

Abstract
Donor lymphocyte infusion (DLI) without prophylactic immunosuppression has been used for relapsed AML after allogeneic stem cell transplant (allo-SCT). However DLI is associated with an increased incidence of acute Graft vs. Host Disease (aGVHD). In mice, administration of azacitidine (AzaC) on days 4, 6, 8, and 10 post DLI increases regulatory T cell (Treg) numbers and prevents GVHD without hindering Graft vs. Leukemia (GVL). Based on these findings, we conducted a phase 1 study of AzaC post DLI for AML relapse post allo-SCT. AzaC was administered on days 4, 6, 8 and 10 post-DLI. Dose escalation was done using a 3+3 design with three AzaC dose levels: 30 mg/m2 (level -1), 45 mg/m2 (level 1) and 75 mg/m2 (level 2). Three patients were treated in the 45 mg/m2 dose level and 5 patients were treated in the 75 mg/m2 dose level; no DLTs or grade 3-5 treatment related toxicities were observed. After a median follow-up of 5.2 months, no patients developed grade III-IV aGVHD and no patients died of aGVHD. Six out of 8 patients in the treatment group responded to treatment including two cytogenetic complete remissions, one hematologic complete remission, and three complete remissions with incomplete count recovery. In conclusion, administration of AzaC early post DLI is well tolerated and can potentially prevent GVHD after DLI. Further studies are required to evaluate the effect of azacitidine early post DLI on GVHD and GVL.
Keywords: allogeneic stem cell transplantation, acute myeloid leukemia, donor lymphocyte infusion, azacitidine, graft versus host disease
1. Introduction
Long-term survival is achieved in about 30–60% of acute myeloid leukemia (AML) patients after allogeneic stem cell transplantation (allo-SCT). Disease relapse is the most common cause of treatment failure following allo-SCT for AML, occurring in 20–70% of patients. Relapse generally carries a poor prognosis with a median survival of only 3–4 months without active treatment [1].
In the event of relapse following allo-SCT, remission may be re-established by administering salvage chemotherapy followed by donor lymphocyte infusion (DLI) without prophylactic immunosuppression. A retrospective analysis of ∼400 AML patients in 41 centers indicates that such therapy increased the estimated survival at 2 years from ∼9% to ∼21%; however many patients developed acute graft-versus-host-disease (aGVHD) (43%), 80% were grade II to IV [2]. More importantly, death secondary to GVHD after DLI occurs in 8% to 16% of patients [2, 3]. Therefore strategies are needed to prevent GVHD without affecting graft versus leukemia (GVL) after DLI. We and others demonstrated that hypomethylating agents convert conventional T cells to Tregs and that using hypomethylating agents after stem cell transplant or DLI in mice prevents GVHD with no effect on GVL [4, 5]. AzaC after allo-SCT and DLI reduces GvHD in these animals by (1) in vivo conversion of alloreactive donor T cells (FOXP3-) into Tregs (CD4+CD25+FOXP3+) and (2) the direct suppressive effects of AzaC on allogeneic T cells [6]. Based on our preclinical experiments, we hypothesize that the administration of AzaC early post DLI (days 4, 6, 8, and 10) will be associated with an increased number of Tregs, reduced rates of aGVHD, and no decrease in GVL in AML patients who relapsed following allo-SCT. The purpose of this phase 1 study was to determine the safety and tolerability and the maximum tolerated dose (MTD) of AzaC when given after salvage chemotherapy and DLI.
2. Methods
2.1. Study Design
This was a single institution, open label, 3+3 dose escalation phase 1 study performed at the Siteman Cancer Center at Washington University School of Medicine in St. Louis, Missouri. The Washington University Institutional Review Board approved the study, and all patients signed an informed consent prior to enrollment. The study was conducted in compliance with the Declaration of Helsinki, and the applicable local and national regulations. The clinical trial was registered at www.clinicaltrials.gov as NCT01390311.
2.2. Eligibility
Patients with AML (according to World Health Organization criteria) with evidence of relapse following allo-SCT that required salvage therapy followed by a DLI were eligible. Previous allo-SCT using a related or unrelated donor was allowed, however the original donor was required to undergo additional apheresis for collection of donor lymphocyte product or authorize the use of cryogenically stored cells from a previous apheresis.
Eligible patients were required to be at least 18 years old, have an ECOG performance status of 3 or below, and have adequate organ function (creatinine < 2.0 mg/dL, total bilirubin < 2.0 mg/dL, aspartate transaminase, alanine transaminase, and alkaline phosphatase < 3.0x the upper limit of normal). Patients were excluded if they had a known hypersensitivity to AzaC or mannitol, were seropositive for HIV, had grade 3-4 aGVHD, or were pregnant or nursing. Prior exposure to AzaC was not exclusionary.
2.3. Treatment Plan
All patients underwent salvage chemotherapy per institutional guidelines. All immunosuppressive medications were stopped before starting salvage chemotherapy. The salvage chemotherapy regimen used was determined by the treating physician. DLI (Day 0) occurred within 1-4 weeks following the salvage chemotherapy (Figure 1). Treatment with AzaC began 4 days after DLI, and consisted of 4 doses administered on days +4, +6, +8 and +10 post DLI. Each dose of AzaC was administered intravenously over 15 minutes. The initial cohort was treated with a dose of 45 mg/m2/day and the subsequent cohort was treated with a dose of 75 mg/m2/day. The possibility of a minus 1 cohort (30 mg/m2) was included. No intra-patient dose escalation was allowed (Figure 1).
Figure 1.
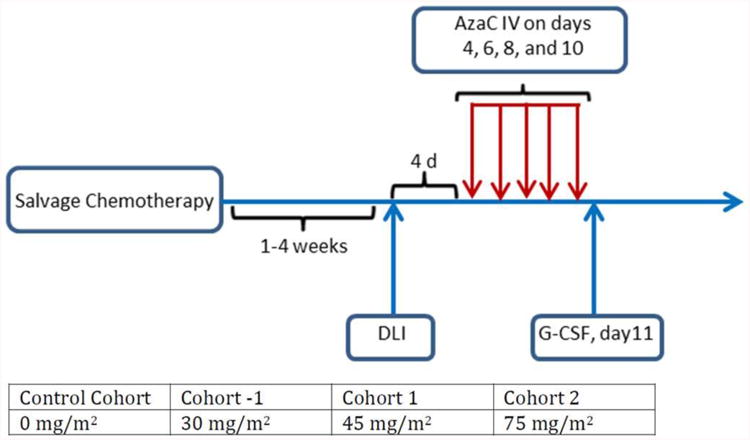
Treatment schema.
Abbreviations: AzaC = Azacitadine; DLI = donor lymphocyte infusion; d = day, G-CSF = granulocyte colony stimulating factor.
Maximum tolerated dose (MTD) was defined as the highest dose level tested in which < 1 of 3 or < 2 of 6 patients experience a dose limiting toxicity (DLT). DLT was defined as any grade 4 hematologic toxicity related to study treatment lasting more than 7 weeks post DLI or any grade 3-5 non-hematologic toxicity related to study treatment occurring within 28 days of AzaC administration.
All patients received standard supportive care including: transfusions, antiemetics, and antibiotics according to institutional guidelines. If neutropenia persisted on day +11 following DLI, G-CSF was started at a dose of 5 μg/kg/day and continued until neutrophil recovery. Any surgery, immunotherapy, biologic therapy, radiotherapy, or chemotherapy used to treat the patient's underlying AML was prohibited during study treatment.
Patients remained on study treatment until one of the following conditions was met: completion of study treatment, unacceptable toxicity, patient withdrawal, treating physician discretion, or death. Following completion of protocol therapy, patients were allowed to proceed to additional treatment at the discretion of their treating physician.
An additional 3 patients who met study eligibility and underwent salvage chemotherapy followed by DLI were enrolled. These patients underwent all study evaluations but did not receive AzaC study treatment. These patients served as a control group for the planned correlative studies.
2.4. Response Evaluation
Blood counts were performed daily following salvage chemotherapy and DLI until neutrophil recovery (neutrophil count >1000 cells/μl for 2 days). Blood counts and bone marrow examinations were performed on Day +32 (+/- 3 days) after DLI to determine response. Karyotyping and fluorescent in situ hybridization (FISH) were performed on bone marrow aspirate samples. Additional blood counts and/or bone marrow examinations were performed at the discretion of the treating physician. No bone marrow examination was performed after pre-DLI chemotherapy and before DLI.
In addition to response rate, disease-free survival (DFS), the interval from the date of the first dose of AzaC to the date of progression or relapse, and overall survival (OS), the interval from the date of the first dose of AzaC to the date of death from any cause were measured.
All patients who completed study treatment and subsequently underwent a bone marrow evaluation for disease response were considered evaluable for response using the revised international working group response criteria for AML [7].
2.5. Acute GVHD Evaluation
Patients were monitored for aGVHD at least weekly through Day +32 following DLI, then at least every other week through Day +100. Acute GVHD was graded according to the modified Glucksberg criteria [8].
2.6. Toxicity Evaluation
Toxicities were evaluated from the start of AzaC study treatment through Day +100, patient withdrawal, start of a subsequent chemotherapy, or death. Toxicities were graded according to version 4.0 of the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE). All patients who received at least one dose of AzaC while on the study were considered evaluable for toxicity.
2.7. Correlative Studies
To determine the effect of AzaC on Treg number and frequency, peripheral blood was collected from study patients at the following time points: baseline, day +11, day +18, day +25, and day +64 following DLI. Treg number and frequency was determined by flow cytometry using CD3, CD4, and CD8 antibodies, and by intracellular staining for FOXP3 expression. Tregs were defined as CD4+FOXP3+ T cells. Treg frequency was calculated by dividing Treg numbers by CD4+ cell counts.
2.8. Statistical Analysis
Data analysis was descriptive in nature. Demographic and clinical characteristics, toxicity, DFS, OS and aGVHD were listed for each patient and each dose level.
3. Results
3.1. Patients Characteristics
From May 2012 through Jan 2014, 11 patients were enrolled in the study: 8 who received AzaC study treatment, and 3 who did not and served as controls for the planned correlative studies. The data reported here applies only to the 8 patients who received AzaC study treatment.
The median age at enrollment was 54.5 years (range 31-68) and 5 out of 8 patients were female. The median time from allo-SCT to enrollment was 4.5 months (range 2.2-37.1). Four had undergone allo-SCT with cells from 10/10 matched sibling donors, while 4 had received cells from a 10/10 matched unrelated donor. Seven patients received intensive salvage regimens (FLAG or FLAG-IDA [4], 7+3 [1], CLAM [1], or MEC [1]), while 1 received a non-intensive regimen (AzaC) prior to DLI. The median number of days between chemotherapy and DLI was 15 (range 10-20). The median number of administered CD3+ Tcells/kg and total nucleated cells/kg were 1.0×107 (range 0.4-4.9 × 107) and 1.2×108 (range 0.4-4.4 × 108). Table 1 summarizes the demographics for each patient.
Table 1.
Patient's characteristics
| ID | Dose Level | Gende r | Age (years) | Donor | BM blast (%) at relapse | Salvage Chemotherapy | CD3+ Dose (×107 cells/kg) | TNC Dose (×108 cells/kg) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 (45mg/m2) | F | 31.1 | MUD | 59 | FLAG-IDA | 0.4 | 0.2 |
| 2 | 1 (45mg/m2) | M | 56.2 | MUD | 15 | 7+3 | 1.2 | 1.9 |
| 3 | 1 (45mg/m2) | F | 53.7 | MUD | 60 | FLAD-IDA | 4.9 | 2.6 |
| 4 | 2 (75mg/m2) | F | 57.8 | MRD | 8 | Azacitidine | 1.0 | 0.5 |
| 5 | 2 (75mg/m2) | F | 49.3 | MRD | 0* | FLAG | 1.0 | 0.4 |
| 6 | 2 (75mg/m2) | M | 68.4 | MUD | 25 | CLAM | 1.0 | 4.4 |
| 7 | 2 (75mg/m2) | F | 52.5 | MRD | 27 | MEC | 1.0 | 3.9 |
| 8 | 2 (75mg/m2) | M | 60.8 | MRD | 85 | FLAG-IDA | 1.0 | 0.4 |
Abbreviations- M = Male; F = Female; TNC = Total nucleated cells ; MUD = matched unrelated donor; MRD = matched related donor.BM = bone marrow.
Patient #5 had extramedullary disease at the time of relapse.
3.2. MTD Determination
Three patients were enrolled into dose level 1 (45 mg/m2 on days 4, 6, 8, and 10 post DLI). None of these experienced a DLT and therefore three more patients were enrolled into dose level 2 (75 mg/m2). No DLTs were observed, so an additional cohort for dose level 2 was opened. Two patients were enrolled and neither had a DLT. Thus in the 5 patients treated at this maximum dose level of 75 mg/m2 no DLTs were observed. As enrollment of a 6th patient in the dose level would not alter the MTD determination, the cohort was closed prematurely.
3.4. Response Assessment
The median duration of neutrophil recovery was 17 days (range 6-34) following DLI and 28 days following salvage chemotherapy (range 21-46). Six out of eight patients achieved remissions, including two cytogenetic complete remissions, one morphologic complete remission, and three complete remissions with incomplete count recovery. Two out of eight patients failed to respond. One responder never achieved full donor chimerism and underwent allo-SCT from another donor. At the time of manuscript preparation, 2 of the 8 patients were alive, however all patients had experienced subsequent relapse/progression. The median DFS was 2.9 months (range 0.9-10.0) and the median OS was 12.5 months (1.6-30.2).
3.5. GVHD
Five of the 8 patients developed aGVHD, all of which were grade 1 or 2. For 3 patients the aGVHD was isolated to skin, while 1 patient had skin and GI involvement, and 1 had liver involvement. Table 2 summarizes the aGVHD experienced by each patient. No patients developed grade 3-4 aGVHD or chronic GVHD and none of the patients died of aGVHD.
Table 2.
Acute GVHD and responses
| ID | Dose Level | DLT | Acute GVHD | Best Response | PFS (months) | OS (months) |
|---|---|---|---|---|---|---|
| 1 | 1 (45mg/m2) | No | None | CRi | 1.5 | 30 |
| 2 | 1 (45mg/m2) | No | Grade 2 (Liver) | CRi | 0.9 | 3.9 |
| 3 | 1 (45mg/m2) | No | Grade 2 (Skin & Gut) | PD | 1.1 | 1.6 |
| 4 | 2 (75mg/m2) | No | None | CCR | 4.3 | 29.5* |
| 5 | 2 (75mg/m2) | No | Grade 1 (Skin) | CCR | 10.0 | 30.2* |
| 6 | 2 (75mg/m2) | No | Grade 1 (Skin) | CRi | 5.5 | 6.1 |
| 7 | 2 (75mg/m2) | No | None | CR | 5.3 | 18.9 |
| 8 | 2 (75mg/m2) | No | Grade 2 (Skin) | PD | 0.9 | 4.4 |
- Ongoing at time of manuscript preparation
Abbreviations- DLT = Dose limiting toxicity; GVHD = Graft-versus-host-disease; PFS = Progression-Free Survival; OS = Overall Survival; CRi = complete remission with incomplete count recovery; PD = Persistent Disease; CCR = Cytogenetic complete remission; CR = Complete remission
3.6. Toxicity
Study treatment was well tolerated. There were no grade 3-5 events related to study treatment. Three patients experienced opportunistic infections and 1 patient experienced febrile neutropenia. No patients required dose reductions, however the day +8 AzaC dose of one patient was held due to hepatic dysfunction (grade 2 elevated bilirubin, grade 2 elevated AST, grade 3 elevated ALT, and grade 3 elevated alkaline phosphatase) that was considered unrelated to study treatment or aGVHD. The patient quickly recovered and received his Day +10 AzaC dose without incident. Table 3 summarizes grade 3-5 adverse events reported regardless of attribution to study treatment. No grade 5 toxicities were observed.
Table 3.
All grade 3-5 adverse events regardless of attribution
| Grade 3 | Grade 4 | Grade 5 | |
|---|---|---|---|
| Anemia | 3 | ||
| Febrile Neutropenia | 2 | ||
| Sepsis | 1 | ||
| Diarrhea | 1 | ||
| Anorexia | 1 | ||
| Alanine aminotransferase increased | 1 | ||
| Hypoalbuminemia | 1 | ||
| Hypocalcemia | 1 | ||
| Hypokalemia | 2 | ||
| Hyponatremia | 3 | ||
| Hypophosphatemia | 1 | ||
| Encephalopathy | 1 |
Within each grade column, the number of patients who experienced each event at that maximum severity is reported
3.7. Effect of AzaC on Tregs post DLI
There was no statistically significant difference in circulating Treg's number or frequency, or the CD3+, CD4+, CD8+, white blood cells, and absolute lymphocyte counts on days 11, 18, 25 and 64 post DLI between recipients of AzaC and the control group (Figure 2 and Figure 3).
Figure 2.
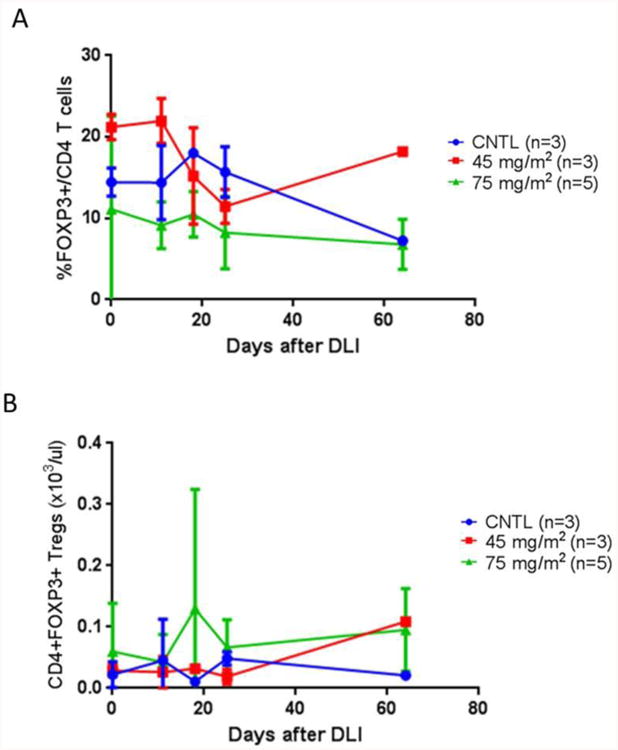
The frequency (A) and number (B) of regulatory T cells in recipients of AzaC (n = 8 patients) compared to control (CNTL) (n = 3 patients).
Figure 3.
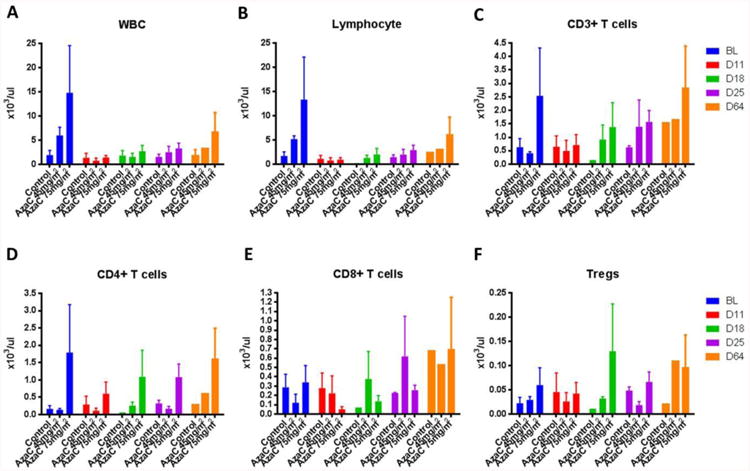
Nucleated white blood cells (A), lymphocytes (B), and CD3+ (C), CD4+ (D), CD8+ (E), and Treg cells/μL (F) at various times and dose levels of AzaC post DLI
4. Discussion
Here we report the results of our phase I clinical trial showing that administration of AzaC at 45 mg/m2 and 75 mg/m2 on days 4, 6, 8, and 10 post DLI is safe and tolerable; it did not result in prolonged cytopenia or other significant toxicities. There was no increase in Treg number or frequency in patients receiving AzaC, but this potentially is attributed to the use of a severe lymphocyte depleting chemotherapy for salvage (FLAG), very low lymphocyte and CD4 counts post DLI and/or the study's small sample size. In addition, it is also possible that AzaC-converted Tregs migrate to GvHD target organs where they suppress GvHD, thereby not being detected in peripheral blood. We are encouraged by the observation of no grade III-IV or deaths related to aGVHD. Although 6 out of 8 patients treated achieved a remission, just 2 patients had durable remission. Schroeder et al reported the outcome of combination of AzaC and DLI as first salvage therapy for relapsed AML or MDS after allo-SCT in a prospective single arm multicenter study. Patients received AzaC on days 1-5 every 28 days and DLI after second cycle, fourth cycle and sixth cycle of AzaC. Overall response rate was 30% including seven CRs (23%). Patients with CR had durable responses with 5 of the 7 patients remaining in CR for a median duration of 777 days (range 461–888 days) [9]. Inferior durable remission in our study compared with Schroeder et al study can be due to: (1) lower GVL related to using azacitidine early post DLI in our study, (2) lower number of patients in this phase 1 study, or (3) using planned several cycles of azacitidine after DLI in Schroeder et al study.
In animal models, regulatory T-cells (Tregs), defined as CD4+CD25+FOXP3+ cells, can mitigate GVHD by suppressing alloreactive donor T cells without sacrificing GVL [10]. Therefore administering Tregs after allogeneic transplant or DLI for prevention of GVHD would seem to be an attractive strategy. There are several major hurdles in using Tregs in an allo-SCT setting: (1) the number of circulating Tregs recoverable from the peripheral blood of donors is very limited (approximately 6% of circulating CD4+ T cells), (2) there is no Treg specific cell surface marker for in vivo or in vitro purification of Tregs, (3) in vitro expansion of Tregs is very inefficient and costly, and (4) in vitro expansion of Tregs often causes loss of function, possibly due to loss of FOXP3 expression [11-18]. Therefore in vivo pharmacologic conversion of conventional T cells to Tregs is a more practical strategy.
FOXP3 expression is required for Treg phenotype. Mutations in the FOXP3 gene result in autoimmune diseases due to the loss of functional Tregs [12-15, 19, 20]. Several studies have demonstrated that in both humans and mice the Foxp3 locus is unmethylated in Tregs, but heavily methylated and silenced in conventional T cells [16, 17, 21-23]. We and others recently demonstrated that hypomethylating agents post transplant or DLI in mice prevent GVHD without affecting GVL by in vivo conversion of alloreactive donor T cells (FOXP3-) into Tregs (CD4+CD25+FOXP3+) and by direct suppressive effects of AzaC on allogeneic T cells [4-6]. It is not completely understood why AzaC does not suppress GVL while it has a suppressive effect on alloreactive T cells. It is possible that the increased expression of leukemia associated antigens on leukemia cells by AzaC may increase the targeting of T cells to these cells and offset the suppressive effect of AzaC on alloreactive T cells [24-27].
Goodyear et al found increased Treg numbers and lower risk of GVHD when AzaC was administered post alemtuzumab based reduced intensity conditioning allo-SCT for AML; however, Treg numbers were significantly higher at only one time point tested, 3 months post allo-SCT [25]. The effect of AzaC on Treg frequency was not reported. None of the patients treated developed > grade II aGVHD.
Interestingly, they demonstrated that AzaC administration induced a cytotoxic CD8 T-cell response to several leukemia associated antigens [25]. There are two main differences between the present study and the Goodyear, et al. study: (1) in the present study AzaC was given after DLI not allo-SCT and (2) in the present study AzaC was given early post DLI before count recovery at doses of 45 mg/m2 and 75 mg/m2 on days +4, +6, +8, +10, while in their study AzaC was given after count recovery at a dose of 36 mg/m2 daily for 5 consecutive days starting at approximately 40 days post allo-SCT and continuing every 28 days for up to a total of 10 cycles. In light of rapid expansion of T cells before count recovery early post transplant especially in allo-SCTs with myeloablative conditioning, starting hypomethylating agents early post allo-SCT could be potentially more beneficial. Currently there is an ongoing phase I clinical trial of using AzaC post allo-SCT prior to count recovery at Washington University in St. Louis to explore these issues (NCT01747499).
No AzaC maintenance post DLI was given in our trial. In light of the short half-life of converted Tregs, administration of additional monthly maintenance AzaC post DLI after count recovery could potentially result in an increased number of Tregs, decreased GVHD, and better leukemia control.
In conclusion, here we report that administration of AzaC at 45 mg/m2 and 75 mg/m2 on days 4, 6, 8, and 10 post-DLI is safe and does not result in prolonged cytopenia or other significant toxicities. The optimal biologic dose (OBD) may be lower than maximal tolerated dose; in the other words dose-related toxicity cannot be regarded as the best surrogate for efficacy [28]. The OBD of AzaC post DLI is a dose that abrogates GVHD with no effect on GVL as measured by remission rate and relapse post DLI. Although here we demonstrated that both 75 mg/m2 and 45 mg/m2 dose levels of AzaC post DLI are safe, to define the efficacy and OBD of AzaC early post DLI both the 75 mg/m2 and 45 mg/m2 dose levels should be tested using greater numbers of patients.
Highlights.
Azacitidine (AzaC) on days 4, 6, 8, and 10 post DLI is safe.
AzaC early post DLI can potentially prevent GVHD.
The effect of AzaC post DLI on GVHD and GVL should be studied in a larger study.
Acknowledgments
This work is supported by NIH P50 CA171963-01. Celgene provided azacitidine.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests
Contribution: A.G., J.C., P.W., J.L., L.E., and J.F.D., designed the study; A.G., J.F.D., and P.W. conducted the study and enrolled patients: A.G., T.F., and M.F. wrote the manuscript; C.A., A.C., R.V., M.S., I.P., K.S.G., M.J., and G.U. enrolled patients, J.C. performed T cell and T subset analyses; and all authors discussed the results and commented on the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Savani BN, Mielke S, Reddy N, Goodman S, Jagasia M, Rezvani K. Management of relapse after allo-SCT for AML and the role of second transplantation. Bone marrow transplantation. 2009;44:769–77. doi: 10.1038/bmt.2009.300. [DOI] [PubMed] [Google Scholar]
- 2.Schmid C, Labopin M, Nagler A, Bornhauser M, Finke J, Fassas A, et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:4938–45. doi: 10.1200/JCO.2007.11.6053. [DOI] [PubMed] [Google Scholar]
- 3.Huff CA, Fuchs EJ, Smith BD, Blackford A, Garrett-Mayer E, Brodsky RA, et al. Graft-versus-host reactions and the effectiveness of donor lymphocyte infusions. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2006;12:414–21. doi: 10.1016/j.bbmt.2005.11.520. [DOI] [PubMed] [Google Scholar]
- 4.Choi J, Ritchey J, Prior JL, Holt M, Shannon WD, Deych E, et al. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood. 2010;116:129–39. doi: 10.1182/blood-2009-12-257253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Abarca LI, Gutierrez-Cosio S, Santamaria C, Caballero-Velazquez T, Blanco B, Herrero-Sanchez C, et al. Immunomodulatory effect of 5-azacytidine (5-azaC): potential role in the transplantation setting. Blood. 2010;115:107–21. doi: 10.1182/blood-2009-03-210393. [DOI] [PubMed] [Google Scholar]
- 6.Cooper MCJ, Ritchey J, et al. Defining The Mechanism Involved In The Inhibition Of GvHD By Azacytidine In Vivo Through The Use Of FoxP3 Diphtheria Toxin Receptor (Foxp3DTR) Donor T Cells. Blood. 2013;122:134. [Google Scholar]
- 7.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the international working group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. Journal of Clinical Oncology. 2003;21:4642–9. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 8.Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. Consensus Conference on Acute Gvhd Grading. Bone marrow transplantation. 1995;15:825–8. [PubMed] [Google Scholar]
- 9.Schroeder T, Czibere A, Platzbecker U, Bug G, Uharek L, Luft T, et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2013;27:1229–35. doi: 10.1038/leu.2013.7. [DOI] [PubMed] [Google Scholar]
- 10.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4(+)CD25(+) regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–50. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 11.Karakhanova S, Munder M, Sehneider M, Bonyhadi M, Ho AD, Goerner M. Highly efficient expansion of human CD4(+)CD25(+) regulatory T cells for cellular immunotherapy in patients with graft-versus-host disease. J Immunother. 2006;29:336–49. doi: 10.1097/01.cji.0000203080.43235.9e. [DOI] [PubMed] [Google Scholar]
- 12.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 13.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 14.Bennett CL, Brunkow ME, Ramsdell F, O'Briant KC, Zhu Q, Fuleihan RL, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53:435–9. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- 15.Chae WJ, Henegariu O, Lee SK, Bothwell ALM. The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. P Natl Acad Sci USA. 2006;103:9631–6. doi: 10.1073/pnas.0600225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. Journal of Experimental Medicine. 2007;204:1543–51. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lal G, Zhang N, van der Touw W, Ding YZ, Ju WJ, Bottinger EP, et al. Epigenetic Regulation of Foxp3 Expression in Regulatory T Cells by DNA Methylation. J Immunol. 2009;182:259–73. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3(+) regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 19.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 20.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 21.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. Plos Biol. 2007;5:169–78. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37:2378–89. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 23.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–63. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 24.Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8(+) T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908–18. doi: 10.1182/blood-2009-11-249474. [DOI] [PubMed] [Google Scholar]
- 25.Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML) Blood. 2012;119:3361–9. doi: 10.1182/blood-2011-09-377044. [DOI] [PubMed] [Google Scholar]
- 26.Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jager E, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leukemia Res. 2010;34:899–905. doi: 10.1016/j.leukres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the Mage-1 Tumor-Antigen Is up-Regulated by the Demethylating Agent 5-Aza-2′-Deoxycytidine. Cancer research. 1994;54:1766–71. [PubMed] [Google Scholar]
- 28.Adjei AA. What is the right dose? The elusive optimal biologic dose in phase I clinical trials. Journal of Clinical Oncology. 2006;24:4054–5. doi: 10.1200/JCO.2006.07.4658. [DOI] [PubMed] [Google Scholar]