

Abstract
Objectives
The Carolina Women’s Care Study (CWCS) at the University of South Carolina followed 467 young women with the goal of identifying biomarkers of human papillomavirus (HPV) persistence. In this study, we analyzed the methylation of HPV16 DNA.
Methods
The aims of this study were to determine the methylation status of the HPV16 L2 gene in DNA isolated from exfoliated cervical cells collected longitudinally as part of the CWCS; and to determine the prevalence of polymorphisms (SNPs) in folate metabolizing enzymes and DNA repair enzymes known to affect DNA methylation in blood-derived genomic DNA from CWCS participants. For methylation studies, DNA samples were bisulfite converted and amplified with the EpiTect® Whole Bisulfitome kit. PCR was performed for amplicons containing five CpG sites in L2. Pyrosequencing was carried out by EpigenDx and analyzed with PyroMark® Software. Taqman® genotyping assays were performed to determine selected SNP alleles in the CWCS cohort.
Results and Conclusion
Methylation data were obtained for 82 samples from 27 participants. Of these, 22 participants were positive for HPV16 for three or more visits (≥ 12 months). Methylation in L2 was detectable but methylation levels varied and were not associated with HPV16 persistence. No linearity of methylation levels over time was observed in participants for whom longitudinal data could be analyzed. Analysis of nine selected SNPs did not reveal an association with persistence. We conclude that at early stages of infection methylation of HPV16 L2 DNA in Pap test samples is not a predictive biomarker of HPV persistence.
Keywords: human papillomavirus, viral persistence, HPV16 L2, DNA methylation, single nucleotide polymorphisms, health disparity
Precis
At early stages of infection, methylation of HPV16 L2 DNA in Pap test samples is not a predictive biomarker of HPV persistence.
Introduction
Human papillomavirus (HPV) is a known etiological cause of cervical cancer [1]. Although cervical cancer is a rare outcome of HPV infection, the widespread prevalence of HPV causes a significant health burden. It is unknown why HPV infections persist. Women with persistent high-risk HPV (HR-HPV) infections are at greatest risk of cervical intraepithelial neoplasia (CIN) and cervical cancer [1]. There are also health disparities because in the United States African Americans are more likely to be diagnosed with, and are more than twice as likely to die from, cervical cancer as European Americans [2].
Methylation of cytosine in CpG DNA sequences is an epigenetic regulator of gene expression and is dysregulated in cancer [3]. Methylation of host genes in CIN lesions and cervical cancer has been measured as potential biomarkers of cervical disease that might progress to cancer [1]. The role of HPV DNA methylation has received less attention. Studies focused on methylation sites in the promoter region of HPV have generally, but not always, found that methylation of this region decreases during progression [4–8]. Studies have also shown evidence that HPV16 DNA undergoes an increase in DNA methylation, especially in the L1 and L2 genes, during progression from CIN to cancer [9–15].
The Carolina Women’s Care Study (CWCS) at the University of South Carolina was a longitudinal study to explore biological and genetic determinants of HPV persistence in African American and European American college-age women (18–22 years old). The CWCS measured HPV prevalence in female college students via biannual visits and HPV testing [16]. The CWCS showed that HR-HPV clearance in African American participants took twice as long as in European Americans [17].
To our knowledge, no study has attempted to correlate methylation of HPV DNA with viral persistence at early stages of infection. Here, we report mapping the methylation of HPV16 L2 DNA in the CWCS cohort. Methylation of five CpG sites in the L2 gene from 222 unique HPV16-positive visits was measured in exfoliated cervical cells. Also, polymorphisms in nine genes associated with biochemical processes related to methylation were determined and analyzed for associations with viral persistence.
Materials and Methods
Study Demographics, Sample Collection, and HPV Typing and Prevalence in the CWCS
The CWCS was a unique prospective study initiated to identify biomarkers of HPV persistence in a college-age cohort (average age 18.8 years at the time of enrollment). The study was conducted at the University of South Carolina Women's Care Clinic and approved by the USC IRB [16]. The CWCS was conducted between 2004 and 2011 and consisted of biannual visits during which Pap test samples were collected. The study included 70% European Americans, 24% African Americans, and the remainder were Hispanic, Pacific Islander, or Asian (ethnicity was self-reported). Because our study began in 2004 and concluded in 2011, only 36 of the 467 women enrolled had received at least one dose of the Gardasil® HPV vaccine at the time of enrollment [16]. Samples were collected from a total of 2,274 participant visits, averaging 4.8 visits per participant. Exfoliated cervical cells were collected in PreservCyt® (Cytyc (now Hologic) Marlborough MA, USA) and DNA was isolated by proteinase K digestion and phenol-chloroform extraction, followed by ethanol precipitation. The DNA was analyzed via real-time PCR with PGMY09/11 primers to identify the presence of HPV [16, 17]. The HPV positive samples were then tested with INNO-LiPa® HPV AMP and Genotyping Extra® kits (Fujirebio Europe, Belgium) to determine the HPV type(s). The prevalence of HR-HPV (all types) was 36.4% (811 out of 2,274 visits). The overall rate of abnormal Pap tests in this young, college-age cohort for the entire CWCS study was low, with ASC-US, LSIL, and HSIL representing 4.9%, 5.8%, and 1.1%, respectively. Please refer to the previously published work for more details of the cohort [16, 17].
Cell lines
The human HPV16-positive cell lines CaSki (approximately 600 HPV16 genome copies/cell) and SiHa (approximately 2 HPV16 genome copies/cell) were used for internal controls on every plate for HPV16 pyrosequencing [18]. The cells were maintained at 37 °C in 5% CO2 in DMEM medium (GIBCO/Invitrogen/Thermofisher, Waltham MA, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Atlanta GA). DNA was extracted from the cells using a phenol-chloroform extraction method [19].
DNA Clean-Up and Quantification
Supplemental Figure 1 shows the workflow of processing HPV16-positive samples for methylation analysis by bisulfite-pyrosequencing, which has the advantages of analyzing several methylation sites, introduces an internal control (DNA sequence including a control for unconverted cytosines), and allows for accurate quantitation of multiple CpG methylation sites in the same reaction [20]. DNA from each sample was further purified using the Genomic DNA Clean & Concentrator® (Zymo Research, Irvine CA, USA). DNA quantitation was carried out using Accuclear® Ultra High Sensitivity dsDNA Quantitation kit (Biotium Inc., Hayward, CA, USA) using human genomic DNA as a calibrator (Bioline, Taunton MA, USA). Samples with sufficient yield of DNA were moved forward into the pyrosequencing work-flow.
Bisulfite Conversion and Whole Genome Amplification
DNA from each sample (250 ng) was bisulfite treated according to the manufacturer’s instructions using the EZ DNA Methylation® kit (Zymo) [21]. Bisulfite treated DNA was eluted from the column and was quantitated as above. Bisulfite-treated DNA (50 ng) was amplified using the EpiTect Whole Bisulfitome® kit [22] and diluted to 20 ng/µl for subsequent PCR reactions. Universal Methylated Human DNA Standard (D5011, Zymo) and human whole genome amplified non-methylated DNA (D5013-1, Zymo) were used with biotin tagged versions of the manufacturer’s primers for pyrosequencing controls.
PCR Amplification for Pyrosequencing Reactions
Amplicons were generated with 0.2 µM forward and reverse PCR primers (one of which was biotinylated), 0.2 mM dNTPs (ThermoFisher), 0.5 M Betaine, 2.5 mM MgCl2, 1X Buffer (Biotium), 1 U Cheetah Taq® (Biotium) using 80 ng of bisulfite-treated, amplified DNA. PCR conditions for all reactions were 95 °C for 10 min; 50 cycles with denaturation at 94 °C for 30 s, annealing at 56 °C for 30 s and elongation at 72 °C for 30 s; 1 cycle at 72 °C for 7 min; and a final hold at 4 °C. Multiple amplifications were performed for each sample to allow for subsequent CpG site analysis by pyrosequencing. Amplicons were analyzed by gel electrophoresis and densitometry was performed using ImageJ [23] and a Perfect Size DNA Molecular Weight Ladder (5Prime, Gaithersburg MD, USA) to quantitate 2–5 pmol of amplicon for pyrosequencing using a calculator provided by the University of Hong Kong Genomics Center [24]. Primer sequences are found in Supplemental Table 1. In addition to the Universal Methylated Human DNA Standard, bisulfite converted CaSki and SiHa gDNA were also included as interplate controls on each plate, and were very similar to methylation levels reported previously for L2 [25]).
Pyrosequencing Reactions and Data Analysis
Single-stranded biotinylated PCR products were sent to EpigenDx (Hopkinton, MA, USA) for pyrosequencing. Sequencing primers to cover the L2 region CpGs were designed using the Pyromark® Assay Design SW 2.0 software (see Supplemental Table 1). A non-CG cytosine in the pyrosequencing region was used as an internal control to assess successful bisulfite conversion (templates show T and not C in this position). Methylation analysis was completed with OpenEpi for chi-squared analysis (Dean AG, Sullivan, K.M., Soe, M.M. 2015. OpenEpi: Open Source Epidemiologic Statistics for Public Health. www.OpenEpi.com) [26] and R with RStudio (2015. R Development Core Team. R Foundation for Statistical Computing; Vienna, Austria) [27] for fitting linear and mixed effects models. Graphpad Prism 5.0 was also used to generate some plots.
Taqman® Genotyping of Single Nucleotide Polymorphisms
ThermoFisher/Life Technologies/Applied Biosystems Taqman® genotyping assays were performed to genotype selected alleles in the total CWCS cohort population of 467 participants from DNA isolated from blood draws collected at the first visit [16, 17]. The assay conditions used genomic DNA (25 ng) and followed the manufacturers’ instructions (www.appliedbiosystems.com). Thermal cycling conditions on an ABI 7900HT for all reactions were 95 °C for 10 min followed by 40 cycles with denaturation at 94 °C for 15 s and annealing/extending at 60 °C for 1 min; and a final hold at 4 °C. ABI PRISM Sequence Detection System 2.1 and Taqman® Genotyper software were used for pre-run set up and post run analysis and genotype calling. Nine individual polymorphisms were determined and are listed with the Applied Biosystems Assay identifier (Supplemental Table 2). Of the original 467 participants, genotyping was attempted for 445 samples from which there was sufficient DNA. Of these 445 samples, successful genotyping calls occurred for all nine SNPs for 432 participants.
Results
Methylation Data and Population Definitions
At the start of the project, all HPV16 positive CWCS exfoliated cervical cell samples (n=369) entered the work flow (Supplemental Figure 1). It was notable that among all the HPV16 positive samples for which we were able to obtain HPV16 L2 methylation data, most were from participants who were HPV16 positive for several visits. Ultimately, methylation data from 82 samples were obtained for 27 participants. Participants who were HPV16 positive for three or more consecutive visits (≥ 12 months) were designated as having a persistent HPV16 infection. Consequently, 22 participants met this criterion and are subsequently called the persistent subset. Abnormal Pap tests for these 82 samples was approximately double the percent of abnormal Pap tests for the entire CWCS study yet remained low, with ASC-US, LSIL, and HSIL representing 12.2%, 11.0%, and 2.4%, respectively. HPV16 is presumed to be episomal at early stages of infection [1].
CpG Methylation Analysis
Prior studies suggested that methylation in the viral L2 region is predictive of high-grade CIN in women with persistent infection [9–13]. Methylation at the CpGs in the L2 region was determined in 82 samples for which we obtained methylation data for at least one of the two amplicons. Data were analyzed separately for the entire group of 27 and then the persistent subset of 22 European American or African American participants. The complete methylation dataset is shown in Supplemental Table 3. Figure 1 shows the percent methylation within the persistent subset for the five CpGs analyzed, as well as mean (+) and median (line within the bar graphs) for each CpG. The methylation data were analyzed with the entire group of 27 and the persistent subset only, and analyzed longitudinally, per total number of visits, per visit or per participant, by individual CpGs, or averaging methylation across the amplicon, as well as by persistence in both the overall set of 27 (data not shown) and the persistent subset of 22 (Figure 1). Data were also analyzed with different thresholds of percent methylation above which a sample would be binned as “high” or “methylated”, e.g., 20%. Of the methylation detected, there was no statistically significant trend or association observed for any of the parameters examined. Table 1 shows the boxplot statistics within the persistent subset.
Figure 1.
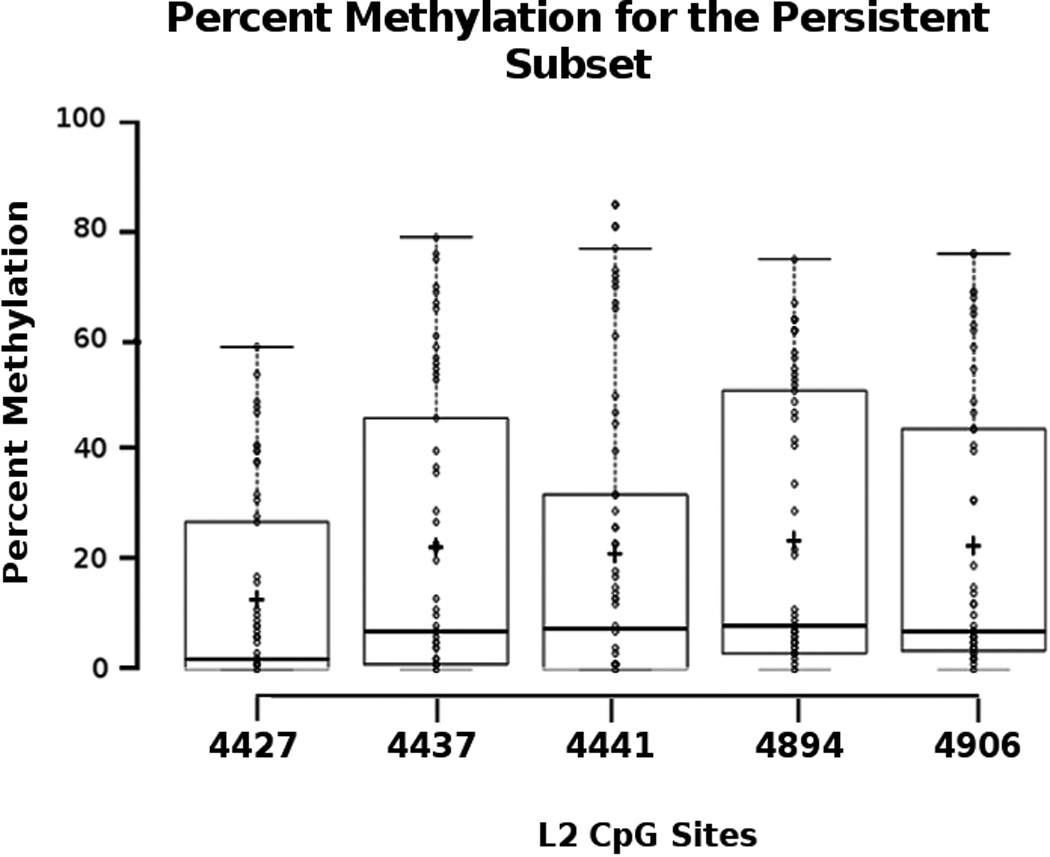
Table 1.
Boxplot statistics of Percent Methylation within the Persistent Subset of 22 participants.
| CpG Position in L2 Region | |||||
|---|---|---|---|---|---|
| 4427 | 4437 | 4441 | 4894 | 4906 | |
|
Upper whisker |
59 | 79 | 77 | 75 | 76 |
|
3rd quartile |
27 | 46 | 32 | 51 | 44 |
|
1st quartile |
0 | 1 | 0 | 3 | 4 |
|
# of data points |
58 | 58 | 58 | 58 | 55 |
| Median | 2.0 | 7.0 | 7.5 | 8.0 | 7.0 |
| Mean | 12.8 | 22.3 | 21.1 | 23.5 | 22.6 |
Analysis by Ethnicity for each CpG
We previously reported that it took about twice as long for African American women in the CWCS to clear HR-HPV than European Americans (601 days versus 316 days) [17]. A categorical methylation analysis across each of the L2 CpGs in the persistent subset was performed by ethnicity (Figure 2 and Table 2). Although there were visual differences between African American and European Americans particularly with site 4427 and 4441 (grey bars versus open bars, Figure 2), these did not reach statistical significance. Table 2 shows the boxplot statistics of percent methylation by ethnicity for the 22 participants in the persistent subset. This is likely due to the low number of African Americans in the persistent population (6 of 22).
Figure 2.
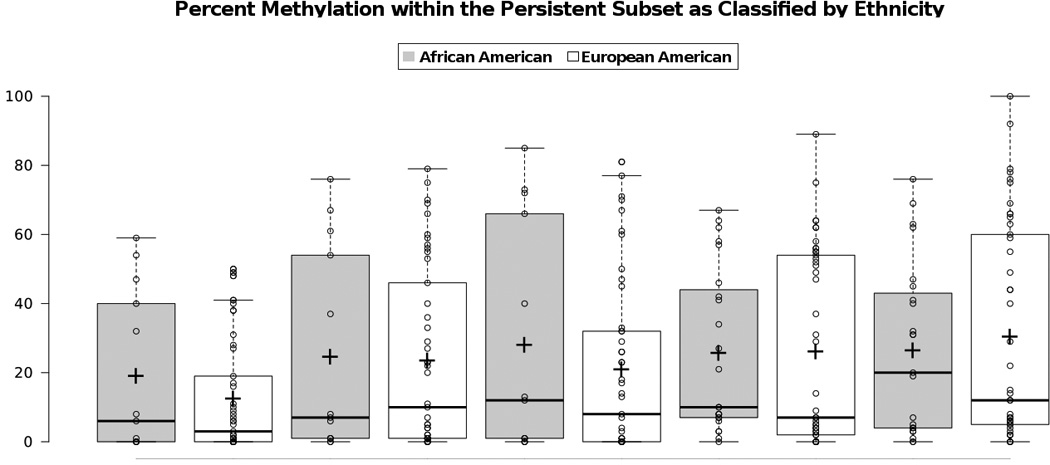
Table 2.
Boxplot statistics of Percent Methylation within the Persistent Subset by Ethnicity (n=6 for AAand n=16 for EA participants).
| CpG Position in L2 Region | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4427 | 4437 | 4441 | 4894 | 4906 | ||||||
| AA | EA | AA | EA | AA | EA | AA | EA | AA | EA | |
|
Upper whisker |
59 | 41 | 76 | 79 | 85 | 77 | 67 | 75 | 76 | 76 |
|
3rd quartile |
40 | 17 | 54 | 46 | 66 | 32 | 44 | 52 | 41 | 49 |
|
1st quartile |
0 | 0 | 1 | 1 | 1 | 0 | 7 | 0 | 4 | 3 |
|
# of data points |
13 | 42 | 13 | 42 | 13 | 42 | 19 | 37 | 19 | 34 |
| Median | 6 | 3 | 7 | 9 | 12 | 8 | 10 | 7 | 7 | 8 |
| Mean | 19 | 11.7 | 24.5 | 22.9 | 28 | 20.2 | 25.9 | 23.3 | 23.5 | 23.2 |
EA – European American participants
AA – African American participants
Analysis of SNPs
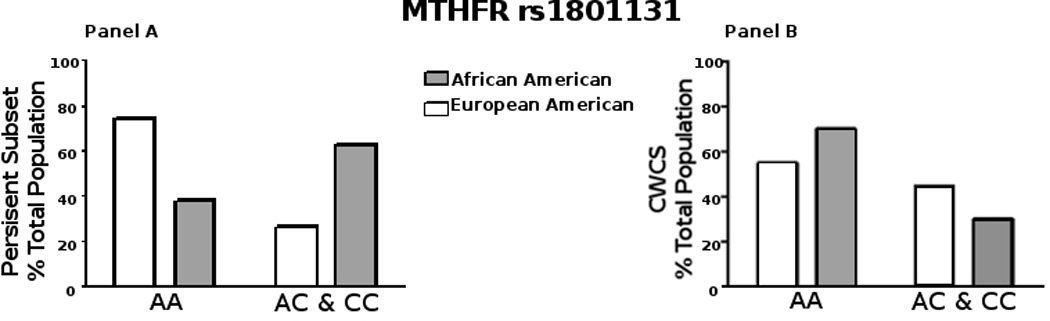
We studied specific SNPs in the following gene sequences: methylene tetrahydrofolate reductase (MTHFR); 5-methyltetrahydrofolate-homocysteine methyltransferase (MTR); betaine-homocysteine methyltransferase (BHMT); thymine DNA glycosylase (TDG); methyl binding domain 4 (MBD4, also known as MED1); and X-ray cross-complementing group 1 (XRCC1) (Supplemental Table 2) in the entire CWCS cohort. The SNPs were chosen because the minor alleles are known to adversely affect the biochemical/cellular activity of these proteins. Of the genotyping attempted for 445 samples, successful genotyping calls were made for all nine SNPs for 432 participants (97%). Table 3 shows each SNP with the minor allele frequency (MAF) as reported in dbSNP (ncbi.nlm.nih.gov) and compared to the MAF calculated in the CWCS cohort. The MAFs for most SNPs calculated in the CWCS cohort were quite similar to those reported in the dbSNP. Within the self-reported ethnicities in CWCS the MAFs were also similar to that reported in the 1000 Genomes Project Phase 3 allele frequencies (ueast.ensemble.org, data not shown). For example, with MTHFR (rs1801131), the MAF in Ensembl was 31% in EUR and 15% in AFR, and in the CWCS the MAF was 26.6% for European Americans and 17.5% for African Americans. We first examined each SNP for an association with HPV persistence for the entire CWCS cohort. In this analysis, we found no association between any of the SNPs and HPV persistence, by Spearman Correlation and logistic regression modeling. We next examined potential associations between the SNPs and methylation status in the persistent subset of 22 participants. Interestingly, we found a lower percentage of the African American participants with the AA homozygous allele of MTHFR (rs1801131) in the persistent subset (Figure 3A) as compared to the total CWCS cohort (Figure 3B). Note that the A to C substitution causes a glutamate to alanine switch in MTHFR that causes a loss of enzyme activity. However, the number of African Americans in this group is small (6 of 22). Therefore, we used a generalized linear mixed model of regression (R-studio glm analysis) against the data set [27]. The regression model found that in this persistent population, the AA allele did not correlate to HPV16 persistence (p value = 0.4730).
Table 3.
MAF for each SNP in the CWCS cohort, compared to the MAF reported in dbSNP.
| SNP | dbSNP | CWCS |
|---|---|---|
| rs1801133 MTHFR C677T | 0.325 | 0.269(n=438) |
| rs1801131 MTHFR A1298C | 0.228 | 0.246(n=440) |
| rs1799782 XRCC1 C580T | 0.130 | 0.073(n=436) |
| rs0025487 XRCC1 A1196G | 0.263 | 0.297(n=436) |
| rs0025489 XRCC1 G839A | 0.061 | 0.036(n=436) |
| rs2307289 MBD4 T1024C | 0.034 | 0.034(n=435) |
| rs3733890 BHMT G821A | 0.301 | 0.297(n=432) |
| rs1805087 MTR A2756G | 0.193 | 0.193(n=436) |
| rs4135113 TDG G595A | 0.105 | 0.054(n=435) |
n= the total number of participants successfully genotyped for that locus.
Figure 3.
Discussion
Epigenetic changes in DNA methylation are receiving much attention as biomarkers of HPV positive cancer [1]. The approach has been to measure methylation of the host genome and viral DNA (presumably after integration) in CIN and cervical cancer specimens. The goal of the CWCS was to explore the biological, genetic, and lifestyle determinants of persistent HPV infection in college-aged women. The average age of enrollment in our cohort was 18.8, and average age of first sexual activity 16.4. Pap test results in all HPV-positive CWCS participants were 69.6% negative, 28.4% ASC-US or LSIL, and only 2% HSIL [16]. By design, the CWCS may provide important insight into the earliest stages of infection (within two years of exposure) that define when persistence begins. In our study, we observed no association of the methylation of HPV16 L2 with HPV16 persistent infection. Overall, the large majority of samples from women with persistent HPV16 infection showed very low methylation (<10%) at all five L2 CpG sites examined (Figure 1). This itself is an interesting observation, as one might have predicted that viral DNA methylation may occur as part of a natural defense against the completion of the HPV life cycle. We compared methylation status from the first HPV16-positive visit through multiple positive visits over time, in individual participants that had persistent HPV16 infection. We observed considerable variation, including examples where HPV16 methylation remained low across multiple visits (e.g., participant # 1, 3, 5, 15; see Supplemental Table 3), others in which it was high across multiple visits (e.g., participant # 18, 23, 25), while in others methylation levels varied (e.g., participant #2, 7, 21). Thus, changes in L2 methylation at early stages of HPV infection do not appear to be predictive of persistent infection. There is no standard cutoff used in the literature that defines a certain percentage as “methylated.” Therefore, data were also analyzed with different threshold cut-offs of % methylation, but this analysis did not reveal an association between L2 methylation and persistence. This study does not address global genomic methylation in the participants. African Americans have been noted to have lower global methylation levels than European Americans [28]. Individual global methylation status is a factor that should be considered in future studies.
Our attempts to amplify and analyze HPV DNA from HPV16 positive samples in participants who became HPV16 negative in subsequent visits almost universally failed, whereas we could more readily detect DNA methylation in the samples from participants with persistent infections. This observation suggests that viral episomal copy numbers are greater in those participants with persistent infections, thus facilitating methylation determination. Note that the viral life cycle is quite dynamic [29]. We did not directly account for viral copy number in our samples. A related question pertains to what epigenetic changes occur to the HPV genome before integration into the host genome versus after integration. Once integrated in a precancerous lesion, it seems more appropriate to consider viral DNA methylation in context with changes in host gene methylation that are associated with cancer progression [30].
We also examined the prevalence of selected common SNPs associated with known biochemical alterations in activity in two processes related to methylation. Polymorphisms were chosen for study because of their link to alterations in biochemical or cellular activity. The lack of association for these SNPs with methylation status in participants with persistent HPV16 infection might be due to the small sample size in which we were able to determine methylation status. In conclusion, our results do not support HPV16 L2 DNA methylation status as a major determinant of HPV16 persistence, at least at early stages of infection.
Supplementary Material
Acknowledgments
The authors thank the Women’s Care Clinic at the University of South Carolina: In particular, the study nurse practitioners Michelle Zager (CFNP) and Julie Steele (WHNP) for collecting the samples and providing care to the study participants; Dr. Debbie Beck for access and use of the facility; and the faculty and staff of the Women’s Care Center for their assistance in assuring the success of the study.
Role of the Funding Source
This study was funded by grant P20MD001770 from the National Institute on Minority Health and Health Disparities, and by grant R21CA169998. The funding source had no involvement.
Abbreviations and Acronyms
- HPV
Human Papillomavirus
- HR-HPV
High Risk HPV
- CWCS
Carolina Women’s Care Study
- SNP
Single Nucleotide Polymorphism
- MAF
Minor Allele Frequency
Footnotes
Disclosure Statement
The authors declare no conflicts of interest.
The CWCS study was approved by the IRB of the University of South Carolina. All participant information and samples were de-identified prior to the study reported here.
References
- 1.Steenbergen RD, Snijders PJ, Heideman DA, Meijer CJ. Clinical implications of (epi)genetic changes in HPV-induced cervical precancerous lesions. Nature reviews Cancer. 2014:395–405. doi: 10.1038/nrc3728. [DOI] [PubMed] [Google Scholar]
- 2.Society AC. Atlanta Georgia: American Cancer Society. 2015 [Google Scholar]
- 3.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature reviews Genetics. 2012:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 4.Badal V, Chuang LS, Tan EH, Badal S, Villa LL, Wheeler CM, Li BF, Bernard HU. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding DC, Chiang MH, Lai HC, Hsiung CA, Hsieh CY, Chu TY. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur J Obstet Gynecol Reprod Biol. 2009:215–220. doi: 10.1016/j.ejogrb.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 6.Hong D, Ye F, Lu W, Hu Y, Wan X, Chen Y, Xie X. Methylation status of the long control region of HPV 16 in clinical cervical specimens. Mol Med Report. 2008:555–560. [PubMed] [Google Scholar]
- 7.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ, Bernard HU. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol. 2004:12762–12772. doi: 10.1128/JVI.78.23.12762-12772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xi LF, Jiang M, Shen Z, Hulbert A, Zhou XH, Lin YY, Kiviat NB, Koutsky LA. Inverse Association between Methylation of Human Papillomavirus Type 16 DNA and Risk of Cervical Intraepithelial Neoplasia Grades 2 or 3. PloS one. 2011:e23897. doi: 10.1371/journal.pone.0023897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK, 3rd, Martel M, Harigopal M, Schofield K, Neapolitano M. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology. 2009:100–107. doi: 10.1016/j.virol.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentnall AR, Vasiljevic N, Scibior-Bentkowska D, Cadman L, Austin J, Szarewski A, Cuzick J, Lorincz AT. A DNA methylation classifier of cervical precancer based on human papillomavirus and human genes. International journal of cancer Journal international du cancer. 2014:1425–1432. doi: 10.1002/ijc.28790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009:438–451. doi: 10.1101/gr.083550.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorincz AT, Brentnall AR, Vasiljevic N, Scibior-Bentkowska D, Castanon A, Fiander A, Powell N, Tristram A, Cuzick J, Sasieni P. HPV16 L1 and L2 DNA methylation predicts high-grade cervical intraepithelial neoplasia in women with mildly abnormal cervical cytology. International journal of cancer Journal international du cancer. 2013:637–644. doi: 10.1002/ijc.28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirabello L, Schiffman M, Ghosh A, Rodriguez AC, Vasiljevic N, Wentzensen N, Herrero R, Hildesheim A, Wacholder S, Scibior-Bentkowska D, et al. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. International journal of cancer Journal international du cancer. 2013:1412–1422. doi: 10.1002/ijc.27750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel DA, Rozek LS, Colacino JA, Van Zomeren-Dohm A, Ruffin MT, Unger ER, Dolinoy DC, Swan DC, Onyekwuluje J, Degraffinreid CR, et al. Patterns of cellular and HPV 16 methylation as biomarkers for cervical neoplasia. J Virol Methods. 2012:84–92. doi: 10.1016/j.jviromet.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecologic oncology. 2011:59–63. doi: 10.1016/j.ygyno.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banister CE, Messersmith AR, Chakraborty H, Wang Y, Spiryda LB, Glover SH, Pirisi L, Creek KE. HPV prevalence at enrollment and baseline results from the Carolina Women's Care Study, a longitudinal study of HPV persistence in women of college age. International journal of women's health. 2013:379–388. doi: 10.2147/IJWH.S45590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banister CE, Messersmith AR, Cai B, Spiryda LB, Glover SH, Pirisi L, Creek KE. Disparity in the persistence of high-risk human papillomavirus genotypes between African American and European American women of college age. The Journal of infectious diseases. 2015:100–108. doi: 10.1093/infdis/jiu394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chardonnet Y, Lizard G, Chignol MC, Schmitt D. Analytical methods for evaluation on whole cells of human papillomavirus infection. Bulletin du cancer. 1995:107–113. [PubMed] [Google Scholar]
- 19.Strauss WM. Preparation of genomic DNA from mammalian tissue. Current protocols in molecular biology / edited by Frederick M Ausubel [et al] 2001:Unit2. doi: 10.1002/0471142727.mb0202s42. [DOI] [PubMed] [Google Scholar]
- 20.Uhlmann K, Brinckmann A, Toliat MR, Ritter H, Nurnberg P. Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. Electrophoresis. 2002:4072–4079. doi: 10.1002/elps.200290023. [DOI] [PubMed] [Google Scholar]
- 21.Izzi B, Binder AM, Michels KB. Pyrosequencing Evaluation of Widely Available Bisulfite Conversion Methods: Considerations for Application. Medical epigenetics. 2014:28–36. doi: 10.1159/000358882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bundo M, Sunaga F, Ueda J, Kasai K, Kato T, Iwamoto K. A systematic evaluation of whole genome amplification of bisulfite-modified DNA. Clinical epigenetics. 2012:22. doi: 10.1186/1868-7083-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sample preparation and submission. < http://cgs.hku.hk/portal/index.php/pyrosequencing/sample-preparation-and-submission>. [Google Scholar]
- 25.Park IS, Chang X, Loyo M, Wu G, Chuang A, Kim MS, Chae YK, Lyford-Pike S, Westra WH, Saunders JR, et al. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer prevention research (Philadelphia, Pa) 2011:207–217. doi: 10.1158/1940-6207.CAPR-10-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dean AG, Sullivan KM, Soe MM. OpenEpi: Open Source Epidemiologic Statistics for Public Health. www.OpenEpi.com [Google Scholar]
- 27.R Development Core Team. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 28.Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, Gamble MV, Susser E. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008:2306–2310. doi: 10.1158/1055-9965.EPI-08-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johannsen E, Lambert PF. Epigenetics of human papillomaviruses. Virology. 2013:205–212. doi: 10.1016/j.virol.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalantari M, Osann K, Calleja-Macias IE, Kim S, Yan B, Jordan S, Chase DM, Tewari KS, Bernard HU. Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1 genes and the cellular DAPK gene: Considerations for use as biomarkers of the progression of cervical neoplasia. Virology. 2014:314–321. doi: 10.1016/j.virol.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.