 4.10–4.05 (m, 3H), 3.93–3.44 (m, 19H), 2.76 (dd, J = 10.8 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 3H), 1.95 (s, 3H), 1.91–1.88 (m, 3H); 13C NMR (200 MHz, D2O) δ 174.42, 174.33, 173.40, 173.10, 103.20, 99.49, 96.97, 80.21, 75.61, 74.90, 72.19, 71.73, 71.48, 71.04, 69.01, 68.55, 67.73, 67.16, 64.82, 62.30, 60.87, 60.39, 52.36, 49.13, 48.04, 36.68, 27.84, 21.92, 21.76, 20.21; HRMS (ESI) calculated for C30H48N5O20 (M-H) 798.2893, found 798.2895.
Neu4,5Ac2α3Galβ3GalNAcβProN3 (32)
22 mg, yield 81%; white solid. 1H NMR (800 MHz, D2O) δ 4.95 (m, 1H), 4.49 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.16 (d, J = 3.2 Hz, 1H), 4.09–4.06 (m, 2H), 4.16–3.59 (m, 16H), 3.52 (dd J = 9.6 and 8.0 Hz, 1H), 3.36 (m, 2H), 2.75 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 3H), 1.94 (s, 3H), 1.92 (t, J = 12.0 Hz, 1H), 1.83 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.62, 174.43, 173.10, 172.92, 104.46, 101.28, 99.22, 79.95, 75.49, 74.62, 74.58, 72.26, 71.49, 70.85, 68.86, 67.76, 67.75, 67.30, 66.88, 62.43, 60.82 (2C), 51.03, 49.10, 47.67, 36.46, 27.99, 22.16, 21.76, 20.19; HRMS (ESI) calculated for C30H48N5O20 (M-H) 798.2893, found 798.2903.
Neu4,5Ac2α3Galβ3GalNAcαProN3 (33)
18 mg, yield 74%; white solid. 1H NMR (800 MHz, D2O) δ 4.94 (m, 1H), 4.89 (d, J = 4.0 Hz, 1H), 4.53 (d, J = 8.0 Hz, 1H), 4.32–3.43 (m, 22H), 2.75 (dd, J = 10.8 and 4.8 Hz, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.95 (s, 3H), 1.91–1.88 (m, 3H); 13C NMR (200 MHz, D2O) δ 174.47, 174.43, 173.43, 173.10, 104.35, 99.55, 97.08, 77.27, 75.60, 74.65, 72.20, 71.73, 71.06, 70.52, 68.95, 68.47, 67.75, 67.29, 64.80, 62.35, 61.11, 60.86, 49.15, 48.57, 48.06, 36.65, 27.85, 21.93, 21.76, 20.21; HRMS (ESI) calculated for C30H48N5O20 (M-H) 798.2893, found 798.2902.
Neu4,5Ac2α3Galβ4GlcNAcβ3Galβ4GlcβProN3 (34)
20 mg, yield 81%; white solid. 1H NMR (800 MHz, D2O) δ 4.93 (m, 1H), 4.67 (d, J = 8.0 Hz, 1H), 4.54 (d, J = 8.0 Hz, 1H), 4.47 (d, J = 8.0 Hz, 1H), 4.41 (d, J = 8.0 Hz, 1H), 4.14 (d, J = 3.2 Hz, 1H), 4.11 (dd, J = 9.6 and 3.2 Hz, 1H), 4.07 (t, J = 10.4 Hz, 1H), 3.98–3.54 (m, 28H), 3.44 (t, J = 6.4 Hz, 2H), 3.29 (t, J = 8.0 Hz, 1H), 2.75 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 3H), 1.94 (s, 3H), 1.91 (t, J = 12.0 Hz, 1H), 1.89 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.78, 174.44, 173.40, 173.08, 102.82, 102.69, 102.41, 101.98, 99.64, 81.93, 78.21, 77.85, 75.41, 75.02, 74.77, 74.65, 74.42, 74.23, 72.65, 72.27, 72.02, 71.62, 71.03, 69.83, 69.25, 68.20, 67.76, 67.36, 67.24, 62.41, 60.91, 60.84, 59.91, 59.70, 55.05, 49.15, 47.73, 36.53, 28.10, 22.04, 21.75, 20.20; HRMS (ESI) calculated for C42H68N5O30 (M-H) 1122.3949, found 1122.3965.
Neu4Ac5Gcα3Galβ4GlcβProN3 (35)
15 mg, yield 67%; white solid. 1H NMR (800 MHz, D2O) δ 4.98 (dt, J = 11.2 and 4.8 Hz, 1H, H-4″), 4.52 (d, J = 8.0 Hz, 1H), 4.47 (d, J = 8.0 Hz, 1H), 4.16–3.55 (m, 21H), 3.44 (t, J = 6.4 Hz, 2H), 3.30 (t, J = 8.8 Hz, 1H), 2.78 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 1.92 (t, J = 12.0 Hz, 1H), 1.89 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.38, 173.45, 173.05, 102.51, 102.01, 99.61, 78.11, 75.39, 75.02, 74.64, 74.21, 72.68, 72.04, 71.67, 70.77, 69.24, 67.72, 67.35, 67.24, 62.39, 60.90, 60.71, 59.92, 48.88, 47.74, 36.65, 28.11, 20.23; HRMS (ESI) calculated for C28H45N4O21 (M-H) 773.2576, found 773.2500.
Neu4Ac5Gcα3Galβ4GlcNAcβProN3 (36)
17 mg, yield 71%; white solid. 1H NMR (800 MHz, D2O) δ 5.02 (dt, J = 11.2 and 4.8 Hz, 1H, H-4″), 4.54 (d, J = 8.0 Hz, 1H), 4.51 (d, J = 8.0 Hz, 1H), 4.15 (t, J = 11.2 Hz, 1H), 4.13 (dd, J = 10.4 and 3.2 Hz, 1H), 4.07–3.55 (m, 20H), 3.37 (m, 2H), 2.80 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.03 (s, 3H), 1.95 (s, 3H), 1.93 (t, J = 12.0 Hz, 1H), 1.82 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.38, 174.38, 173.45, 173.05, 102.45, 101.03, 99.63, 78.17, 75.39, 75.02, 74.62, 72.22, 72.04, 71.67, 70.77, 69.26, 67.73, 67.38, 67.04, 62.39, 60.91, 60.71, 59.90, 54.96, 48.88, 47.65, 36.64, 27.98, 22.05, 20.24; HRMS (ESI) calculated for C30H48N5O21 (M-H) 814.2842, found 814.2870.
Neu4Ac5Gcα3Galβ3GlcNAcβProN3 (37)
14 mg, yield 65%; white solid. 1H NMR (800 MHz, D2O) δ 5.02 (dt, J = 11.2 and 4.8 Hz, 1H, H-4″), 4.54 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.15–3.34 (m, 24H), 2.78 (dd, J = 12.0 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.02 (s, 3H), 1.90 (t, J = 12.0 Hz, 1H), 1.83 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.34, 174.45, 173.43, 173.05, 103.31, 100.78, 99.43, 82.35, 75.54, 75.25, 74.95, 71.96, 71.75, 70.75, 68.94, 68.59, 67.67, 67.14, 67.04, 62.25, 60.89, 60.71, 60.59, 54.33, 48.85, 47.66, 36.79, 27.97, 22.18, 20.22; HRMS (ESI) calculated for C30H48N5O21 (M-H) 814.2842, found 814.2869.
Neu4Ac5Gcα3Galβ3GlcNAcαProN3 (38)
12 mg, yield 56%; white solid. 1H NMR (800 MHz, D2O) δ 5.02 (dt, J = 10.4 and 4.8 Hz, 1H, H-4″), 4.85 (d, J = 3.2 Hz, 1H), 4.51 (d, J = 8.0 Hz, 1H), 4.15 (t, J = 10.4 Hz, 1H), 4.12–3.42 (m, 23H), 2.79 (dd, J = 12.8 and 4.8 Hz, 1H), 2.05 (s, 3H), 2.02 (s, 3H), 1.91 (t, J = 12.8 Hz, 1H), 1.89 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.35, 174.32, 173.43, 173.06, 103.19, 99.48, 96.98, 80.19, 75.60, 74.90, 71.96, 71.76, 71.48, 70.76, 69.02, 68.54, 67.67, 67.14, 64.83, 62.27, 60.87, 60.71, 60.39, 52.36, 48.85, 48.05, 36.78, 27.84, 21.93, 20.23; HRMS (ESI) calculated for C30H48N5O21 (M-H) 814.2842, found 814.2875.
Neu4Ac5Gcα3Galβ3GalNAcβProN3 (39)
10 mg, yield 51%; white solid. 1H NMR (800 MHz, D2O) δ 5.02 (m, 1H), 4.50 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.16–3.37 (m, 24H), 2.75 (dd, J = 12.0 and 4.8 Hz, 1H), 2.06 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 1.90 (t, J = 12.0 Hz, 1H), 1.83 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.34, 174.61, 173.48, 173.04, 104.49, 101.28, 99.44, 79.90, 75.49, 74.64, 74.63, 71.94, 71.73, 70.77, 68.85, 67.75, 67.70, 67.20, 66.87, 62.28, 60.86, 60.82, 60.70, 51.04, 48.85, 47.66, 36.78, 27.99, 22.17, 20.21; HRMS (ESI) calculated for C30H48N5O21 (M-H) 814.2842, found 814.2872.
Neu4Ac5Gcα3Galβ3GalNAcαProN3 (40)
11 mg, yield 52%; white solid. 1H NMR (800 MHz, D2O) δ 5.02 (dt, J = 10.4 and 4.8 Hz, 1H, H-4″), 4.89 (d, J = 4.0 Hz, 1H), 4.53 (d, J = 8.0 Hz, 1H), 4.30 (dd, J = 11.2 and 4.0 Hz, 1H), 4.23 (d, J = 3.2 Hz, 1H), 4.15 (t, J = 10.4 Hz, 1H), 4.10–3.41 (m, 21H), 2.78 (dd, J = 12.8 and 4.8 Hz, 1H), 2.04 (s, 3H), 2.01 (s, 3H), 1.90 (t, J = 12.8 Hz, 1H), 1.88 (m, 2H); 13C NMR (200 MHz, D2O) δ 175.35, 174.45, 173.44, 173.05, 104.34, 99.51, 97.07, 77.24, 75.58, 74.64, 71.97, 71.75, 70.77, 70.51, 68.95, 68.47, 67.69, 67.27, 64.79, 62.31, 61.10, 60.85, 60.70, 48.85, 48.56, 48.05, 36.74, 27.84, 21.93, 20.22; HRMS (ESI) calculated for C30H48N5O21 (M-H) 814.2842, found 814.2874.
Neu4Ac5Gcα3Galβ4GlcNAcβ3Galβ4GlcβProN3 (41)
10 mg, yield 62%; white solid. 1H NMR (800 MHz, D2O) δ 5.04 (dt, J = 10.4 and 4.8 Hz, 1H, H-4‴), 4.70 (d, J = 8.8 Hz, 1H), 4.56 (d, J = 8.0 Hz, 1H), 4.49 (d, J = 8.0 Hz, 1H), 4.44 (d, J = 8.0 Hz, 1H), 4.18–3.57 (m, 33H), 3.46 (t, J = 6.4 Hz, 2H), 3.12 (t, J = 8.0 Hz, 1H), 2.80 (dd, J = 12.0 and 4.8 Hz, 1H), 2.07 (s, 3H), 2.03 (s, 3H), 1.91 (t, J = 12.0 Hz, 1H), 1.91 (m, 2H); 13C NMR (200 MHz, D2O) δ 174.78, 174.44, 173.40, 173.08, 102.82, 102.69, 102.41, 101.98, 99.64, 81.93, 78.21, 77.85, 75.41, 75.02, 74.77, 74.65, 74.42, 74.23, 72.65, 72.27, 72.02, 71.62, 71.03, 69.83, 69.25, 68.20, 67.76, 67.36, 67.24, 62.41, 60.91, 60.84, 59.91, 59.70, 55.05, 49.15, 47.73, 36.53, 28.10, 22.04, 21.75, 20.20; HRMS (ESI) calculated for C42H68N5O31 (M-H) 1138.3898, found 1138.3938.
Gram-scale OP2E synthesis of monotreme milk trisaccharide Neu4,5Ac2α3Lac
Lactose (1.09 g, 3.05 mmol), Neu4,5Ac2 (1.07 g, 3.05 mmol), and CTP (1.93 g, 3.66 mmol) were dissolved in water (30 mL) and the pH of the solution was adjusted to 7.1 using NaOH solution (4 M). MgCl2 (20 mM), Tris-HCl buffer (100 mM, pH 7.1), NmCSS (15 mg), and PmST3 (20 mg) were then added and the total volume of the reaction mixture was brought up to 100 mL by adding H2O. The reaction mixture was incubated in an incubator shaker with shaking (100 rpm) at 30 °C for overnight. The reaction was monitored by mass spectrometry (MS). Additional CTP (0.96 g) was added to the reaction mixture and the pH was carefully adjusted to pH 7.1 using NaOH solution (4 M). The reaction was allowed to continue for another 20 h at room temperature until an optimal yield was achieved. After adding ethanol (100 mL), the reaction mixture was incubated at 4 °C for 1 h. The precipitates were removed by centrifuge and the supernatant was concentrated. The residue was purified using a ODS-SM column (51 g, 50 μM, 120 Å, Yamazen) on a CombiFlash® Rf 200i system. Mobile phase A: water; Mobile phase B: acetonitrile; Gradient: 10% B for 6 minutes, 10% to 100% B over 12 minutes, 100% B for 6 minutes, then 100% to 80% B over 1 minutes. The fractions containing the pure product were collected and concentrated to produce the final purified product Neu4,5Ac2α3Galβ4Glc (1.33 g, 71%) as a white powder. The identity and the purity of the product were confirmed by NMR and HRMS.
Sialidase assays
Sialidase assays were carried out in duplicate at 37 °C for 40 min (for bacterial sialidases) or 30 min (for human influenza A viruses) in a 384-well plate (Fisher Scientific, Chicago, IL) in a final volume of 20 μL containing Neu4,5Ac2α3GalβpNP (0.3 mM) and Aspergillus oryzae β-galactosidase (12 μg, 126 mU). The amount of the β-galactosidase required to completely hydrolyze GalβpNP (0.3 mM) within the time frame of the assay was predetermined and confirmed by control assays with GalβpNP (0.3 mM). The reactions were stopped by adding 40 μL of N-cyclohexyl-3-aminopropane sulfonic acid (CAPS) buffer (0.5 M, pH 11.5). The amount of para-nitrophenolate formed was determined by measuring the A405 nm of the reaction mixtures using a microtiter plate reader. Neu5Acα3GalβpNP50 was used as a control.
The assay conditions for bacterial sialidases were: A. ureafaciens sialidase (1.5 mU) in sodium acetate buffer (100 mM, pH 5.5); C. perfringens sialidase (3 mU) in MES buffer (100 mM, pH 5.0); V. cholerae sialidase (1.5 mU) in sodium acetate buffer (100 mM, pH 5.5) containing NaCl (150 mM) and CaCl2 (10 mM); S. pneumoniae sialidase (1.5 mU) in sodium acetate buffer (100 mM, pH 6.0); PmST1 (10 μg) in sodium acetate buffer (100 mM, pH 5.5). Viral sialidase assays were carried out in a MES buffer (100 mM, pH 5.0) containing a purified human influenza A virus (1×105 HAU mL−1) selected from A/Puerto Rico/34/8 H1N1 (A/PR8), A/Philippines/2/82/X-79 H3N2 (A/Philips), A/Memphis/71 H3N1 (A/Mem71), and A/Udorn/307/72 H3N2 (A/Udorn72).51
To test whether the cleavage of Neu4,5Ac2 by human influenza A viruses was caused by de-O-acetylation or direct de-sialylation, Neu4,5Ac2α3Lac (10 mM) was incubated with purified human influenza A viruses (1×105 HAU mL−1) in a MES buffer (100 mM, pH 5.0) for 30 min at 37 °C. The reaction mixture was then subjected to high-resolution mass spectrometry analysis.
Supplementary Material
Acknowledgments
This work was supported by US NIH grants R01HD065122, R01GM076360-04S1 (to X.C.), and U01CA199792 (to A.V.) as well as scholarships from China Scholarship Council (to J.Z. and B.S). Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538. We would like to thank Professor Nicole Baumgarth at the University of California-Davis for providing purified human influenza viruses as kind gifts.
H.Y., Y.L., and X.C. are co-founders of Glycohub, Inc., a company focused on the development of carbohydrate-based reagents, diagnostics, and therapeutics. Glycohub, Inc. played no role in the design, execution, interpretation, or publication of this study.
Footnotes
Electronic Supplementary Information (ESI) available: HRMS assay data, 1H and 13C NMR spectra for products. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Varki A. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schauer R. Glycoconj J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angata T, Varki A. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Varki A. ACS Chem Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schauer R. Adv Carbohydr Chem Biochem. 1982;40:131–234. doi: 10.1016/s0065-2318(08)60109-2. [DOI] [PubMed] [Google Scholar]
- 6.Hanaoka K, Pritchett TJ, Takasaki S, Kochibe N, Sabesan S, Paulson JC, Kobata A. J Biol Chem. 1989;264:9842–9849. [PubMed] [Google Scholar]
- 7.Schauer R, Kamerling JP. In: Glycoproteins II. Montreuil J, Vliegenthart JFG, Schachter H, editors. Elsevier; Amsterdam: 1997. pp. 243–402. [Google Scholar]
- 8.Kamerling JP, Dorland L, van Halbeek H, Vliegenthart JF, Messer M, Schauer R. Carbohydr Res. 1982;100:331–340. doi: 10.1016/s0008-6215(00)81046-0. [DOI] [PubMed] [Google Scholar]
- 9.Inoue S, Iwasaki M, Ishii K, Kitajima K, Inoue Y. J Biol Chem. 1989;264:18520–18526. [PubMed] [Google Scholar]
- 10.Lochnit G, Geyer R. Eur J Biochem. 1995;228:805–816. [PubMed] [Google Scholar]
- 11.Klein A, Roussel P. Biochimie. 1998;80:49–57. doi: 10.1016/s0300-9084(98)80056-4. [DOI] [PubMed] [Google Scholar]
- 12.Oftedal OT, Nicol SC, Davies NW, Sekii N, Taufik E, Fukuda K, Saito T, Urashima T. Glycobiology. 2014;24:826–839. doi: 10.1093/glycob/cwu041. [DOI] [PubMed] [Google Scholar]
- 13.Urashima T, Inamori H, Fukuda K, Saito T, Messer M, Oftedal OT. Glycobiology. 2015;25:683–697. doi: 10.1093/glycob/cwv010. [DOI] [PubMed] [Google Scholar]
- 14.Chen X. Adv Carbohydr Chem Biochem. 2015;72:113–190. doi: 10.1016/bs.accb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyoshi I, Higashi H, Hirabayashi Y, Kato S, Naiki M. Mol Immunol. 1986;23:631–638. doi: 10.1016/0161-5890(86)90100-8. [DOI] [PubMed] [Google Scholar]
- 16.Iwasaki M, Inoue S, Troy FA. J Biol Chem. 1990;265:2596–2602. [PubMed] [Google Scholar]
- 17.Iwersen M, Vandamme-Feldhaus V, Schauer R. Glycoconj J. 1998;15:895–904. doi: 10.1023/a:1006911100081. [DOI] [PubMed] [Google Scholar]
- 18.Pritchett TJ, Paulson JC. J Biol Chem. 1989;264:9850–9858. [PubMed] [Google Scholar]
- 19.Levinson B, Pepper D, Belyavin G. J Virol. 1969;3:477–483. doi: 10.1128/jvi.3.5.477-483.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regl G, Kaser A, Iwersen M, Schmid H, Kohla G, Strobl B, Vilas U, Schauer R, Vlasak R. J Virol. 1999;73:4721–4727. doi: 10.1128/jvi.73.6.4721-4727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langereis MA, Zeng Q, Heesters BA, Huizinga EG, de Groot RJ. PLoS Pathog. 2012;8:e1002492. doi: 10.1371/journal.ppat.1002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellebo A, Vilas U, Falk K, Vlasak R. J Virol. 2004;78:3055–3062. doi: 10.1128/JVI.78.6.3055-3062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogura H, Furuhata K, Sato S, Anazawa K, Itoh M, Shitori Y. Carbohydr Res. 1987;167:77–86. doi: 10.1016/0008-6215(87)80269-0. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X. Appl Microbiol Biotechnol. 2008;79:963–970. doi: 10.1007/s00253-008-1506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Yu H, Cao H, Muthana S, Chen X. Appl Microbiol Biotechnol. 2012;93:2411–2423. doi: 10.1007/s00253-011-3579-6. [DOI] [PubMed] [Google Scholar]
- 28.Higa HH, Paulson JC. J Biol Chem. 1985;260:8838–8849. [PubMed] [Google Scholar]
- 29.Yu H, Chokhawala H, Karpel R, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 30.Thon V, Lau K, Yu H, Tran BK, Chen X. Glycobiology. 2011;21:1206–1216. doi: 10.1093/glycob/cwr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thon V, Li Y, Yu H, Lau K, Chen X. Appl Microbiol Biotechnol. 2012;94:977–985. doi: 10.1007/s00253-011-3676-6. [DOI] [PubMed] [Google Scholar]
- 32.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem Int Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding L, Zhao C, Qu J, Li Y, Sugiarto G, Yu H, Wang J, Chen X. Carbohydr Res. 2015;408:127–133. doi: 10.1016/j.carres.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X. J Am Chem Soc. 2009;131:18467–18477. doi: 10.1021/ja907750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng J, Yu H, Lau K, Huang S, Chokhawala HA, Li Y, Tiwari VK, Chen X. Glycobiology. 2008;18:686–697. doi: 10.1093/glycob/cwn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malekan H, Fung G, Thon V, Khedri Z, Yu H, Qu J, Li Y, Ding L, Lam KS, Chen X. Bioorg Med Chem. 2013;21:4778–4785. doi: 10.1016/j.bmc.2013.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Chen X. Org Biomol Chem. 2016;14:2809–2818. doi: 10.1039/c6ob00058d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu H, Lau K, Thon V, Autran CA, Jantscher-Krenn E, Xue M, Li Y, Sugiarto G, Qu J, Mu S, Ding L, Bode L, Chen X. Angew Chem Int Ed. 2014;53:6687–6691. doi: 10.1002/anie.201403588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem Commun. 2010;46:6066–6068. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu H, Thon V, Lau K, Cai L, Chen Y, Mu S, Li Y, Wang PG, Chen X. Chem Commun. 2010;46:7507–7509. doi: 10.1039/c0cc02850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen C, Zhang Y, Xue M, Liu XW, Li Y, Chen X, Wang PG, Wang F, Cao H. Chem Commun. 2015;51:7689–7692. doi: 10.1039/c5cc01330e. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Yu H, Chen Y, Lau K, Cai L, Cao H, Tiwari VK, Qu J, Thon V, Wang PG, Chen X. Molecules. 2011;16:6396–6407. doi: 10.3390/molecules16086396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X. Chem Commun. 2011;47:10815–10817. doi: 10.1039/c1cc14034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai L, Ban L, Guan W, Mrksich M, Wang PG. Carbohydr Res. 2011 doi: 10.1016/j.carres.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blixt O, van Die I, Norberg T, van den Eijnden DH. Glycobiology. 1999;9:1061–1071. doi: 10.1093/glycob/9.10.1061. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Xue M, Sheng X, Yu H, Zeng J, Thon V, Chen Y, Muthana MM, Wang PG, Chen X. Bioorg Med Chem. 2016;24:1696–1705. doi: 10.1016/j.bmc.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X, Liu Z, Zhang J, Zhang W, Kowal P, Wang PG. Chembiochem. 2002;3:47–53. doi: 10.1002/1439-7633(20020104)3:1<47::AID-CBIC47>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 49.Muthana MM, Qu J, Li Y, Zhang L, Yu H, Ding L, Malekan H, Chen X. Chem Commun. 2012;48:2728–2730. doi: 10.1039/c2cc17577k. [DOI] [PubMed] [Google Scholar]
- 50.Chokhawala HA, Yu H, Chen X. Chembiochem. 2007;8:194–201. doi: 10.1002/cbic.200600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Cao H, Dao N, Luo Z, Yu H, Chen Y, Xing Z, Baumgarth N, Cardona C, Chen X. Virology. 2011;415:12–19. doi: 10.1016/j.virol.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.