

Abstract
Synucleinopathies, including Parkinson’s disease (PD), are neurodegenerative diseases characterized by accumulation of α-synuclein (SYN), a small neuronal protein with prion like properties that plays a central role in PD pathogenesis. SYN can misfold and generate toxic oligomers/aggregates, which can be cytotoxic. Environmental arsenic (As)-containing pesticide use correlates with increased incidence of PD. Moreover, because As exposure can lead to inhibition of autophagic flux we hypothesize that As can facilitate the accumulation of toxic SYN oligomers/aggregates and subsequent increases in markers of autophagy. We therefore examined the role of As in the oligomerization of SYN, and the consequences thereof. Chronic exposure of SH-SY5Y cells overexpressing SYN to As caused a dose-dependent oligomerization of SYN, with concomitant increases in protein ubiquitination and expression of other stress markers (protein glutathione binding, γ-GCS, light chain 3 (LC3)-I/II, P62, and NAD(P)H dehydrogenase quinone 1), indicative of an increased proteotoxic stress. Immunocytochemical analyses revealed an accumulation of SYN, and it’s colocalization with LC3, a major autophagic protein. Mice exposed to As (100 ppb) for 1 month, exhibited elevated SYN accumulation in the cortex and striatum, and elevations in protein ubiquitination and LC3-I and II levels. However, tyrosine hydroxylase (TH), an indicator of dopaminergic cell density, was upregulated in the As exposed animals. Because SYN can inhibit TH function, and As can decrease monoamine levels, As exposure possibly leads to compensatory mechanisms leading to an increase in TH expression. Our findings suggest that susceptible individuals may be at higher risk of developing synucleinopathies and/or neurodegeneration due to environmental As exposure.
Keywords: arsenic, α-synuclein, Parkinson’s disease, autophagy.
Parkinson’s disease (PD) is a multifactorial neurodegenerative disorder affecting approximately 2% of the population above the age of 60. PD is a consequence of the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), with clinical manifestations of major motor dysfunction appearing only after a significant population (< 60%) of dopaminergic neurons have already been lost (Jankovic, 2008). The underlying cause of such a drastic and selective loss of dopaminergic neurons in PD is poorly understood, due to the multifactorial nature of the disease. However, it is estimated that approximately 10% of PD cases are due to genetic factors, with additional cases arising as a consequence of gene (x) and/or environmental exposures (Wirdefeldt et al., 2011). The most distinctive pathological feature of PD at the cellular level is the development of intracellular inclusions known as Lewy Bodies (LBs), enlarged aggrosomes formed for cellular protection (Tanaka et al., 2004). The major component of LBs is α-synuclein (SYN), a small neuronal protein with prion like properties (Herva and Spillantini, 2015; Prusiner et al., 2015). The normal function of SYN is not well understood. However, SYN does play a role in dopamine (DA) homeostasis via inhibition of the activity and expression of the rate-limiting enzyme in the biosynthesis of DA, tyrosine hydroxylase (TH) (Alerte et al., 2008; Perez et al., 2002; Yu et al., 2004). Although physiological levels of SYN monomers themselves are not toxic, SYN oligomers, aggregates, and fibrils do exhibit the potential to be cytotoxic, with oligomers and soluble aggregates apparently being the principal culprits (Conway et al., 2000, 2001; Sharon et al., 2003; Waxman and Giasson, 2009).
SYN oligomerization and aggregation are accelerated by DA. DA-modified SYN stabilizes SYN oligomer/protofibril formation and DA-modified SYN has the ability to block chaperone-mediated autophagy (CMA), thus augmenting the neurotoxic process (Conway et al., 2001; Martinez-Vicente et al., 2008). By relieving the inhibition on DA synthesis, DA-modified SYN may stimulate the biosynthesis of DA, which can subsequently further modify SYN. Interestingly, environmental arsenic (As) exposure plays an important role in protein aggregation pathways (Jacobson et al., 2012; Lau et al., 2013). As is a naturally occurring element found in food and water sources around the world, reaching μM concentrations in some parts of the world. At these levels, As interferes with the cellular antioxidant response, including thiol content depletion, increased reactive oxygen species (ROS) production, reduced superoxide dismutase levels, and inhibition of chaperone function, resulting in cellular protein aggregation (Ellinsworth, 2015; Jacobson et al., 2012; Jomova et al., 2011; Kim et al., 2014; Liu et al., 2000). Thus, the modus operandi of As toxicity closely resembles the cellular changes seen during PD progression. Moreover, As crosses the blood brain barrier and reduces neurotransmitter levels, including monoamines (Yadav et al., 2010). Indeed, coexposure to As and DA has synergistic neurotoxic effects in cellular models of PD, such as SH-SY5Y neuroblastoma cells (Shavali and Sens, 2008). Critically, epidemiological studies revealed significant associations between As pesticide exposure and late onset PD (Elbaz et al., 2009). Although animals exposed to As exhibit neurotoxic pathology, no studies have examined the role of As in the development of SYN oligomers/aggregates nor their role in the development of PD. We hypothesize that low level As exposure will lead to increases in the oligomerization of SYN, causing increased cellular proteotoxicity and the engagement of autophagy concomitant with an inhibition of autophagic flux, leading to cell death.
MATERIALS AND METHODS
Generation of SH-SY5Y Cells Overexpressing SYN
SH-SY5Y cells were obtained from the American Type Culture Collection (CRL-2266, ATCC, Manassas, Virginia). Cells were grown in DMEM/F12 50/50 supplemented with 10% fetal bovine serum (FBS). Human SNCA plasmid bacterial stock (EX-G0543-M02) was obtained from GeneCopoeia (Rockville, Maryland). The bacterial stock was grown on agar plates containing 100 µg/ml ampicillin. Selected colonies were grown in LB broth containing 100 µg/ml ampicillin. Bacteria were pelleted by centrifugation. Plasmid was purified using a Qiagen plasma mega kit (Qiagen, Valencia, California). SH-SY5Y cells were transfected with SNCA Plasmid using Lipofectamine 2000 (ThermoFisher Scientific, Waltham, Massachusetts), at a 1:3 ratio, using OPTI-MEM medium. Cells were selected using 400 µl/ml G418/Geneticin over a period of 1 month. Media was changed every 3–4 days, or as needed. Selected cells were analyzed by Western blot to determine SYN expression levels. For long-term storage, cells were placed in DMEM/F12 50/50 supplemented with 10% FBS and 5% DMSO and maintained in liquid nitrogen.
In vitro Immunocytochemistry
SH-SY5Y cells were plated on 4-well chamber slides, 25K cells per well with DMEM/F12 media supplemented with 10% FBS. After 24 h, cells were differentiated by DMEM/F12 supplemented with 1% FBS and 10 µM all trans retinoic acid (ATRA) for 24 h. After differentiation cells were treated with 25 ppb sodium arsenite for 72 h. All in vitro immunocytochemistry (ICC) steps were conducted at room temperature, unless mentioned otherwise. Cells were fixed in 4% paraformaldehyde for 20 min. Slides were washed in PBS and blocked with 10% goat serum (0.3% Triton X in PBS) for 45 min. Slides were incubated with anti-SYN primary antibody (S-3062, Sigma, St Louis, Missouri) 1:1000 dilution (1% goat serum, 1% bovine serum albumin (BSA), 0.1% triton X in PBS) overnight in 4°C. Slides were washed 3 times with PBS (0.1% Triton X, 0.1% BSA) and incubated with Alexa Fluor 594 goat anti-rabbit secondary antibody 1: 3000 (A-11037, Life Technologies, Carlsbad, California) for 1 h. After washing slides 3 times, they were incubated with nuclear stain Hoeschst 33342 (1 µg/ml) for 5 min and washed once with PBS and water. For colocalization staining of SYN and microtubule-associated protein 1A/1B-light chain 3 (LC3), slides were simultaneously incubated with anti-SYN primary antibody (S-3062, Host-Rabbit, Sigma) 1: 1000, and anti-LC3 primary antibody (2G6, Host-Mouse, Enzo Lifesciences, Farmingdale, New York) 1: 1000 (1% goat serum, 1% BSA, 0.1% Triton X in PBS) overnight at 4°C. After washing, slides were incubated with Alexa Fluor 594 goat anti-rabbit secondary antibody 1:3000 (A-11037, Life Technologies) and goat anti-mouse secondary antibody 1:3000 (A-11029, Life Technologies) for 1 h. Slides were visualized by fluorescence microscopy with channels selected for Texas Red (EX 589 nm, EM 615 nm), FITC (EX 490 nm, EM 525 nm), and DAPI nuclear stain (EX 345 nm, EM 455 nm).
In vitro As exposure and Western Blots
Stable SH-SY5Y cell lines overexpressing SYN were cultured on 6-well plates (1 million cells per well) with DMEM/F12 media supplemented with 10% FBS, G418/Geneticin (400 µg/ml), and ampicillin (100 µg/ml). After 24 h, fresh media was provided (without antibiotics) and cells were exposed to various concentrations of sodium arsenite (5, 10, 25, and 50 ppb). After 24, 48, and/or 72 h, cells were lysed using 1X Bolt LDS sample buffer (ThermoFisher Scientific). Lysate mixtures were boiled (10 min), followed by sonication (approximately 4 Watts power, 3 × 10 s). Lysate mixtures were centrifuged for 1 min (14 000 RCF) and mixed with 3:1 4× XT sample buffer (Bio-Rad Laboratories, Hercules, California) with 5% 2-beta mercaptoethanol (reducing agent was not used for anti-glutathione [GSH] antibody). Samples were boiled for an additional 5 min and samples loaded onto 12% acrylamide gels. Protein from gels was transferred to a PVDF membrane and blocked with 5% milk (TBST, 0.1% tween 20) for 30 min. Membranes were incubated with primary antibody (anti-SYN antibody; 1: 1000, [S-3062, Sigma], anti-actin antibody; 1: 10000, [8H10D10, Cell Signaling Technology Inc, Danvers, Massachusetts], anti-ubiquitin [UB] antibody; 1: 5000, [REF: 07/2012, Cell Signaling Technology Inc], anti-GSH antibody; 1: 200, [sc-52399, Santa Cruz Biotechnology, Dallas, Texas] anti-LC3 antibody [L-7543, Sigma], anti-SQSTM1/P62 antibody [sc-28359, Santa Cruz Biotechnology], anti-NQO1 antibody [sc-32793, Santa Cruz Biotechnology], anti-γ-GCS antibody [sc-55586, Santa Cruz Biotechnology]) in 4°C overnight, followed by 6 × 10 min washes (TBST, 0.1% Tween 20). Membranes were incubated in appropriate secondary antibody (goat anti-mouse IgG–HRP, 1: 2500, [sc-2005, Santa Cruz Biotechnology], goat anti-rabbit Ig–HRP; 1: 3000, [656120, Life Technologies]) for 1 h and washed as previously described. Gels were imaged with Pierce ECL Western blotting substrate using BIORAD Imager, with the Image Lab software (Beta 2, Version 3.0.1, Bio-Rad Laboratories).
In vivo As Exposure and Behavioral Studies
Wild-type female Swiss Webster mice were obtained from Harlan Sprague-Dawley Inc (Indianapolis, Indiana). Animals were housed in the Animal Care Facility at the University of Arizona under a 12:12 h light/dark cycle (lights on 07:00 am) in temperature-controlled rooms. All procedures were approved by the University of Arizona Institutional Animal Care and Use Committee and conformed to National Institutes of Health guidelines. Food and water were provided ad libitum. Animals at 2 month age were exposed to 100 ppb arsenic (NaAsO2) through the drinking water for 2 weeks and/or 5 weeks (Control n = 3, As 2 weeks n = 2 [1 animal was found moribund prior to reaching geriatric age], As 5 weeks n = 4). After the exposure, animals were maintained under identical conditions, until geriatric age (18 months old). At geriatric age, animals were subject to a motor function beam test. Beam length was set to 1 m, with the start point being 40 mm wide (narrowing uniformly toward endpoint) and endpoint being 4 mm wide immediately prior to home cage. For training, animals were placed on the start point under intense light and directed toward home cage. After 4 training sessions, animals underwent the beam test once per day for 4 days. Distance traveled and other behavioral factors, such as fine motor coordination, pertaining to PD were recorded. One animal was excluded from the behavioral tests (unable to train). Therefore, for behavioral tests the final animal number was: Control n = 3, As 2 weeks n = 2, As 5 weeks n = 3.
In vivo Immunohistochemistry
Animals were dosed with 100 mg/kg pentobarbital. After full anesthesia was achieved animals were subjected to transcardial perfusion, initially with 50 ml PBS (pH 7.4; room temperature) followed by 50 ml 4% paraformaldehyde in PBS (pH 7.4). Brains were removed and fixed in 4% paraformaldehyde overnight at room temperature, followed by 24 h at 4°C. After fixation, fixative solution was removed and replaced with 70% ethanol until further processing. Brains were embedded in paraffin, and 7 µm thick coronal sections were obtained from Bregma Zero and placed on microscope slides. Sections were deparafinized (xylene, 3 × 4 min), followed by hydration (100% ethanol, 3 × 4 min, 95% ethanol, 2 × 4 min, 70% ethanol, 4 min, DI water 2 × 2 min). Antigen retrieval was performed by incubating the sections in 80°C citrate buffer (10 mM Citric acid + 0.05% Tween 20, pH 6) for 20 min. Buffer was replaced with fresh room temperature buffer and placed on ice for 20 min, followed by buffer removal and placement in running tap water for 10 min. Slides were washed with TBS (3 × 3 min) and water (3 × 3 min). An endogenous peroxidase blocking step was performed (1:10 hydrogen peroxide: methanol, 20 min), followed by TBS (3 × 3 min) and water (3 × 3 min) washes. The blocking step was performed (1.5% Normal Goat Serum [S-1000, Vector Laboratories Inc Burlingame, California] in Normal Antibody Diluent [1X TBST 0.5% Triton X, 1% BSA]) for 1 h. Blocking buffer was removed and slides were incubated with TH primary antibody at a concentration of 1:2000 (AB 152, EMD Millipore, Billerica, Massachusetts) and/or anti-SYN primary antibody at a concentration of 1:1000 (S-3062, Sigma) for 90 min at room temperature, and 36 h in 4°C. Primary antibody was removed and slides were washed with TBST (3 × 3 min). Slides were incubated with secondary antibody (goat anti-rabbit-IgG, BA-1000, Vector Laboratories Inc) at room temperature for 1 h, followed by TBS washes (5 × 5 min). For visualization, VECTASTAIN Elite ABC Kit (PK-6100 Vector Laboratories Inc) and Betazoid DAB chromogen Kit (BDB2004L, Biocare Medical, Concord, California) were used as per manufacturer recommendations. Slides were counterstained with hematoxylin/eosin and dehydrated in the reverse order mentioned earlier. Slides were cover-slipped with cytoseal mounting medium and analyzed by a blinded third party neuroscientist with bright-field microscopy to avoid experimenter bias.
In vivo Western Blots
The sectioned brain samples were deparafinized and hydrated as previously described, subsequently removed from the microscope slide, and placed in 100 µl of 1X Bolt LDS sample buffer (ThermoFisher Scientific). Sections were boiled for 30 min and sonicated with a tip sonicator (4 Watts power, 6 × 10 s). Sample buffer (33 µl of 4× XT; Bio-Rad Laboratories) with 5% 2-β-mercaptoethanol was added, and samples were boiled for another 10 min. Samples were centrifuged for 1 min (14 000 RCF) and 30 µl aliquots loaded on 12% acrylamide gels. The remainder of the Western blot procedure was performed as previously described. Glyceraldehyde-3-phosphate dehydrogenase ([GAPDH], anti-GAPDH antibody, 8245, Abcam, Cambridge, Massachusetts) was used as the loading control for the in vivo Western blots.
Densitometry Analysis and Statistical Analysis
Densitometry was performed using the Image Lab software (Beta 2, Version 3.0.1, Bio-Rad Laboratories), with Actin as the in vitro, and GAPDH as the in vivo loading control/normalizing control. Proteins with 75 kDa molecular weight or above were considered as high molecular weight oligomers for UB and SYN. For GSH-binding to proteins, bands between 37 and 50 kDa were chosen for densitometry. Column statistics with 2 tailed T-test were used for in vitro Western blot samples. Due to 2 exposure time points in the in vivo experiments (2 and 5 weeks), 1-way ANOVA with post hoc Tukey’s multiple comparison tests were performed on the in vivo Western blot samples. All statistical analysis was performed using GraphPad Prism (Version 6.07, GraphPad Software Inc, La Jolla, California). Results are expressed as mean values (behavioral test), absolute values (weight), normalized values (densitometry), and represent the mean ± SE (in vitro; n = 3–5, in vivo; Control n = 3, As 2 weeks n = 2, As 5 weeks n = 4).
RESULTS
Arsenic Induces Accumulation of SYN in SH-SY5Y Cells
SH-SY5Y cells are a neuroblastoma cell line with robust research applications, extensively used for PD and other neurological disease investigations. These cells can differentiate into multiple neuronal phenotypes, including dopaminergic neurons (Korecka et al., 2013). SH-SY5Y cells predifferentiated with ATRA for 24 h developed intracellular inclusions, which resembled LBs, 72 h after exposure to As (Figure 1B), and which were absent from their untreated counterparts (Figure 1A). To better identify the origin of intracellular inclusions, potential colocalization of SYN with LC3, which plays a major role in autophagy (Lau et al., 2013), was investigated. Colocalization of SYN and LC3 (Figure 1C) confirmed the formation of autophagic vacuoles 72 h after exposure to 25 ppb As, similar to those observed by Lau et al. (2013). Western blot analysis of SH-SY5Y cells overexpressing SYN demonstrated a comparable results 72 h after exposure to As. However, we were unable to reliably detect these oligomers after 24, and 48 h of exposure to As (data not shown). Nonetheless, SYN monomers accumulated 72 h after exposure to As, and high molecular weight SYN oligomers were visible at longer Western blot imaging times (Figure 2), with 25 and 50 ppb exposed samples being significantly different (P < .05) from control (Figure 3). Other proteotoxic stress related parameters were also assessed, including UB-bound proteins (Figure 2), and glutathionylated proteins (Figure 2), an indirect measure of oxidative stress. UB-bound proteins increased with all concentrations, with significant (P < .05) increases at 50 ppb As exposure. γ-glutamylcysteine synthetase (γ-GCS), the rate-limiting enzyme in GSH synthesis, was also elevated after As exposure, reaching statistical significance (P < .05) at higher concentrations of As (10 and 25 ppb). LC3 I/II and its binding partner, UB-binding protein P62 (P62) also known as sequestosome 1, were also elevated in a dose-dependent manner in the As-treated samples (Figure 2). Thus, LC3 I was significantly (P < .01) elevated at 25 and 50 ppb As, while LC3 II and p62 were significantly elevated at all 3 As concentrations (10, 25, and 50 ppb; Figure 3). NAD(P)H dehydrogenase quinone 1 (NQO1) protein was not detectable at 10 or 25 ppb As (Figs. 2 and 3) but 50 ppb As exposure resulted in a > 10-fold increase in protein levels (P < .01, Figure 3). All stress markers; SYN oligomers, protein ubiquitination, protein GSH binding, γ-GCS, LC3 I/II, P62, and NQO1 increased in a concentration-dependent manner, with increases being especially visible following chronic 72 h 25 and 50 ppb As exposure.
FIG. 1.
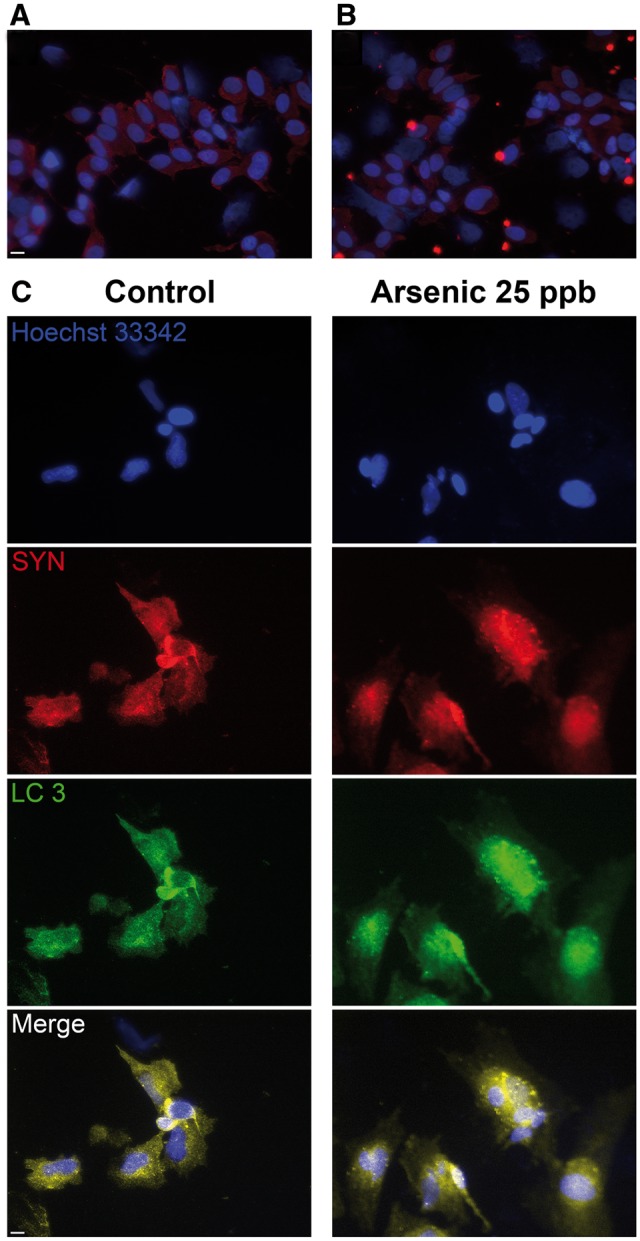
As exposure stimulates the accumulation of α-synuclein (SYN) into intracellular inclusions and it’s colocalization with the autophagy marker light chain 3 (LC3). All trans retinoic acid differentiated SH-SY5Y cells were exposed to 25 ppb As for 72 h, and immunocytochemical analysis performed on (A) control, (B) As-exposed cells, (C) Colocalization of SYN (red) and LC3 (green) in As-exposed cells. Cells were stained for SYN (red), nuclear stain (blue), or LC3 (green), Scale bar = 10 μm.
FIG. 2.

Exposure to low level As increases the levels of ubiquitin (UB) and glutathionylated proteins, SYN oligomerization, and the expression of various stress markers. SH-SY5Y cells overexpressing SYN were exposed to increasing As concentrations (5, 10, 25, and 50 ppb) for 72 h, followed by Western blot analysis of UB and glutathionylated proteins, and the stress protein markers γ-GCS, LC3, p62, and NAD(P)H dehydrogenase quinone 1 (NQO1); Note, for SYN different imaging exposure times were employed.
FIG. 3.

Densitometry analysis of in vitro Western blot samples displaying a dose-dependent increase in UB and glutathionylated proteins, SYN oligomerization and increased expression of stress markers (γ-GCS, LC3, p62, and NQO1). Column statistics with 2-tailed T-tests; *significant difference from control samples (P < .05), **significant difference from control samples (P < .01), and ***significant difference from control.
Arsenic Induces Accumulation of SYN in Wild-Type Swiss Webster Mice
Wild-type Swiss Webster mice were exposed to 100 ppb As via drinking water to determine the effects of chronic As exposure (2 and 5 weeks) on motor function, SYN oligomerization, accumulation of SYN, and loss of TH reactive dopaminergic neurons in the geriatric age mice (18 months old). No treatment effect of chronic As exposure on motor function was observed (data not shown). However, animals exposed to As for 5 weeks exhibited a significant increase in SYN immunoreactivity in the cortex (Figure 4C), as demonstrated by brown in vivo immunohistochemistry (IHC) DAB staining of SYN, relative to their control counterparts (Figure 4A). Exposure to As for 2 weeks resulted in a more modest increase in SYN staining (Figure 4B), as rated by the blinded analysis. SYN staining resembled Lewy neurites (Spillantini et al., 1998), which are abnormal SYN filaments, and no structures similar to LBs (intracellular protein field inclusions, surrounded by a membrane, resembling a halo) were detected. The increase in SYN immunoreactivity due to As exposure was less apparent in the caudate putamen, an area rich in dopaminergic SNcp projections (Figs. 4D–F). For determination of dopaminergic neuronal loss, TH staining was employed, which provides a common measure of dopaminergic cell loss. Low magnification images (Figs. 5A–C) revealed that the brown DAB staining for TH was clearly localized to the striatum. Surprisingly, TH immunoreactivity was higher in the animals exposed to As for 5 weeks (Figure 5F) compared with their control and shorter As exposed littermates (Figs. 5D and E). Although animals were only exposed to As temporarily (2 and 5 weeks), Western blot analysis coupled to densitometry revealed the 5 week As exposed animals accumulated a 2-fold increase in SYN monomers, with the accumulation of SYN oligomers also being significantly higher than in control animals (1-way ANOVA P = .0327, F = 6.382, R2 = 0.6802, Tukey’s P < .05, 95% CI of diff = −4.889 to −0.1970), with a > 3-fold increase (Figure 6). Although UB monomer and UB oligomer levels (protein ubiquitination) also increased following As exposure (Figs. 6A and B), the difference did not reach statistical significance. LC3-I staining increased in parallel with increases in the length of As exposure, with a 2-fold increase in LC3-I at 2 weeks exposure, and an approximately 3-fold increase following 5 weeks of exposure, with statistical significance reached between control and 5 weeks of As exposure (1-way ANOVA P value = .0006, F = 32.56, R2 = 0.9156, Tukey’s P < .001, 95% CI of diff = −2.561 to −1.069) and a significant difference between 2 and 5 weeks of As exposure (1-way ANOVA P value = .0006, F = 32.56, R2 = 0.9156, Tukey’s P < .05, 95% CI of diff = −1.874 to −0.1830) (Figure 6B). Animals exposed to As exhibited LC3-II staining similar to LC3-I, with a > 2-fold increase after 2 week exposure to As and > 3-fold increase with 5 week exposure to As, which was significantly different from control littermates (1-way ANOVA P value = .0053, F = 14.25, R2 = 0.8261, Tukey’s P < .01, 95% CI of diff = −4.145 to −0.9750). Western blot densitometry of TH revealed elevations in animals exposed to As for 5 weeks, consistent with the results obtained by IHC immunoreactivity; however, the Western blot TH surge did not reach statistical significance (Figs. 6A and B).
FIG. 4.

Short-term (2 or 5 weeks) As exposure causes changes in SYN expression in mouse brain. Immunohistochemical analysis shows increased SYN (brown) staining in the cortex (A–C) and striatum (D–F) (Bregma Zero) of mice exposed to 100 ppb As, counterstained with hematoxylin/eosin (purple/pink). A, Control cortex, B, 2 week As exposure [cortex], C, 5 week As exposure [cortex], D, control striatum, E, 2 week As exposure [striatum], and F, 5 week As exposure [striatum]. Scale bar = 10 μm.
FIG. 5.

Short-term (2 or 5 weeks) As exposure causes changes in tyrosine hydroxylase (TH) expression in mouse brain. Immunohistochemistry of TH (brown) in the cortex and striatum from mice at low magnification (A–C) and immunohistochemistry showing increased TH (brown) staining in the striatum D–F) (Bregma Zero) of mice exposed to 100 ppb As, counterstained with hematoxylin/eosin (purple/pink). A, Control, B, 2 week As exposure, and C, 5 week As exposure; D, control striatum, E, 2 week As exposure striatum, and F, 5 week As exposure striatum. Low magnification scale bar = 10 μm (A–C) and high magnification scale bar = 10 μm (D–F).
FIG. 6.

Short-term (2 or 5 weeks) As exposure results in the oligomerization of SYN, and increases in ubiquitinylated proteins and LC3 expression in mouse brain. Western blot analysis with densitometry of tissue showing increased SYN oligomerization and increased UB and LC3 from mice exposed to 100 ppb arsenic for 2 weeks or 5 weeks. A, Western blot staining for SYN, UB, LC3, TH, and GAPDH. B, Densitometric analysis of the Western blots, normalized to GAPDH. One-way ANOVA with Tukey’s multiple comparison tests; *significant difference from control animals (*P < .05, **P < .01, ***P < .001) and #significant difference from 2 week treated animals (#P < .05).
DISCUSSION
Short-term (2–5 week window in vivo; 24–72 h in vitro) exposure to low levels of As can induce the accumulation of SYN. Thus, ICC of ATRA-differentiated SH-SY5Y cells exposed to 25 ppb As revealed the formation of intracellular inclusions, positively staining for SYN (Figure 1), and which resembled LBs. LBs are enlarged intracellular inclusions, and represent 1 of the main pathological hallmarks of PD, and other synucleinopathies, including dementia with Lewy bodies (DLB). Such features appear at later stages of the disease and are primarily localized in the brain stem in PD but widespread throughout the brain in DLB (Beyer et al., 2009). The inclusions formed in this study appear to be autophagic vacuoles, because SYN and LC3 colocalize in these inclusions, and LC3 is major component of autophagic vacuoles. As inhibits autophagic flux, and because the principal degradation pathway of SYN is via CMA (Cuervo and Wong, 2014), our observations of fluorescent puncta filled with SYN (Figure 1) are consistent with prior findings (Lau et al., 2013; Teng et al., 2015). Protein UB and glutathionylation were also elevated in SH-SY5Y cells overexpressing SYN (Figs. 2 and 3), further implicating As exposure as augmenting protein damage in this model. γ-GCS, the rate-limiting enzyme in GSH synthesis (Franklin et al., 2009) was also elevated in the As-treated samples (Figs. 2 and 3). The low, environmentally relevant levels of As used in this study preclude the detection of ROS by conventional means; GSH binding to proteins and γ-GCS were therefore used as surrogate measures of oxidative stress. Consequently, the increase in protein ubiquitination and glutathionylation is indicative of proteotoxic stress subsequent to As exposure.
Exposure to As is known to increase the expression of LC3 I/II and its binding partner, P62 (sequestosome 1). This increase is due to initiation of autophagy processes in combination with an inhibition of autophagic flux (Bjorkoy et al., 2009; Lau et al., 2013), resulting in accumulation of LC3 I/II and P62 (Figs. 2 and 3), a consequence of the inability of the autophagosome to fuse with the lysosome to complete autophagy. LC3 I/II and P62 elevations confirm the ICC experiments which revealed the accumulation of autophagic vacuoles, suggesting an increase in autophagy. In combination with a 10-fold increase in NQO1 expression (Figs. 2 and 3), a protein upregulated during oxidative stress, we conclude that exposure of SH-SY5Y cells to As induces cell stress pathways, which, although initially activate autophagy, result in the subsequent accumulation of SYN and UB oligomers, and other stress-response associated proteins (γ-GCS, LC3 I/II, P62, and NQO1).
In addition to SYN, ubiquitinated proteins are also one of the main components of LBs (Leverenz et al., 2007), and both pathways contribute to the etiology of neurodegenerative disease. Elevated oligomerization of SYN has been demonstrated at low levels of As exposure and provides a novel mechanism underlying the neurotoxic potential of As. In contrast to this study, Teng et al. (2015) suggests that SYN monomers decrease upon As exposure. In this study, an increase in SYN monomers occurred, although at 50 ppb As there were lower levels of SYN monomers relative to 25 ppb. It is therefore important to emphasize that the 50 ppb As used in this studies is approximately 7.5-fold lower than the concentrations used by Teng et al. (2015). Therefore, the possibility that higher levels of As decrease SYN monomer expression or increase degradation cannot be excluded. Variations in As concentrations, duration of exposure, detection methodology, and other variables all contribute to differences in the response to As, as emphasized by Singh et al. (2011). Because SYN oligomers and soluble aggregates represent the toxic SYN species (Conway et al., 2000, 2001; Sharon et al., 2003; Waxman and Giasson, 2009), and we employed environmentally relevant concentrations of As, our data support the potential role of As in neurodegeneration, and perhaps especially in individuals with genetic predisposition. Redox active metals, including iron, copper, and manganese can stimulate SYN oligomerization/aggregation (Bisaglia et al., 2009; Cai et al., 2010; Verina et al., 2013). Our data suggest that accumulation of SYN occurs primarily as a consequence of the inhibition of autophagic flux by As. It is not yet known if As can directly stimulate SYN oligomerization/aggregation through SYN metal binding sites.
Mice at 2 month of age, and only briefly (relative to overall life time) exposed to 100 ppb As (2 and 5 weeks) exhibited increased SYN immunoreactivity (Figs. 4 and 6), despite the fact that animals were removed from As exposure prior to geriatric age (18 months), enabling sufficient time to recover from any possible adverse consequences of As exposure. Thus, the effect of relatively short-term (2–5 weeks) exposure to As on SYN is either (1) immediate and persistent with little effective compensatory response postexposure or (2) the effects of the relative short-term exposure to As initiate events that develop over the remainder of the lifetime of the animal and are evidenced at autopsy. More detailed temporal studies will be required to resolve these alternatives.
No behavioral changes associated with decreased motor function were observed (data not shown). However, motor symptoms in PD only become noticeable after the loss of > 60% of dopaminergic neurons (Jankovic, 2008), which may indicate that the As exposure levels and duration used in this study were insufficient to induce motor deficits. Again, more detailed studies will be necessary to expand on our initial findings. In particular, behavioral tests require a large number of animals due to significant interindividual variations in animal personality (Brooks and Dunnett, 2009). Nonetheless, we did observe the accumulation of SYN in the cortex of animals exposed to As for 5 weeks, and to a lesser extent in the striatum (Figure 5). The localization of SYN in the cortex is indicative of a more DLB-like pathology, rather than PD-like pathology (Beyer et al., 2009). TH immunoreactivity, the rate-limiting enzyme in DA production, was also increased in the animals exposed to As for 5 weeks (Figs. 5 and 6). SYN can inhibit TH expression and function (Alerte et al., 2008; Perez et al., 2002; Yu et al., 2004), and As can decrease DA levels (Yadav et al., 2010). Thus, the increased TH expression in As exposed animals is likely due to the engagement of compensatory mechanisms to overcome the combined negative effects of SYN and As on dopaminergic function. Widely unrecognized behavioral symptoms of PD appear decades earlier, which include sensory deficits, cognitive issues, and sleep disturbances. Moreover, the brain pathology during the period preceding an official PD diagnosis is not well studied (Postuma et al., 2012). It is possible that pathological changes observed in mice precede visible PD and/or DLB clinical symptoms. LC3-I and LC3-II were also significantly elevated in the cortex and striatum of As treated animals (Figure 6) consistent with the findings of Teng et al. (2015) and with our in vitro studies. LC3 elevations, coupled to increased protein UB, suggest increased autophagy vacuole accumulation in the brain of As exposed animals. Taken together, the results suggest that transient exposure to As can induce stress-response pathways in the brain, and the effects persist even after removal of the toxicant. Mutations that result in SYN overexpression are known to lead to generation of PD and DLB (Nuytemans et al., 2010). Thus, it is logical to expect generation of synucleinopathies from accumulation of SYN, the protein being in the monomeric form, oligomeric form, or aggregated form.
In summary, our findings provide novel insights into how low level As exposure may contribute to the accumulation of SYN. Moreover, acute exposure to As is sufficient to induce changes that may contribute to neurodegeneration, and in particular to synucleinopathies.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the assistance of Samantha Y. Andrade and Bianca G. Reilly.
FUNDING
This work was supported by awards from the National Institute of Environmental Health Sciences, (P30 ES006694 to S.S.L., T32 ES016652 to T.J.M. and A.B.C., and T32 ES007091 to A.B.C.).
REFERENCES
- Alerte T. N., Akinfolarin A. A., Friedrich E. E., Mader S. A., Hong C. S., Perez R. G. (2008). Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: Lessons from viral transduction of knockout mice. Neurosci. Lett. 435, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer K., Domingo-Sabat M., Ariza A. (2009). Molecular pathology of Lewy body diseases. Int. J. Mol. Sci. 10, 724–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaglia M., Tessari I., Mammi S., Bubacco L. (2009). Interaction between alpha-synuclein and metal ions, still looking for a role in the pathogenesis of Parkinson’s disease. Neuromolecular Med. 11, 239–251. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G., Lamark T., Pankiv S., Overvatn A., Brech A., Johansen T. (2009). Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 452, 181–197. [DOI] [PubMed] [Google Scholar]
- Brooks S. P., Dunnett S. B. (2009). Tests to assess motor phenotype in mice: A user’s guide. Nat. Rev. Neurosci. 10, 519–529. [DOI] [PubMed] [Google Scholar]
- Cai T., Yao T., Zheng G., Chen Y., Du K., Cao Y., Shen X., Chen J., Luo W. (2010). Manganese induces the overexpression of alpha-synuclein in PC12 cells via ERK activation. Brain Res. 1359, 201–207. [DOI] [PubMed] [Google Scholar]
- Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Harper J. D., Williamson R. E., Lansbury P. T., Jr. (2000). Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 920, 42–45. [DOI] [PubMed] [Google Scholar]
- Conway K. A., Rochet J. C., Bieganski R. M., Lansbury P. T., Jr. (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294, 1346–1349. [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Wong E. (2014). Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 24, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz A., Clavel J., Rathouz P. J., Moisan F., Galanaud J. P., Delemotte B., Alperovitch A., Tzourio C. (2009). Professional exposure to pesticides and Parkinson disease. Ann. Neurol. 66, 494–504. [DOI] [PubMed] [Google Scholar]
- Ellinsworth D. C. (2015). Arsenic, reactive oxygen, and endothelial dysfunction. J. Pharmacol. Exp. Ther. 353, 458–464. [DOI] [PubMed] [Google Scholar]
- Franklin C. C., Backos D. S., Mohar I., White C. C., Forman H. J., Kavanagh T. J. (2009). Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 30, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herva M. E., Spillantini M. G. (2015). Parkinson’s disease as a member of prion-like disorders. Virus Res. 207, 38–46. [DOI] [PubMed] [Google Scholar]
- Jacobson T., Navarrete C., Sharma S. K., Sideri T. C., Ibstedt S., Priya S., Grant C. M., Christen P., Goloubinoff P., Tamas M. J. (2012). Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell. Sci. 125, 5073–5083. [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2008). Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. [DOI] [PubMed] [Google Scholar]
- Jomova K., Jenisova Z., Feszterova M., Baros S., Liska J., Hudecova D., Rhodes C. J., Valko M. (2011). Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 31, 95–107. [DOI] [PubMed] [Google Scholar]
- Kim M., Seo S., Sung K., Kim K. (2014). Arsenic exposure in drinking water alters the dopamine system in the brains of C57BL/6 mice. Biol. Trace Elem. Res. 162, 175–180. [DOI] [PubMed] [Google Scholar]
- Korecka J. A., van Kesteren R. E., Blaas E., Spitzer S. O., Kamstra J. H., Smit A. B., Swaab D. F., Verhaagen J., Bossers K. (2013). Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS One 8, e63862.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A., Zheng Y., Tao S., Wang H., Whitman S. A., White E., Zhang D. D. (2013). Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 33, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz J. B., Umar I., Wang Q., Montine T. J., McMillan P. J., Tsuang D. W., Jin J., Pan C., Shin J., Zhu D., et al. (2007). Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 17, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Kaduska M., Liu Y., Qu W., Mason R. P., Walker M. P. (2000). Acute arsenic induced free radical production and oxidative stress related gene expression in mice. Toxicologists 54, 280–281 (Abstract). [Google Scholar]
- Martinez-Vicente M., Talloczy Z., Kaushik S., Massey A. C., Mazzulli J., Mosharov E. V., Hodara R., Fredenburg R., Wu D. C., Follenzi A., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuytemans K., Theuns J., Cruts M., Van Broeckhoven C. (2010). Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum. Mutat. 31, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F., Zigmond M. J. (2002). A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma R. B., Aarsland D., Barone P., Burn D. J., Hawkes C. H., Oertel W., Ziemssen T. (2012). Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov. Disord. 27, 617–626. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B., Woerman A. L., Mordes D. A., Watts J. C., Rampersaud R., Berry D. B., Patel S., Oehler A., Lowe J. K., Kravitz S. N., et al. (2015). Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R., Bar-Joseph I., Frosch M. P., Walsh D. M., Hamilton J. A., Selkoe D. J. (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37, 583–595. [DOI] [PubMed] [Google Scholar]
- Shavali S., Sens D. A. (2008). Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol. Sci. 102, 254–261. [DOI] [PubMed] [Google Scholar]
- Singh A. P., Goel R. K., Kaur T. (2011). Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 18, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M. (1998). Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Kim Y. M., Lee G., Junn E., Iwatsubo T., Mouradian M. M. (2004). Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 279, 4625–4631. [DOI] [PubMed] [Google Scholar]
- Teng Y. C., Jeng C. J., Huang H. J., Lin A. M. (2015). Role of autophagy in arsenite-induced neurotoxicity: The involvement of alpha-synuclein. Toxicol. Lett. 233, 239–245. [DOI] [PubMed] [Google Scholar]
- Verina T., Schneider J. S., Guilarte T. R. (2013). Manganese exposure induces alpha-synuclein aggregation in the frontal cortex of non-human primates. Toxicol. Lett. 217, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman E. A., Giasson B. I. (2009). Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim. Biophys. Acta 1792, 616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt K., Adami H. O., Cole P., Trichopoulos D., Mandel J. (2011). Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 26(Suppl. 1), S1–58. [DOI] [PubMed] [Google Scholar]
- Yadav R. S., Shukla R. K., Sankhwar M. L., Patel D. K., Ansari R. W., Pant A. B., Islam F., Khanna V. K. (2010). Neuroprotective effect of curcumin in arsenic-induced neurotoxicity in rats. Neurotoxicology 31, 533–539. [DOI] [PubMed] [Google Scholar]
- Yu S., Zuo X., Li Y., Zhang C., Zhou M., Zhang Y. A., Ueda K., Chan P. (2004). Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 367, 34–39. [DOI] [PubMed] [Google Scholar]