

Abstract
Oncolytic virotherapy, a type of nanomedicine in which oncolytic viruses (OVs) are used to selectively infect and lyse cancer cells, is an emerging field in cancer therapy. Some OVs exhibit a specific tropism for cancer cells, whereas others require genetic modification to enhance their binding with and entry into cancer cells. OVs both kill tumor cells and induce the host’s immune response against tumor cells. Armed with antitumor cellular molecules, antibodies, and/or in combination with anticancer drugs, OVs can accelerate the lysis of cancer cells. Among the OVs, vaccinia virus has been the focus of preclinical and clinical research because of its many favorable properties. In this review, the basic mechanisms of action of OVs are presented, including their entry, survival, tumor lysis, and immune activation, and the latest research in vaccinia virus-based virotherapy and its status as an anticancer nanomedicine in prospective clinical trials are discussed.
Keywords: oncolytic viruses, cancer therapy, vaccinia virus, virotherapy
Introduction
Conventional cancer treatments aimed at killing tumor cells, such as chemotherapy and radiotherapy, have some limitations, such as chemo- and radioresistance and cytotoxicity.1–3 A successful therapy should target only cancer cells without harming normal cells; however, this cannot be achieved with conventional anticancer drugs. The creation of cancer-targeting drugs is a complex process, and the complicated environment in which cancer cells reside must also be considered.4 These difficulties can be overcome by gene therapy and oncolytic virotherapy.5
Oncolytic viruses (OVs) are viruses capable of specifically infecting and killing cancer cells. The use of these live viruses in cancer treatment is called oncolytic virotherapy.6 Based on their manner of entry and tropism toward cancer cells, different viruses are employed in the treatment of different cancers. The specific tropism of OVs for particular types of cancer cells has facilitated their therapeutic use as a nano-medicine in oncolytic virotherapy.7 OVs comprise both enveloped and nonenveloped viruses, with genomes composed of either DNA or RNA. Selective binding and entry into cancer cells are inherent among viruses; however, some viruses bear modifications in their envelope structures that enhance their selectivity. Both enveloped and nonenveloped viruses are used in oncolytic virotherapy.8 Modifications in OVs introduced through genetic engineering, including insertions and deletions in the genome, can deliver additional therapeutic molecules to cancer cells and make OVs nonpathogenic to normal cells. OVs have been armed with cellular molecules that induce apoptosis, and their efficacy of killing tumor cells has been studied both in vitro and in vivo.9
Several OVs are currently used in oncolytic virotherapy, and their safety and efficacy have been well characterized. For example, the effects of the adenovirus strains Ad1 and Ad5 have been investigated in clinical trials for the treatment of various cancers. Recently completed trials of oncorine (H101) and onyx-015 have proven the safety and clinical efficacy of these adenovirus strains.10 The safety and tolerability of strains of the herpes simplex virus (HSV), including talimogene laherparepvec (T-VEC; Amgen Inc., Thousand Oaks, CA, USA), G207, G47 Delta, and HSV 1716, have also been evaluated in several trials.11 In addition, other OVs such as the measles virus, Newcastle disease virus (NDV), parvovirus, poliovirus, Seneca Valley virus, and retroviruses have been studied in various trials for their ability to treat cancer. Interim reports show good efficacy and positive predictions for future trials.12 This review focuses on vaccinia virus (VV) because of its unique characteristics such as its infectivity and efficacy in mice and human cells. VV exhibits good oncolytic activity and efficacy in both models, whereas other OVs exhibit limited activity in human cells.13 Moreover, VV-based clinical trials have yielded promising results in patients with various cancers.14
In this review, the authors first focus on the general mechanisms of action of OVs, including their entry into and survival within cancer cells and their activation of the immune response. Next, the current trends, clinical trial outcomes, and future perspectives of research on the use of VV in oncolytic virotherapy are discussed.
OVs in cancer cells
How do OVs recognize cancer cells?
The mechanism by which OVs enter cancer cells varies depending on the receptors present on host cells.8 For example, adenoviruses exhibit tropism toward integrins and the coxsackievirus–adenovirus receptor (CAR); internalization through the αvβ3 or αvβ5 integrins leads to entry into host cells.15 Adenovirus strains B1, B2, and Ad11 use DSG2 and cluster of differentiation (CD) 46 as a receptor for entry. However, these strains have limitations to be used as anticancer vectors since they stimulate epithelial-to-mesenchymal transition, which is a central event in carcinogenesis.16 Overexpression of the receptors for intercellular adhesion molecule 1 (ICAM-1) and decay-accelerating factor (DAF) by multiple myeloma, melanoma, and breast cancer cells aids the entry of coxsackievirus into these cells.17 HSV uses herpesvirus entry mediator (HVEM) and nectin as receptors for entry; these are highly expressed in melanoma and carcinoma cells.18 CD46 and signaling lymphocytic activation molecule are used by the measles virus for entry.19 NDV and VV enter cancer cells by endocytosis, because of the absence of specific receptors for their attachment.20,21 Poliovirus exhibits greater tropism for neurons, in which it uses CD155 as a receptor (Table 1).22 Although the receptors such as adenovirus receptors integrin and CAR can be found in normal cells, cancer selectivity is further governed by oncogenes and tumor suppressor genes. Aberrant expressions of these genes in cancer cells allow OVs’ replication. On the other hand, defect in the antiviral immunity of cancer cells also favors OVs’ replication. In normal cells, antiviral immunity attenuates OVs’ survival, as a result of clearance, despite their entry into the cells.8
Table 1.
Oncolytic viruses: tumor cell entry mechanisms
| Virus | Receptors | Entry mechanism/cell types |
|---|---|---|
| Adenovirus | Integrin and CAR | Internalization through αvβ3 or αvβ5 integrins15 |
| Coxsackievirus | ICAM-1 and DAF | High-level expression in multiple myeloma, melanoma, and breast cancer cells17 |
| Herpesvirus | HVEM and Nectin | Overexpressed in melanoma and carcinomas18 |
| Measles virus | CD46 and SLAM | Prevents cell elimination in a complement pathway19 |
| Newcastle disease virus | No receptor | Endocytosis by cancer cells20 |
| Poliovirus | CD155 | Overexpressed in cancer cells and prevents pathogenesis in neurons22 |
| Vaccinia virus | No receptor | Endocytosis: endosomal or direct delivery21 |
Abbreviations: CAR, coxsackievirus–adenovirus receptor; CD, cluster of differentiation; DAF, decay-accelerating factor; HVEM, herpesvirus entry mediator; ICAM-1, intercellular adhesion molecule 1; SLAM, signaling lymphocytic activation molecule.
Survival of OVs in cancer cells
Cell cycle–related genes govern the cell cycle–transition process in normal cells. Aberrant expression of these genes in cancer cells creates a more suitable environment for the replication and life cycle of OVs. The expression of some cell cycle–related genes, such as p16, retinoblastoma (Rb), and p53, regulates the cell cycle.23 Rb regulates the transition from G1–S phase through the E2 factor (E2F) and cyclin-dependent kinases.24 p53, termed the “guardian of the genome”, is upregulated and activated by cellular stress, such as DNA damage or viral infection, and this results in stimulation of the proteins p21 and Bax, which induce apoptosis.25 The overall function of these genes is to promote apoptosis when the cell cycle is dysfunctional or disrupted in normal cells. In contrast, the downregulation of tumor suppressor genes and upregulation of oncogenes interrupt the cell cycle, leading to uncontrolled cell proliferation of cancer cells.26 In addition, impaired antiviral activation is also evident in tumor cells. Mutations in the small GTPase RAS can cause upregulated proliferation, which is affected by an increase in the proteins involved in cell division.27 NDV and vesicular stomatitis virus (VSV) capitalize on these changes in the environment surrounding cancer cells to achieve more efficient replication. Viruses such as reoviruses, HSV-1, adenoviruses, VV, and influenza viruses use hyperactivated RAS, which blocks PKR, for selective and efficient replication in cancer cells.8 In addition, p53 mutations in cancer cells make them a suitable target for adenoviruses, reoviruses, and parvovirus, thereby enhancing viral replication in cancer cells. Similarly, abnormal expressions of Rb and p16, which control cell-cycle entry, make cancer cells vulnerable to OVs such as adenoviruses, HSV-1, VV, and reoviruses.28 Also, the upregulation of antiapoptotic protein B-cell lymphoma-XL in cancer cells allows increased viral replication and confers a selective advantage for NDV.20
In addition, tumor microenvironment (TME) plays an important role in oncolytic activity of OVs. Cytokines produced by the cells in TME can also affect the efficacy of OVs. Transforming growth factor-β (TGF-β) produced by cancer-associated fibroblasts diminishes the level of antiviral transcripts and makes them sensitive to virus infection. In contrast, high levels of fibroblast growth factor 2 (FGF2) produced also by cancer-associated fibroblasts reduce retinoic acid-inducible gene I (RIG-I) expression. Thus, low expression of RIG-I delays the ability of malignant cells to detect and respond to virus.29
Cancer cell lysis by OVs
After entry, most OVs replicate and assemble in the cytosol and nucleus, which causes viral spreading across the cell, resulting in the lysis of tumor cells. The lysis of infected tumor cells releases multiple viruses, which then infect surrounding tumor cells. However, viral infection also causes inflammation and the recruitment of dendritic cells (DCs) to cancer cells, inducing both adaptive (such as B- and T-cell activation) and innate (natural killer [NK]-cell activation) immune responses against tumor cells. Therefore, virus-induced cell death enhances the activity of DCs and cytotoxicity of NK cells toward tumor cells. Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns (PAMPs), which activate TNF-associated factor (TRAF) 3, IFN-related factor (IRF) 3, IRF7, and RIG-I as a defense mechanism against invading microbial pathogens. This activation leads to stimulation of the Janus kinase–signal transducer and activator of the transcription pathway, resulting in the production of interferons (IFNs) involved in antiviral immunity to inactivate the pathogen. However, this IFN-mediated cell-signaling pathway is blocked in OV-infected cancer cells. This inhibition allows the survival of OVs and lysis of tumor cells.30
Activation of the immune response by OVs against cancer cells
As stated in the “Cancer cell lysis by OVs” section, the therapeutic efficacy of OVs depends on the combined effect of direct cancer cell lysis and indirect activation of antitumor immune responses. The immune responses induced by OVs have a range of effects. The antiviral immune response is activated upon OV infection. Genotoxicity and endoplasmic reticulum stress lead to the upregulation of reactive oxygen species generation and the initiation of antiviral cytokine production.31 Consequently, infected cancer cells produce reactive oxygen species, antiviral cytokines, and Type I IFNs, thereby stimulating antigen-presenting cells, CD8+ T-cells, and NK cells (Figure 1). However, viral progeny, PAMPs, danger-associated molecular patterns (DAMPs), and tumor-associated antigens (TAAs) including neoantigens are released after oncolysis. The released viral progeny propagates the infection. The PAMPs, consisting of viral particles and DAMPs, induce host cell proteins, trigger the activation of TLRs, and stimulate the immune system. As a result of these immune-stimulatory occurrences, antigen-presenting cells capture the released TAAs and neoantigens. Together, these events instigate immune responses toward both virally infected and uninfected cancer cells through de novo immune responses.32
Figure 1.

Infection with OVs enhances immune activation against cancer cells.
Notes: Infection with OVs induces genotoxicity and ER stress in cancer cells. IFN-1 released from cancer cells activates NK cells and CD8+ T-cells. This event induces cytotoxicity and cell death in surrounding uninfected cancer cells. Conversely, oncolysis induces the production of neoantigens by cancer cells, which are presented by APCs to CD4+ and CD8+ T-cells for activation. Activated CD4+ T-cells secrete IL-2 and enhance the cytotoxicity of CD8+ T-cells. These mechanisms enhance immune activation against cancer cells. Data from Kaufman et al.8
Abbreviations: APC, antigen-presenting cell; CD, cluster of differentiation; ER, endoplasmic reticulum; IFNs, interferons; IL, interleukin; NK, natural killer; OVs, oncolytic viruses; ROS, reactive oxygen species.
The negative impact of humoral immunity on the systemic delivery of OVs can be overcome by abolishing antiviral immunity, thus enhancing the efficacy of OVs. The antiviral immunity against OVs could be compromised by administration of chemotherapeutic drugs. For instance, coadministration of cyclophosphamide with reovirus serotype 3 dearing (Reolysin®; Oncolytics Biotech Inc.) in advanced cancer patients showed the safety result, without decreasing titer of neutralizing antireovirus antibody. This study shows viable virus that is associated with peripheral blood mono-nuclear cells.33 In addition, immune cells such as DCs and T-cells have been used for systemic delivery of OVs. Virus could be protected from neutralizing antibodies after systemic administration by immune cell carriage. A single cycle of intravenous (IV) administration of reovirus in colorectal cancer patients (before the surgery) was done and the presence of viral genome was found in metastatic liver tumors and immune cells.34 The efficacy of DCs and T-cells as “the cellular carriers” was shown in the lymph node B16tk melanoma metastases model. Reovirus was used with different groups as either neat or loaded onto DCs or T-cells, delivered intravenously into reovirus-naive or reovirus immune C57Bl/6 mice. Complete clearance of metastases was reported only when the virus was delivered on T-cells or mature DC in reovirus-immune mice, whereas neat reovirus or loaded immature DC gave only partial early tumor clearance. The efficacy of tumor killing of DCs and T-cells against preexisting antiviral immunity was demonstrated in this study.35
The treatment of permissive tumors with OVs triggers a robust antitumor T-cell response, thereby contributing to their efficacy; for example, VSV harboring a deletion in the M protein at position 51 (VSV-Δ51) exerts a significant antitumor response. VSV-Δ51 is sensitive to IFNs and neutralizing antibodies, which act to clear the virus from the host. Therefore, a robust antitumor T-cell response was induced in permissive tumors treated with VSV-Δ51.36 Rapid infiltration of IFN-γ-producing NK and T-cells was observed in VSV-Δ51-challenged tumors. Infected cell vaccine (ICV), consisting of tumor cells infected with VSV-Δ51 that present a multitude of tumor antigens, was used as a potent oncolytic vaccine platform. In addition, increased expression of granulocyte macrophage colony stimulating factor (GM-CSF) prompted by an ICV made with VSV-Δ51-GM-CSF (VSVgm-ICV) prevented B16-F10 tumor engraftment in >95% of mice tested.37
OV-based virotherapy
The use of OVs in cancer therapy research
Because of their characteristics, almost all types of OVs have been used in oncolytic virotherapy. Depending on their genomic type and size, various therapeutic molecules can be inserted into OVs for treatment applications. The efficacy, tolerance, and toxicity of some viruses have already been studied. The modified viruses and their mechanisms of action are summarized in Table 2. Some have been used in clinical trials.30,38 Although other OVs also have benefits, this review mainly focuses on VV.
Table 2.
Oncolytic viruses for cancer therapy: modification and mechanism of target replication
| Virus | Modification | Mechanism | References |
|---|---|---|---|
| Adenovirus | Deletion of E1A and E1B | E1A and E1B target the tumor suppressors p53 and the retinoblastoma-associated protein pRb promoted cell-cycle entry | Andtbacka et al71 |
| Hexon modification: insertion of TGFBR targeting peptide CKS17 in the HVR5 of the capsid protein | Decreased binding of coagulation factor X to CKS17-modified adenovirus particles | Lucas et al88 | |
| Herpesvirus | Deletions in neurovirulence gene ICP34.5 and the inhibitor of antigen presentation ICP47 | Improves cancer cell selectivity and prevents infection of neurons Induces the early activation of the US11 promoter |
Poppers et al89 |
| Coxsackievirus | T-cell checkpoint inhibitors | Promoting the release of DAMPs against cancer cells (HMGB-1, calreticulin, and ATP) | Miyamoto et al90 |
| Poliovirus | Replacing the viral IRES | Binds DRBP76–NF4 heterodimer (comprising cellular double-stranded RNA-binding protein 76 and nuclear actor of activated T-cells 45 kDa). This structure blocks viral replication in normal cells, but does not occur in glioma cells | Merrill et al22 |
| Measles virus | Encoding synthetic miRTS | Binds cellular miRNAs and represses viral replication | Leber et al91 |
| Newcastle disease virus | GM-CSF insertion at noncoding regions | High sensitivity of NDV to Type I IFNs enhances cancer cell specificity | Janke et al92 |
| Vaccinia virus | Disruption of vTK, deletion of VGF, and B18R | Replication occurs in cytosol. Disruption of vTK, deletion of VGF, and B18R led to selective replication in cancer cells | Parviainen et al65 |
Abbreviations: DAMPs, danger-associated molecular patterns; DRBP76, double-stranded RNA-binding nuclear protein 76; HMGB-1, high mobility group box 1; HVR5, hypervariable region 5; IFNs, interferons; IRES, internal ribosome entry site; miRTS, microRNA-target site; miRNAs, microRNAs; NDV, Newcastle disease virus; TGFBR, transforming growth factor-β receptor.
The VV and cancer therapy
VV belongs to the Poxviridae family and its genetic material consists of double-stranded DNA ∼190 kbp in length. Infection with this virus causes the formation of pock lesions on the skin. Three major strains of this virus have been characterized to date: Lister, Western Reserve, and Wyeth.39 The safety and immunogenicity of VV were reported in the US smallpox vaccination program.40 Disruption of the viral thymidine kinase (vTK) and deletion of vaccinia growth factor (VGF) genes of VV instill selectivity for cancer cells while retaining its natural oncolytic ability. These two genes are vital for the replication of the virus in normal cells; however, the mutated virus can survive and replicate in cancer cells because of the abundance of nucleotides available for DNA synthesis inside cancer cells.41 Moreover, the replication of VV occurs in the cytoplasm of cells, preventing the integration of its genome into nucleus. These properties of VV make it a good candidate for oncolytic virotherapy.21 VV differs from other OVs in several ways: for instance, it can infect both mice and human tumor cells. This ability makes VV suitable for a wide range of oncolytic virotherapy research. The large genome can be modified by either the deletion of virulence genes or insertion of genes for therapeutic uses through genetic engineering.14 It also exhibits tropism to a variety of cancer cells. Both preclinical and clinical studies have proven its efficacy. Moreover, it can be administered through IV and intratumoral (IT) methods. Administration of the virus through IV methods has benefits for metastatic cancers, whereas direct injection into the tumor reduces tumor size.12
VV entry
Four forms of the virus exist during the life cycle of VV: intracellular mature virion (IMV), cell-associated enveloped virion (CEV), and extracellular enveloped virion (EEV). Among them, IMV and EEV are seen most often during assembly, often during assembly.42 VV enters tumor cells by endocytosis, during which direct fusion of the virus to the plasma membrane leads to it being engulfed. Glycosaminoglycan, such as heparan sulfate that is located on the cell surface, mediates VV and host cell interaction in which VV A27L protein plays a role in binding with heparin sulfate. Heparan sulfate–virus interaction might induce conformation rearrangements that may enhance the subsequent fusion events.43 For the direct fusion of the mature virions released from infected cells, four proteins (including H3, A26, A27, and D8) are necessary. These proteins form a complex with laminin, integrin, and CD98. A cytoplasmic adapter protein, TRAF2, facilitates VV entry through direct fusion at the plasma membrane. Enclosure within endocytic vesicles protects the virus from circulating antibodies.44 The low pH of the TME facilitates endosomal-mediated entry of the virus into cancer cells. Endoplasmic reticulum acts as a factory for viral replication in the cytosol (Figure 2).45,46 Upon virus infection, the mitogen-activated protein kinase- and cAMP-dependent protein kinase-mediated cell-signaling pathways are induced to achieve viral multiplication inside the cells. In general, the coordination of viral proteins and host cell factors facilitates the endocytosis of VV.47
Figure 2.
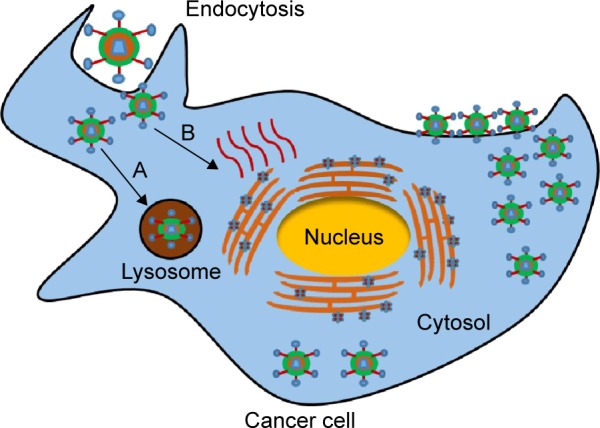
Vaccinia virus entry mechanism.
Notes: The vaccinia virus enters through endocytosis via two different mechanisms: A, lysosome-mediated entry; B, direct replication in the cytosol. ER acts as a factory for virus replication. Data from Tolonen et al.46
Abbreviation: ER, endoplasmic reticulum.
VV replication
Its replication cycle makes VV the most favorable candidate for oncolytic virotherapy. The replication of VV is fast and efficient; within 4–6-hours postinfection, infectious virions are produced from the host cells. The direct lysis of infected tumor cells contributes numerous virions, propagating the infection to the surrounding cancer cells. Rapid spreading of the virus is evident within 2 hours of oncolysis. Moreover, replication of VV occurs in the cytoplasm of infected cells; thus, no integration of the viral genome with the host genome occurs. In addition, VV infection almost completely halts protein synthesis in the host cell. These features of the replication cycle of VV make it a suitable vector for oncolytic therapy.21,39
VV engineering: preclinical studies
Although VV induces tumor cell lysis, various molecules including cytokines, single-chain antibodies (scAbs), and drugs can be engineered into VV to accelerate its antitumor activity (Figure 3). Preclinical studies have proved the efficacy of VV armed with cytokines. Most researchers used GM-CSF to increase tumor cell lysis. GM-CSF is an immune modulator that acts in a paracrine manner on various cells and recruits circulating neutrophils, monocytes, and lymphocytes to kill cancer cells.48
Figure 3.

Vaccinia virus armed with transgenes for cancer therapy.
Notes: Insertion and deletion in the genome of the vaccinia virus makes it suitable for antitumor activity. Disruption of the viral thymidine kinase and deletion of vaccinia growth factor genes favors viral replication in cancer cells. Similarly, the virus can be equipped with various transgenes for specific activities that enhance the destruction of cancer cells.
Abbreviation: scAbs, single-chain antibodies.
Lister strains
A recombinant VV expressing the colorectal tumor suppressor Klf4 was generated to evaluate the response of HT-29 cells. Normally, HT-29 cells do not show any response to GLV1h-68 strains, but a single injection of the recombinant VV expressing Klf4 inhibited tumor growth in a xenograft model. To improve its antitumoral effects, the virus-mediated expression of a membrane-permeable Klf4–TAT fusion protein was utilized. TAT, transacting activator of transcription fusion protein domain, mediates the transduction of Klf4 into HT cells. As a result, antitumoral effects were further improved.49 The antitumor efficacy of VV engineered to include a secretory biospecific T-cell engager and EphA2-TEA-VV (Epha2) was examined both in vitro and in vivo. To allow sufficient replication before T-cell activation, the inserted T-cell engagers were expressed under transcriptional control of the F17R late promoter. This “bystander killing” effect of EphA2-TEA-VV activated T-cells that eliminated both virally infected and uninfected tumor cells. Secretion of interleukin-2 and IFN-γ in animal models was used as a marker of T-cell activation.50
VV, in combination with scAbs against vascular endothelial growth factor (VEGF), such as GLAF-1 and GLAF-2, has been used in oncolytic virotherapy research. The combined effect of GLAF-1 encoding VV (GLV-1h164) with fractionated irradiation on tumor-associated endothelial cells was evaluated. This combination drastically reduced the endothelial cells associated with glioma cells.51 In addition, the combined effect of anti-VEGF, anti-epidermal growth factor receptor (EGFR), and anti-fibroblast activation protein (FAP) with VV was also studied. VEGF, EGFR, and FAP are vital factors for the regulation of angiogenesis, proliferation, and stromagenesis. A combination of scAbs (anti-VEGF and anti-EGFR or anti-FAP) caused a significant reduction in the tumor cell population.52
GLV-1h153 is a Lister strain; the antitumor properties of this strain have been characterized in models of triple-negative breast cancer (TNBC). The infection, replication, and regression of tumor cells were examined both in vitro and in vivo. A 5-week treatment with the virus caused the disappearance of metastatic cells in harvested lymph nodes and organs.53 In addition, the efficacy of GLV-1h153 encoding human sodium iodide symporter (hNIS) in combination with radioiodine (I131) was studied in murine cell lines and animal models. The expression of hNIS and uptake of iodine were confirmed in orthotopic xenograft mice models. Consequently, a sixfold regression was seen when compared with individuals treated with virus alone.54 Furthermore, the antivascular properties of VV armed with anti-VEGF were analyzed in TNBC cell lines, because VEGF expression is higher in TNBC cells than in any other tumor cells. GLV-1h164 was generated using an scAb for VEGF to suppress VEGF activity, and its efficacy was tested in TNBC cell lines and an orthotopic murine model.55 The therapeutic efficacy of GLV-1h153 that expresses NIS was also evaluated in prostate cancer models as different combinations with external beam radiotherapy and NIS-mediated radioiodine therapy. Human prostate cancer cell lines – PC3, DU145, LNCaP, and WPMY-1 were used as in vitro models. Both xenograft and immunocompetent transgenic adenocarcinoma of the mouse prostate mouse models were used as in vivo models. Using immunocompetent models as well as xenograft models shows that the immune system plays an important role in OV efficacy. Among the combinations, addition of radioiodide to VV-NIS-infected tumors was more effective than each single-agent-treated group.56
The Lister strain series – GLV-1h68, GLV-1h285, and GLV-1h289 – were generated through the deletion of three genes, respectively: J2R, which encodes vTK; F14.5L, which encodes a secretory signal peptide; and A56R, which encodes hemagglutinin. Inactivation of J2R causes selective replication in cancer cells, whereas the deletion of the F14.5L and A56R genes made these strains attenuated viruses. The oncolytic efficacy of these strains was tested in animal models, and significant regression was reported. In addition, this study showed the antitumor and antivascular characteristics of the GLV-1h68 strain in human hepatocellular carcinoma (HCC) cell lines such as PLC and HuH7. A PLC tumor xenograft mouse model showed inhibition of tumor growth following treatment with the virus. Furthermore, the infiltration of neutrophils, macrophages, DCs, and B-cells was evident in their tumors. Upregulation of 13 proinflammatory cytokines was also reported in this study.57
The antivascular effects of VV were not only demonstrated in mouse models but also in canine cancer models. The GLAF-2 scAb-encoding GLV-5b451 strain was evaluated in different canine cancer cell lines. Efficient infection and destruction of cancer cells by this modified virus strain were achieved. Furthermore, a significant reduction in and long-term inhibition of tumor growth were reported in a canine soft-tissue sarcoma xenograft mouse model. Also, CD31 immunostaining indicated a notable reduction in neoangiogenesis.58
Wyeth strains
The systemic armed oncolytic and immunologic efficacies of the vaccinia poxvirus JX-594 were investigated in animal models. JX-594 was engineered by the addition of the GM-CSF gene and disruption of the vTK gene. It was evaluated in two immune-competent models: a rabbit model with metastases and a rat liver cancer model. IV administration of JX-594 was well tolerated and highly effective against primary intrahepatic tumors in both models. In addition, no detectable metastases were reported in either model. This study documented tumor-specific virus replication and gene expression, GM-CSF detection, and tumor-infiltrating cytotoxic T-lymphocytes.59 The effect of JX-594 on the tumor-associated vasculature was tested; the results showed a significant regression in the tumor-associated vasculature in xenograft models as well as in patients. This study demonstrated that a biologic agent can be used for infection and selective replication in endothelial cells.60
Recently, modified forms of VV termed “evolved Wyeth strain VV” and the vTK-deleted evolved Wyeth strain VV “cancer-favoring oncolytic VV” showed significant suppression of stem cell-like cancer cells. Drug resistance is a major obstacle to cancer treatment, and stem cell-like cancer cells have shown significant resistance, making them very difficult to treat. In this study, the therapeutic efficacy and cytotoxicity of these strains were evaluated in humans, mouse colon cancer spheres, and a mouse model. The virus-treated group was compared with a fluorouracil-treated group, and a significant reduction in tumor size was evident in cancer-favoring oncolytic VV-treated mice. Moreover, viral treatment in combination with fluorouracil achieved greater tumor regression than any other group.61
Thorough investigation of the characteristics of VV strains in preclinical studies has shown that VV represents a promising nanomedicine for cancer treatment, and thus suitable for clinical trials as a translational medicine.
Western Reserve strains
Combination therapy with Western Reserve stains has also been investigated. Recently, a study evaluated the combined effect of the B18R viral stain and anti-CTLA-4 in mice. Renca (murine kidney adenocarcinoma) and MC38 (murine colon adenocarcinoma) cells were used in this experiment to monitor the antitumor effect. C57/BL6 and BALB/c mice were used for tumor formation. For treatment, two strains, B18R and the double-deleted Western Reserve VV (vvDD) strain, were used. Both the vTK and VGF genes were deleted in vvDD. Anti-CTLA-4 treatment was administered from 0 day to 4 days, but very poor viral replication was observed. It was discovered that anti-CTLA-4 treatment elicited the antiviral response against the virus. However, delayed administration of anti-CTLA-4 treatment (after 4 days) caused significant viral replication and tumor regression in mice. This study revealed the roles of CD4+, CD8+, and CD25+ T-cells in tumor regression and showed the potential of the combination of antibody and virus and optimum timing of their administration to achieve a synergistic antitumor effect.62
The roles of CD4+ and CD8+ T-cells in the TME were elucidated using a VV encoding FCU1 derived from the yeast cytosine deaminase and uracil phosphoribosyltransferase genes. This modified viral strain was evaluated in renal carcinoma (Renca) cells, and orthotopic tumor growth inhibition was observed with systemic administration of VV-FCU1. Furthermore, it was associated with an infiltration of tumors by CD8+ T-lymphocytes and a reduction in the proportion of infiltrating Tregs; consequently, the ratio of CD8+:CD4+ Tregs favored CD8+ cytotoxic T-cells. Depletion of CD4+ T-cells enhanced antitumor efficacy, whereas CD8+ T-cell depletion abolished therapeutic efficacy.63
The combinatorial effect of VV and other viruses was also studied in vitro as well as in vivo. A study showed a synergistic interaction between the VSV and VV, using a deleted B18R Western Reserve virus in combination with the AV3 strain of the VSV. The purpose of this combination was to establish local areas of infection within an infected host (as with vvDD) and to inhibit IFN synthesis in cancer cells. Enhanced VSV replication and infection were reported in a variety of tumor types in vitro and in mouse models. Moreover, explanted tissues from cancer patients also showed the same results.64
The biodistribution and antitumor properties of a vTK-inactivated vaccinia strain have been well characterized. They were assessed in a variety of cell lines from humans and mice to clarify the replication efficiency of the mutated virus. The biodistribution of viruses was monitored by luciferase gene expression, which was encoded by synthetic promoter.65
VV-based nanomedicine: clinical trials
Earlier clinical trials
VV was first used in the treatment of cancer in 1964. It was administered to a patient with disseminated melanomatosis. Treatment with repeated injections of VV was performed in 69-year-old patient. The authors reported the regression of both visceral and cutaneous tumors.66 The efficiency of vaccinia oncolysate has also been shown in patients with advanced metastatic cancer. This study showed an immune response triggered by VV against tumors. Additionally, vaccinia oncolysate was used as an immunotherapeutic agent, and the beneficial effects of the treatment were proven in two women with metastatic melanoma and colon cancer, respectively.67
Another study showed the efficacy of the attenuated vaccinia AS strain in two patients with cancer: 2×108 units of virus were intravenously administered to these patients with advanced adenocarcinoma, resulting in a reduction in the size of their lung and bone tumors.68 Meanwhile, VV showed good efficacy in terms of biodistribution and gene delivery for cancer treatment. Various studies have indicated the treatment efficacy of VV.69,70
Recent clinical trials
The recent outcomes of OV-based clinical trials have boosted studies in oncolytic virotherapy. An improved durable response rate of a strain of HSV-1, ie, T VEC, which encodes GM-CSF, was successfully demonstrated in recent randomized Phase III clinical trials.71 The US Food and Drug Administration approved T-VEC in October 2015 for melanoma. It is the first oncolytic virotherapy approved by the US Food and Drug Administration for the treatment of cancer available in the US, and it was also approved by Europe in January 2016. However, the People’s Republic of China had approved the world’s first oncolytic virotherapy for cancer treatment. Two viruses, namely T-VEC and H101, have achieved regulatory review. In combination with chemotherapy, oncolytic adenovirus-based H101 was approved in the People’s Republic of China for the treatment of head and neck cancer in November 2005.72 These results have stimulated interest in clinical trials for almost all OVs.
The oncolytic efficacy and safety of engineered VV strains were recently evaluated in patients with various types of cancers. Most of the clinical trials were completed at the Phase I level. JX-594, encoding GM-CSF with the disruption of vTK, caused significant regression of tumors in patients with various cancers. The main outcomes of these clinical trials are shown in Table 3.73,74 Other than that, the GL-ONC1 (GLV-1h68) from Benelux Corporation is currently studied in Phase I and Phase I/II clinical trials on human cancer patients GL-ONC.75 Important events in these clinical trials are summarized in Figure 4. In a Phase I clinical trial, an IT injection of JX-594 into the primary or metastatic liver tumors of 14 patients was well tolerated. The safety profile of JX-594 was evaluated in terms of viral replication, GM-CSF expression, and systemic dissemination. However, direct hyperbilirubinemia was observed as a dose-limiting toxicity.76 Another clinical trial was conducted in patients with advanced liver cancer to monitor the antivascular properties of JX-594. Interestingly, antihepatitis B virus (HBV) activity of JX-594 was observed in three patients with advanced refractory HBV-associated HCC. IT application of the virus (3×108 PFU) induced antivascular cytokines and targeted distant tumors in these patients. The suppression of HBV replication in the presence of JX-594 was first reported in this study. However, further studies are warranted because the results were shown in only three patients.77 The selective replication and infection of JX-594 in tumor tissues were confirmed in other clinical trials. IV infusion was shown to cause viral infection of metastatic tumors. These results were confirmed by pharmacokinetic and quantitative polymerase chain reaction analyses. The overall results revealed an antitumor effect of JX-594 in metastatic tumors in a dose-limiting manner.60
Table 3.
Major outcomes of vaccinia virus-based clinical trials
| Vaccinia strain | Clinical trial phase | Major outcomes | Status | References |
|---|---|---|---|---|
| JX-594, Wyeth | Phase I | Systemic dissemination and tolerance in primary or metastatic liver tumors | Completed | Park et al76 |
| JX-594, Wyeth | Phase I | Intravenous infusion led to selective replication in metastatic solid tumors, but not in normal cells | Completed | Breitbach et al60 |
| JX-594, Wyeth | Phase II | Dose effect on overall survival duration in patients with liver cancer | Completed | Heo et al79 |
| JX-594, Wyeth | Phase Ib | Safety profile of multiple intravenous Pexa-Vec infusions in patients with treatment-refractory colorectal cancer | Completed | Park et al85 |
| vvDD, Western Reserve | Phase I | Safety, systemic spread, and antitumor activity in various tumors | Completed | Zeh et al80 |
Figure 4.

Important events in clinical trials of the vaccinia virus.
Notes: This chronologic graph represents key achievements in oncolytic virotherapy. The first trial was started in 1964, and various subsequent trials are in different phases. Recently, the safety and efficacy of the vaccinia virus were proven by the results of Phases I and II clinical trials.
The combinatorial effects of JX-594 and other therapeutic drugs have also been documented. A combination of JX-594 and sorafenib was evaluated in patients with HCC. As a safety precaution, preclinical studies were first performed in various HCC cell lines and in mice models. Concurrent treatment with both agents reduced JX-594 replication but caused a significant reduction in cell numbers or tumor size when administered sequentially. Subsequently, sequential therapy with these two therapeutic agents was carried out in three patients with HCC. A significant reduction in tumor perfusion and size was reported. These results were confirmed by the Choi criteria (up to 100% necrosis). This study documented the tolerance to and antitumor efficacy of JX-594 in combination with sorafenib.78 The effects of a randomized dose of JX-594 were further studied in patients with liver cancer at the Phase II level. This study indicated that the duration of patient survival was significantly related to dose rather than tumor response rate and immune end points. The oncolytic and immunomodulatory mechanisms of action and tumor response to JX-594 were also revealed in this trial.79
A Phase I trial was conducted on the Western Reserve strain-based vvDD: the outcomes of IT dose escalation of vvDD in 16 patients with advanced solid tumors were evaluated, and recovery of the virus in adjacent tumors was reported. This study was conducted in patients with metastatic melanoma, breast cancer, and colon cancer. Elevated levels of aspartate transaminase, alkaline phosphatase, and lactate dehydrogenase were reported as toxicities of vvDD injection.80
Current challenges: VV-based oncolytic virotherapy
Although VV-based therapies have shown promise, challenges must be resolved to increase their efficacy and safety; its bioavailability, the host immune response, and resistance to the virus all have an influence on the efficacy of virotherapy. Both IV and IT injections of the viruses have limitations. When using IV administration, viruses can be sequestered by the liver and spleen. Host immune mechanisms, such as neutralizing antibodies against viruses, reduce their availability to cancer cells. Also, some types of tumor cells exhibit resistance to the viruses, causing a drastic reduction in oncolysis.81–83 To improve the safety and efficacy of oncolytic virotherapy, researchers have expended much effort to overcome these hurdles.
To deliver the virus to the tumor and evade the host immune system, cytokine-induced killer (CIK) cells were infected with VV. VV-infected CIK cell-treated mice showed a significant reduction in tumor size compared with mice treated with virus or untreated CIK cells alone.84 At the clinical level, various types of toxicity have been reported to date. Liver toxicities such as elevated serum enzymes related to liver function and hyperbilirubinemia were observed.85 To avoid such toxicities, a constant dose must be defined. Moreover, adverse events of VV were reported during the smallpox vaccination program, ranging from mild flu-like symptoms to encephalitis. The severity of these adverse effects depends on the type of virus used. The Western Reserve virus strain is usually associated with more severe adverse events than other strains.40 In general, VV is used for oncolytic virotherapy at the preclinical as well as the clinical level, but some problems remain unresolved.
To improve the efficacy and safety of VV and reduce the toxicity associated with treatment, more trials have been performed. The efficacy of oncolytic virotherapy has been enhanced through isolated limb perfusion and intravesical therapy. Isolated limb perfusion administration of GLV-1h68 in combinations with biochemotherapy and radiotherapy showed increased treatment efficacy in an animal model of extremity soft tissue sarcoma.86 Intravesical therapy of bladder cancer with oncolytic HSV also showed enhanced treatment efficacy.87 Good treatment efficacy of aforementioned methods might be linked with avoidance of immune system.
Conclusion and further insights
Oncolytic virotherapy based on VV has proven its benefits in both clinical and preclinical studies. Because of the large size of the VV genome, genetic engineering (such as addition and deletion) is highly beneficial. Combination therapy with cell-cycle regulators, immune-checkpoint inhibitors, apoptosis inducers, and/or other molecules should be the focus of future research. Combinatorial treatment with monoclonal antibodies against immune checkpoint blockades has also been evaluated in recent clinical trials and has shown promising results. In addition, approaches with low toxicity and good efficacy must be taken into consideration. Although virologic and immunologic aspects of the mechanism of virotherapy have been well characterized, future studies should investigate the positive outcomes of oncolytic virotherapy using VV. At present, most of the preclinical studies are using xenograft models to investigate oncolytic effect of OVs; this approach should be replaced by immunocompetent animals, in order to take the immune system together to understand the actual coordinated efficacy OVs. This is very important for further translational research in oncolytic virotherapy as well as for the efficient combination therapy of OVs with immune checkpoint blockades (Figure 5). To elucidate immune interactions in cancer cells by OVs, more translational trials should be well designed and conducted. These approaches should also be considered in translational medicine, especially in future clinical trials.
Figure 5.

Future prospective: mAbs (anticytotoxic T-lymphocyte-associated protein-4/anti-programmed death-1/anti-programmed death-ligand 1) against immune checkpoints encoded by the VV.
Notes: mAbs engineered with the VV may enhance tumor cell death. After the replication of the virus in the cytosol, synthesis of mAbs would occur, followed by the inhibition of cytotoxic T-lymphocyte-associated protein 4, programmed death-1, and programmed death-ligand 1 may take place. It also causes inhibition of immune checkpoints that expressed by other uninfected cancer cells.
Abbreviations: APCs, antigen-presenting cells; mAbs, monoclonal antibodies; VV, vaccinia virus.
Acknowledgments
This study was supported by a grant from the Korean Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare, Republic of Korea (HI13C0259 and HI16C1067); supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2014S1A2A2027641); and by the Ministry of Science, ICT, and Future Planning (NRF-2015R1A2A2A01004489).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Vardy J, Rourke S, Tannock IF. Evaluation of cognitive function associated with chemotherapy: a review of published studies and recommendations for future research. J Clin Oncol. 2007;25(17):2455–2463. doi: 10.1200/JCO.2006.08.1604. [DOI] [PubMed] [Google Scholar]
- 2.Kim Y, Joo KM, Jin J, Nam D-H. Cancer stem cells and their mechanism of chemo-radiation resistance. Int J Stem Cells. 2009;2(2):109–114. doi: 10.15283/ijsc.2009.2.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vardy J, Tannock I. Cognitive function after chemotherapy in adults with solid tumours. Crit Rev Oncol Hematol. 2007;63(3):183–202. doi: 10.1016/j.critrevonc.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Amer MH. Gene therapy for cancer: present status and future perspective. Mol Cell Ther. 2014;2(1):1–19. doi: 10.1186/2052-8426-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2(12):938–950. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 7.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15(3):260–265. doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161(4):355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Guo ZS, Bartlett DL. Oncolytic viruses as platform for multimodal cancer therapeutics: a promising land. Cancer Gene Ther. 2014;21(7):261–263. doi: 10.1038/cgt.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 13.Jefferson A, Cadet VE, Hielscher A. The mechanisms of genetically modified vaccinia viruses for the treatment of cancer. Crit Rev Oncol Hematol. 2015;95(3):407–416. doi: 10.1016/j.critrevonc.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther. 2011;11(5):595–608. doi: 10.1517/14712598.2011.558838. [DOI] [PubMed] [Google Scholar]
- 15.You Z, Fischer DC, Tong X, Hasenburg A, Aguilar-Cordova E, Kieback DG. Coxsackievirus-adenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression. Cancer Gene Ther. 2001;8(3):168–175. doi: 10.1038/sj.cgt.7700284. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Li ZY, Liu Y, et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 2011;17(1):96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shafren DR, Dorahy DJ, Ingham RA, Burns GF, Barry RD. Coxsacki-evirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol. 1997;71(6):4736–4743. doi: 10.1128/jvi.71.6.4736-4743.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farassati F, Yang AD, Lee PW. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol. 2001;3(8):745–750. doi: 10.1038/35087061. [DOI] [PubMed] [Google Scholar]
- 19.Dörig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75(2):295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 20.Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol. 2011;85(12):6015–6023. doi: 10.1128/JVI.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt FI, Bleck CK, Mercer J. Poxvirus host cell entry. Curr Opin Virol. 2012;2(1):20–27. doi: 10.1016/j.coviro.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro Oncol. 2004;6(3):208–217. doi: 10.1215/S1152851703000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degos L, Linch DC, Löwenberg B. Textbook of Malignant Haematology. CRC Press; London: 1999. [Google Scholar]
- 24.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2(4):E65–E67. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 25.Martinez J, Georgoff I, Martinez J, Levine AJ. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes Dev. 1991;5(2):151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- 26.Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proc Natl Acad Sci U S A. 1997;94(7):2776–2778. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81(1):153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 28.Kim M, Williamson CT, Prudhomme J, et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene. 2010;29(27):3990–3996. doi: 10.1038/onc.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ilkow CS, Marguerie M, Batenchuk C, et al. Reciprocal cellular crosstalk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015;21(5):530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 30.Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8(10):1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alvarez-Breckenridge C, Kaur B, Chiocca EA. Pharmacologic and chemical adjuvants in tumor virotherapy. Chem Rev. 2009;109(7):3125–3140. doi: 10.1021/cr900048k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roulstone V, Khan K, Pandha HS, et al. Phase I trial of cyclophosphamide as an immune modulator for optimizing oncolytic reovirus delivery to solid tumors. Clin Cancer Res. 2015;21(6):1305–1312. doi: 10.1158/1078-0432.CCR-14-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4(138):138ra177. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilett EJ, Prestwich RJ, Kottke T, et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009;16(5):689–699. doi: 10.1038/gt.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Power AT, Wang J, Falls TJ, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15(1):123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 37.Lemay CG, Rintoul JL, Kus A, et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol Ther. 2012;20(9):1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan ZY, Zhang L, Li S, Qian XZ, Guan ZZ. Safety of an E1B deleted adenovirus administered intratumorally to patients with cancer. Ai Zheng. 2003;22(3):310–313. Chinese. [PubMed] [Google Scholar]
- 39.Fields BN, Knipe DM, Howley PM. Fields Virology. 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 40.Poland GA, Grabenstein JD, Neff JM. The US smallpox vaccination program: a review of a large modern era smallpox vaccination implementation program. Vaccine. 2005;23(17–18):2078–2081. doi: 10.1016/j.vaccine.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 41.Puhlmann M, Brown CK, Gnant M, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000;7(1):66–73. doi: 10.1038/sj.cgt.7700075. [DOI] [PubMed] [Google Scholar]
- 42.Locker JK, Kuehn A, Schleich S, et al. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell. 2000;11(7):2497–2511. doi: 10.1091/mbc.11.7.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung C-S, Hsiao J-C, Chang Y-S, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol. 1998;72(2):1577–1585. doi: 10.1128/jvi.72.2.1577-1585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci U S A. 2005;102(51):18572–18577. doi: 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Townsley AC, Moss B. Two distinct low-pH steps promote entry of vaccinia virus. J Virol. 2007;81(16):8613–8620. doi: 10.1128/JVI.00606-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolonen N, Doglio L, Schleich S, Locker JK. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell. 2001;12(7):2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Paolo NC, Miao EA, Iwakura Y, et al. Virus binding to a plasma membrane receptor triggers interleukin-1α-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31(1):110–121. doi: 10.1016/j.immuni.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749–758. doi: 10.1038/mt.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrig K, Chen NG, Stritzker J, et al. Vaccinia virus-mediated expression of transcription factor Klf4 enhances oncolytic virotherapy of colorectal cancer. J Hum Virol Retrovirol. 2014;2(4):00051. [Google Scholar]
- 50.Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22(1):102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buckel L, Advani SJ, Frentzen A, et al. Combination of fractionated irradiation with anti-VEGF expressing vaccinia virus therapy enhances tumor control by simultaneous radiosensitization of tumor associated endothelium. Int J Cancer. 2013;133(12):2989–2999. doi: 10.1002/ijc.28296. [DOI] [PubMed] [Google Scholar]
- 52.Huang T, Wang H, Chen NG, Frentzen A, Minev B, Szalay AA. Expression of anti-VEGF antibody together with anti-EGFR or anti-FAP enhances tumor regression as a result of vaccinia virotherapy. Mol Ther – Oncol. 2015;2:15003. doi: 10.1038/mto.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gholami S, Chen CH, Lou E, et al. Vaccinia virus GLV-1h153 is effective in treating and preventing metastatic triple-negative breast cancer. Ann Surg. 2012;256(3):437–445. doi: 10.1097/SLA.0b013e3182654572. [DOI] [PubMed] [Google Scholar]
- 54.Gholami S, Chen CH, Lou E, et al. Vaccinia virus GLV-1h153 in combination with 131I shows increased efficiency in treating triple-negative breast cancer. FASEB J. 2014;28(2):676–682. doi: 10.1096/fj.13-237222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gholami S, Chen CH, Belin LJ, et al. Vaccinia virus GLV-1h153 is a novel agent for detection and effective local control of positive surgical margins for breast cancer. Breast Cancer Res. 2013;15(2):R26. doi: 10.1186/bcr3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mansfield DC, Kyula JN, Rosenfelder N, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther. 2016;23(4):357–368. doi: 10.1038/gt.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gentschev I, Muller M, Adelfinger M, et al. Efficient colonization and therapy of human hepatocellular carcinoma (HCC) using the oncolytic vaccinia virus strain GLV-1h68. PLoS One. 2011;6(7):e22069. doi: 10.1371/journal.pone.0022069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adelfinger M, Bessler S, Frentzen A, et al. Preclinical testing oncolytic vaccinia virus strain GLV-5b451 expressing an anti-VEGF single-chain antibody for canine cancer therapy. Viruses. 2015;7(7):4075–4092. doi: 10.3390/v7072811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14(3):361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 61.Yoo SY, Bang SY, Jeong SN, Kang DH, Heo J. A cancer-favoring oncolytic vaccinia virus shows enhanced suppression of stem-cell like colon cancer. Oncotarget. 2016;7(13):16479–16489. doi: 10.18632/oncotarget.7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. 2015;21(24):5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fend L, Remy-Ziller C, Foloppe J, et al. Oncolytic virotherapy with an armed vaccinia virus in an orthotopic model of renal carcinoma is associated with modification of the tumor microenvironment. Oncoimmunology. 2016;5(2):e1080414. doi: 10.1080/2162402X.2015.1080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le Boeuf F, Diallo JS, McCart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18(5):888–895. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parviainen S, Ahonen M, Diaconu I, et al. GMCSF-armed vaccinia virus induces an antitumor immune response. Int J Cancer. 2015;136(5):1065–1072. doi: 10.1002/ijc.29068. [DOI] [PubMed] [Google Scholar]
- 66.Burdick KH, Hawk WA. Vitiligo in a case of vaccinia virus-treated melanoma. Cancer. 1964;17(6):708–712. doi: 10.1002/1097-0142(196406)17:6<708::aid-cncr2820170604>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 67.Wallack MK, Steplewski Z, Koprowski H, et al. A new approach in specific, active immunotherapy. Cancer. 1977;39(2):560–564. doi: 10.1002/1097-0142(197702)39:2<560::aid-cncr2820390227>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 68.Arakawa S, Jr, Hamami G, Umezu K, Kamidono S, Ishigami J, Arakawa S. Clinical trial of attenuated vaccinia virus AS strain in the treatment of advanced adenocarcinoma. J Cancer Res Clin Oncol. 1987;113(1):95–98. doi: 10.1007/BF00389974. [DOI] [PubMed] [Google Scholar]
- 69.Mastrangelo MJ, Maguire HC, Eisenlohr LC, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6(5):409–422. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 70.Gomella LG, Mastrangelo MJ, McCue PA, Maguire HC, Mulholland SG, Lattime EC. Phase I study of intravesical vaccinia virus as a vector for gene therapy of bladder cancer. J Urol. 2001;166(4):1291–1295. [PubMed] [Google Scholar]
- 71.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 72.Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. doi: 10.1093/jnci/djj111. [DOI] [PubMed] [Google Scholar]
- 73.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 74.Jebar AH, Errington-Mais F, Vile RG, Selby PJ, Melcher AA, Griffin S. Progress in clinical oncolytic virus-based therapy for hepatocellular carcinoma. J Gen Virol. 2015;96(pt 7):1533–1550. doi: 10.1099/vir.0.000098. [DOI] [PubMed] [Google Scholar]
- 75.GENELUX [webpage on the Internet]. Clinical Trails [Accessed August 23, 2016]. Available from: http://www.genelux.com/clinical-trials/
- 76.Park B-H, Hwang T, Liu T-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 77.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16(9):1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 78.Heo J, Breitbach CJ, Moon A, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19(6):1170–1179. doi: 10.1038/mt.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeh HJ, Downs-Canner S, McCart JA, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther. 2015;23(1):202–214. doi: 10.1038/mt.2014.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aghi M, Martuza RL. Oncolytic viral therapies – the clinical experience. Oncogene. 2005;24(52):7802–7816. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- 82.Evgin L, Acuna SA, Tanese de Souza C, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther. 2015;23(6):1066–1076. doi: 10.1038/mt.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311(5768):1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- 84.Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17(10):1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park SH, Breitbach CJ, Lee J, et al. Phase 1b trial of biweekly intravenous Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther. 2015;23(9):1532–1540. doi: 10.1038/mt.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilkinson MJ, Smith HG, Pencavel TD, et al. Isolated limb perfusion with biochemotherapy and oncolytic virotherapy combines with radiotherapy and surgery to overcome treatment resistance in an animal model of extremity soft tissue sarcoma. Int J Cancer. 2016;139(6):1414–1422. doi: 10.1002/ijc.30162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simpson GR, Horvath A, Annels NE, et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br J Cancer. 2012;106(3):496–507. doi: 10.1038/bjc.2011.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lucas T, Benihoud K, Vigant F, et al. Hexon modification to improve the activity of oncolytic adenovirus vectors against neoplastic and stromal cells in pancreatic cancer. PLoS One. 2015;10(2):e0117254. doi: 10.1371/journal.pone.0117254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poppers J, Mulvey M, Khoo D, Mohr I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol. 2000;74(23):11215–11221. doi: 10.1128/jvi.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyamoto S, Inoue H, Nakamura T, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72(10):2609–2621. doi: 10.1158/0008-5472.CAN-11-3185. [DOI] [PubMed] [Google Scholar]
- 91.Leber MF, Bossow S, Leonard VH, et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol Ther. 2011;19(6):1097–1106. doi: 10.1038/mt.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Janke M, Peeters B, de Leeuw O, et al. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007;14(23):1639–1649. doi: 10.1038/sj.gt.3303026. [DOI] [PubMed] [Google Scholar]