

Abstract
Background
Modulation of inflammation and oxidative stress appears to limit sepsis-induced damage in experimental models. The kidney is one of the most sensitive organs to injury during septic shock. In this study, we evaluated the effect of N-acetylcysteine (NAC) administration in conjunction with fluid resuscitation on renal oxygenation and function. We hypothesized that reducing inflammation would improve the microcirculatory oxygenation in the kidney and limit the onset of acute kidney injury (AKI).
Methods
Rats were randomized into five groups (n = 8 per group): (1) control group, (2) control + NAC, (3) endotoxemic shock with lipopolysaccharide (LPS) without fluids, (4) LPS + fluid resuscitation, and (5) LPS + fluid resuscitation + NAC (150 mg/kg/h). Fluid resuscitation was initiated at 120 min and maintained at fixed volume for 2 h with hydroxyethyl starch (HES 130/0.4) dissolved in acetate-balanced Ringer’s solution (Volulyte) with or without supplementation with NAC (150 mg/kg/h). Oxygen tension in the renal cortex (CμPO2), outer medulla (MμPO2), and renal vein was measured using phosphorimetry. Biomarkers of renal injury, inflammation, and oxidative stress were assessed in kidney tissues.
Results
Fluid resuscitation significantly improved the systemic and renal macrohemodynamic parameters after LPS. However, the addition of NAC further improved cortical renal oxygenation, oxygen delivery, and oxygen consumption (p < 0.05). NAC supplementation dampened the accumulation of NGAL or L-FABP, hyaluronic acid, and nitric oxide in kidney tissue (p < 0.01).
Conclusion
The addition of NAC to fluid resuscitation may improve renal oxygenation and attenuate microvascular dysfunction and AKI. Decreases in renal NO and hyaluronic acid levels may be involved in this beneficial effect. A therapeutic strategy combining initial fluid resuscitation with antioxidant therapies may prevent sepsis-induced AKI.
Keywords: Acute kidney injury, Sepsis, N-acetylcysteine, Kidney oxygenation, Inflammation
Background
Inflammation is a key process in the pathophysiology of septic shock [1]. The whole activation of leukocytes, the cascade of inflammation, the associated cytokine storm, and endothelial cell dysfunction collaborate to alter the microcirculation [2, 3]. Subsequently, tissue hypoxia and dysoxia due to heterogeneity of the microcirculation will occur [4]. Fluid resuscitation may not only help resolve some of these issues but also lead to the activation of oxidative pathways in itself, resulting in a heterogeneous distribution of blood flow and tissue oxygenation, especially in the renal cortex [5, 6]. Because reactive oxygen species (ROS) participate in the pathophysiology of endotoxemic shock, it has been suggested that moderating the oxidative stress and inflammatory reaction would translate into improving the microcirculation and oxygenation of the tissues. Previous studies have demonstrated an interest in the use of antioxidants for preventing sepsis-induced damage in these organs [7–9]. Indeed, the experimental literature is full of studies of drugs that target and are effective at dampening inflammation and oxidative stress. Some trials have reported several anti-inflammatory or antioxidant drugs with discordant effects on major outcomes [10, 11]. In fact, antioxidant therapies for specific organs might be of interest. Acute kidney injury (AKI) and acute lung injury are particularly common complications of sepsis, and the development of either increases mortality probably because these organs are more sensitive to inflammation and oxidative stress insults. Thus, the kidney could benefit from antioxidant drugs.
A common approach for the inhibition of oxidant-mediated injury is the use of glutathione-modulating agents such as sulfhydryl or thiol compounds. Among all the drugs used to interact with this pathway, N-acetylcysteine (NAC) is the most studied for its lung and renal protective effects [12–16]. NAC is a thiol compound with antioxidant and vasodilatory properties [12]. NAC is regarded as an important antioxidant as it is a source of sulfhydryl and glutathione groups in cells and, due to its interaction with ROS, is a scavenger of free radicals. In septic patients, the endogenous antioxidant glutathione is depleted [17]. Decreased levels of glutathione may lead to decreased protection of cell membranes against oxygen radicals. NAC serves as a precursor of glutathione and can replenish the intracellular glutathione stores [12]. Moreover, NAC targets kidney microcirculatory blood flow [18, 19]. NAC has also been widely studied for its nephroprotective effects in various settings.
Hypoxia and inflammation have an interdependent relationship. Several molecular pathways of cross-talk between hypoxia and inflammation in the kidney have been identified [20–22]. From a physiological perspective, although hypoxia may lead to inflammation and vice versa, it is unclear whether correcting or modulating either of these states would translate into better tissue oxygenation and improved outcomes. NAC would be interesting for testing whether the modulation of inflammation could correct tissue hypoxia during sepsis.
To date, no study has focused on tissue oxygenation in specific organs such as the kidney but rather demonstrated that either pretreatment or post-treatment with NAC decreased the markers of organ injury. In this study, we assessed kidney tissue oxygenation in an endotoxemic shock model resuscitated with balanced hydroxyethyl starch—Ringer’s acetate either with or without supplementation with NAC. We sought to promote blood flow and oxygenation to the organs by the means of reducing inflammation and oxidative stress.
Methods
Animals
All experiments in this study were approved by the institutional Animal Experimentation Committee of the Academic Medical Center of the University of Amsterdam (DFL102538). The care and handling of the animals were in accordance with the guidelines for Institutional Animal Care and Use Committees. The study was conducted in accordance with the Declaration of Helsinki. Experiments were performed on albino Wistar rats (Harlan Netherlands BV, Horst, The Netherlands) with a mean ± SD body weight of 325 ± 6 g.
Surgical preparation
All animals were anesthetized with an intraperitoneal injection of a mixture of 90 mg/kg ketamine (Nimatek®, Eurovet, Bladel, The Netherlands), 0.5 mg/kg dexmedetomidine (Dexdomitor, Pfizer Animal Health BV, Capelle aan den IJssel, The Netherlands), and 0.05 mg/kg atropine-sulfate (Centrafarm Pharmaceuticals BV, Etten-Leur, The Netherlands). After a tracheotomy was performed, the animals were mechanically ventilated with a fraction of inspired oxygen (FiO2) of 0.4. Body temperature was maintained at 37 ± 0.5 °C during the entire experiment by an external thermal heating pad. Ventilator settings were adjusted to maintain an arterial partial pressure of carbon dioxide (PaCO2) between 35 and 40 mmHg. For drug and fluid administration as well as hemodynamic monitoring, vessels were cannulated with polyethylene catheters with an outer diameter of 0.9 mm (Braun, Melsungen, Germany). A catheter in the right carotid artery was connected to a pressure transducer to monitor the mean arterial blood pressure (MAP) and heart rate. The right jugular vein was cannulated for continuous infusion of Ringer’s lactate (Baxter, Utrecht, The Netherlands) at a rate of 15 ml/kg/h and for the maintenance of anesthesia. The right femoral artery was cannulated for drawing blood samples; the right femoral vein, for drug administration. The left kidney was exposed, decapsulated, and immobilized in a Lucite kidney cup (K. Effenberger, Pfaffingen, Germany) via an ~4-cm incision in the left flank in each animal. Renal vessels were carefully separated to preserve the nerves and adrenal gland. A perivascular ultrasonic transient time flow probe was placed around the left renal artery (type 0.7 RB Transonic Systems Inc., Ithaca, NY, USA) and connected to a flow meter (T206, Transonic Systems Inc., Ithaca, NY, USA) to continuously measure renal blood flow (RBF). The left ureter was isolated, ligated, and cannulated with a polyethylene catheter for urine collection. After the surgical preparation, one optical fiber was placed 1 mm above the decapsulated kidney, and another optical fiber was placed 1 mm above the renal vein to measure renal microvascular and venous oxygenation using phosphorimetry. A small piece of aluminum foil was placed on the dorsal side of the renal vein to prevent the underlying tissues from contributing to the phosphorescence signal in the venous PO2 measurements. The surgical field was covered with a humidified gauze compress throughout the entire experiment to prevent drying of the exposed tissues.
Experimental protocol
After a 30-min stabilization, the rats were randomized into the five following groups at baseline: (1) control group, (2) control + NAC, (3) endotoxemic shock with lipopolysaccharide (LPS) without fluid resuscitation, (4) LPS + fluid resuscitation, and (5) LPS + fluid resuscitation + NAC (150 mg/kg/h). The groups received either an intravenous bolus of 5 mg/kg LPS (LPS group; Escherichia coli 0127:B8, Sigma, Paris, France; three groups of eight rats each) or vehicle (control group, two groups of eight rats each). Animals were observed or kept in shock for over 120 min. Fluid resuscitation (15 ml/kg/h) was then started and maintained for 180 min in the LPS groups with 6 % hydroxyethyl starch (HES130/0.4) dissolved in Ringer’s acetate (HES-RA; Volulyte® 6 %, Fresenius Kabi Deutschland GmbH, Germany) as a balanced colloid solution. NAC was administered to the appropriate groups at a rate of 150 mg/kg/h as previously reported [15]. An LPS group was not resuscitated to serve as a shock control. Time points for the measurements were baseline (T0), during shock 120 min after administration of LPS (T1), 30 min after initiating fluid resuscitation (early reperfusion phase) (T2), and 120 min after starting fluid resuscitation (late reperfusion phase) (T3), which was the final endpoint of the experiment (Fig. 1).
Fig. 1.
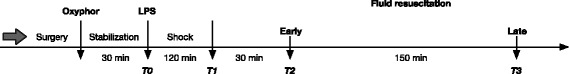
Timeline of the experimental protocol
Blood gas measurements and biochemistry
Arterial blood samples of 0.5 ml were collected from the femoral artery at T0, T1, T2, and T3. The blood samples were replaced by the same volume of balanced colloid solution. The samples were used to determine blood gas parameters (Radiometer ABL 505 Blood Gas Analyzer, Copenhagen, Denmark). The hematocrit and the levels of potassium, bicarbonate, and the anion gap were recorded by the analyzer.
Measurement of renal microvascular oxygenation and venous PO2
The renal microvascular partial pressure of oxygen (μPO2) and renal venous PO2 (rvPO2) were measured by oxygen-dependent quenching of phosphorescence lifetimes of the phosphorescent dye Oxyphor G2 (Oxygen Enterprises Ltd., Philadelphia, PA, USA) as described previously [23, 24]. A total of 6 mg/kg IV over 5 min was administered followed by 30 min of stabilization before recording baseline measurements.
Calculation of derivatives of oxygenation parameters and renal vascular resistance
Renal oxygen delivery was calculated using the following formula: DO2ren (ml/min) = RBF × arterial oxygen content (1.31 × hemoglobin × SaO2) + (0.003 × PaO2), where SaO2 is the arterial oxygen saturation and PaO2 is the arterial partial pressure of oxygen. Renal oxygen consumption was calculated using the following formula: VO2ren (ml/min/g) = RBF × (CaO2 − CvO2), where the renal venous oxygen content (CvO2) was calculated as (1.31 × hemoglobin × SrvO2) + (0.003 × rvPO2). The SrvO2ren was calculated using the Hill equation with P50 = 37 Torr (4.9 kPa) and the Hill coefficient = 2.7. An estimation of the renal vascular resistance (RVR) was defined as RVR (dynes sec cm−5) = (MAP/RBF) × 100.
Assessment of kidney function
Creatinine clearance (Clcrea [ml/min]) was assessed as an index of the glomerular filtration rate. Clearance was calculated using the following formula: Clcrea = (Ucrea × V)/Pcrea, where Ucrea is the concentration of creatinine in urine, V is the urine volume per unit time, and Pcrea is the concentration of creatinine in the plasma. The renal energy efficiency for sodium transport (VO2/TNa) was assessed using a ratio calculated from the total amount of VO2 over the total amount of sodium reabsorbed (TNa, mmol/min) according to the following formula: (Clcrea × PNa) − UNa × V.
NO metabolism
The index of total nitric oxide (NO) production is the sum of both nitrite and nitrate accumulated in tissue samples. To determine this index, a saturated solution of vanadium (III) chloride (VCl3) in 1 mol/l HCl was used as a reducing agent. At a temperature of 90 °C, the VCl3 reagent quantitatively converts nitrite, nitrate, and S-nitroso compounds to NO in a glass reaction vessel. NO was then flushed out of the reaction vessel by the flow of helium gas and was then measured using a Sievers NO analyzer (General Electric Company, GE Water & Process Technologies Analytical Instruments) to detect chemiluminescence as the amount of light from the ozone-NO reaction in the measurement chamber of the analyzer. NO levels were determined in homogenized frozen kidney tissues. A ratio of tissue NO to tissue protein content was used for standardization of NO release per gram of protein.
Glycocalyx component assessments
Hyaluronan is the main component of the endothelial glycocalyx, and alterations in its concentration are attributed to glycocalyx volume loss. Inhibition of tumor necrosis factor-alpha protects against endotoxin-induced endothelial glycocalyx perturbation. Plasma hyaluronan concentrations were determined using a Corgenix hyaluronic acid test kit (Corgenix Inc., Westminster, CO, USA) based on an enzyme-linked hyaluronic acid-binding protein assay.
Measurement of oxidative stress and inflammatory cytokines
All kidneys were homogenized in cold 5 mM sodium phosphate buffer. The homogenates were centrifuged at 12,000g for 15 min at 4 °C, and the supernatants were used to determine TNF-α, IL-6, hyaluronic acid, malondialdehyde (MDA), and protein carbonyl levels. The levels of these markers were expressed as per gram of protein (Bradford assay). To determine the oxidative stress and inflammatory cytokines levels, enzyme-linked immunosorbent assay (ELISA) kits were used. Tumor necrosis factor-α (TNF-α) (DY510, R&D system, Inc. Minneapolis, USA), interleukin-6 (IL-6) (DY506, R&D system, Inc. Minneapolis, USA), tissue MDA, and protein carbonyl were determined in homogenized tissue samples.
Immunohistochemical analysis
Kidney tissues were fixed in 4 % formalin and embedded in paraffin. After preparation, kidney sections were incubated with a neutrophil gelatinase-associated lipocalin (NGAL) antibody (NGAL antibody 41105, Abcam, Cambridge, UK) and a polyclonal antibody to rat liver-type fatty acid protein (L-FABP) (HP8010, Hycult Biotect, Uden Holland). Antibodies were diluted in a large volume of UltrAb Diluent (TA-125-UD, Thermo Fisher Scientific, Breda, Holland). The slides were counterstained with Mayer’s hematoxylin (LabVision TA-125-MH Thermo Fisher Scientific, Breda, Holland) and mounted in a vision mount (LabVision, TA-060-UG, Thermo Fisher Scientific, Breda, Holland) after washing in distilled water. Both the intensity and the distribution of L-FABP and NGAL staining were scored. For each sample, a histological score (HSCORE) value was derived by summing the percentages of cells that were stained at each intensity multiplied by the weighted intensity of the staining: HSCORE = S Pi (i + 1), where i is the intensity score and Pi is the corresponding percentage of the cells.
Statistical analyses
The results are expressed as the mean ± SD. Statistical significance was calculated by one-way and two-way analysis of variance (ANOVA) followed by either Tukey’s or Bonferroni’s multiple comparison tests using GraphPad Prism (GraphPad Prism, Version 5, Software Program, San Diego, CA, USA). p < 0.05 was considered statistically significant.
Results
Systemic and renal hemodynamic parameters
The evolution of systemic and renal hemodynamics is presented in Table 1. Infusion of LPS induced an early drop in the MAP (76.8 ± 9.3 mmHg versus 45.8 ± 7.9 mmHg, p < 0.001) and RBF (4.5 ± 1.5 ml/min versus 0.7 ± 0.6 ml/min, p < 0.001) in the control group versus the LPS group, respectively, at T3. Fluid resuscitation with HES-RA both with and without NAC significantly improved RBF compared to the LPS alone group (p < 0.05). Both HES-RA and HES-RA + NAC significantly decreased the RVR compared to LPS alone (p < 0.001). After LPS, the addition of NAC to the fluid did not result in improved hemodynamic parameters compared to fluid resuscitation alone. The infusion of NAC led to a decrease in the MAP in the absence of LPS (57.3 ± 5.6 mmHg versus 76.8 ± 9.3 mmHg in the control group, p < 0.001).
Table 1.
Evolution of systemic and renal hemodynamics parameters during the experiment
| T0 (baseline) | T1 (shock) | T2 (30 min) | T3 (120 min) | |
|---|---|---|---|---|
| MAP (mmHg) | ||||
| Time control | 102 ± 9.0 | 86.3 ± 13.4 | 80.1 ± 8.8 | 76.8 ± 9.3 |
| Control + NAC | 87.7 ± 7.6 | 75.3 ± 10.5 | 70.5 ± 8.0 | 57.3 ± 5.6*** |
| LPS | 101 ± 11.1 | 56.8 ± 10.5*** | 53.7 ± 9.4*** | 45.8 ± 7.9*** |
| LPS + HES-RA | 92.1 ± 8.7 | 55.6 ± 9.5*** | 65.5 ± 4.9 * , + | 51.6 ± 3.7*** |
| LPS + HES-RA + NAC | 98.8 ± 8.7 | 49.8 ± 8.5*** | 64.1 ± 4.7** | 52.6 ± 6.1*** |
| RBF (ml/min) | ||||
| Time control | 5.4 ± 0.6 | 4.8 ± 0.8 | 5.3 ± 0.9 | 4.5 ± 1.5 |
| Control + NAC | 5.7 ± 0.7 | 4.3 ± 0.9 | 4.8 ± 1.5 | 4 ± 1.1 |
| LPS | 5.7 ± 1.4 | 1.3 ± 0.7*** | 1.4 ± 0.5*** | 0.7 ± 0.6*** |
| LPS + HES-RA | 6.6 ± 0.3 | 1.9 ± 1.5*** | 5.6 ± 1.8 +++ | 5.0 ± 0.4 +++ |
| LPS + HES-RA + NAC | 5.5 ± 0.4 | 1 ± 0.4*** | 3.5 ± 0.9*, ++ | 4.8 ± 2.2 +++ |
| RVR )dyn.s.sec−5) | ||||
| Time control | 1910.2 ± 260.5 | 1793.7 ± 227.4 | 1524.3 ± 188.2 | 1861.3 ± 751.2 |
| Control + NAC | 1548.6 ± 249.2 | 1782.6 ± 368.6 | 1606.7 ± 583.2 | 1521.9 ± 511.1 |
| LPS | 1774.8 ± 302.4 | 5911.5 ± 3333.4*** | 4182.9 ± 829.7* | 10,372 ± 5182.2 *** |
| LPS + HES-RA | 1410 ± 152.2 | 3293.2 ± 1978.9 + | 1130.2 ± 348.1 ++ | 1004.3 ± 48.3 +++ |
| LPS + HES-RA + NAC | 1787.2 ± 236 | 5795.8 ± 2517.4*** | 1909.8 ± 375.7 | 1199.1 ± 318.3 +++ |
LPS lipopolysaccharide, HES-RA hydroxyethyl starch-ringer acetate, NAC N-acetylcysteine
Values are presented as mean ± SD *p < 0.05, **p < 0.01, and ***p < 0.001 control versus other groups; + p < 0.05, ++ p < 0.01, and +++ p < 0.001 LPS versus other groups
Renal microvascular oxygenation
The percentage variations in CμPO2, MμPO2, DO2ren, and VO2ren between baseline (T0) and the end of the experiment (T3) are shown in Fig. 2. Compared to the control groups, LPS infusion induced a significant decrease in CμPO2 (40.6 ± 8.8 mmHg versus 68.2 ± 4.1 mmHg in the control group at T3, p < 0.001) and MμPO2 (32.2 ± 7.9 mmHg versus 51.6 ± 3.2 mmHg in the control group, p < 0.001). Fluid resuscitation with HES-RA alone did not improve either CμPO2 or MμPO2. HES-RA combined with NAC significantly improved CμPO2 during sepsis (p < 0.01). LPS induced a significant decrease in DO2ren and VO2ren (8.3 ± 6.1 ml O2/min in the LPS group versus 67.2 ± 23.2 ml O2/min in the control group at T3 and 7.8 ± 6.5 ml O2/min in the LPS group versus 32.9 ± 10 ml O2/min in the control group at T3, p < 0.05, respectively). Fluid resuscitation with or without NAC significantly improved VO2ren compared to the LPS alone group (p < 0.05). Of note, the addition of NAC to the control group also increased VO2ren compared to the control group alone (p < 0.05). The hematocrit values are reported in Table 3. A significant decrease in hematocrit occurred after fluid resuscitation in both groups compared to the control and LPS alone groups (p < 0.001). The magnitude of hemodilution in both groups was in the same range.
Fig. 2.

Percentage change of renal microvascular oxygen tension, oxygen delivery, and consumption from baseline to T3. In the renal cortex (CμpO2) (a), in the medulla (MμpO2) (b), renal oxygen delivery (DO2ren) (c), and renal oxygen consumption (VO2ren) (d). *p < 0.05, **p < 0.01, *p < 0.001 versus control; + p < 0.05 LPS versus LPS group
Table 3.
Time-course of acid-base status, lactate levels, and hematocrit resuscitated with and without the addition of NAC
| T0 (baseline) | T1 (shock) | T2 (30 min) | T3 (120 min) | |
|---|---|---|---|---|
| pH | ||||
| Time control | 7.37 ± 0.03 | 7.39 ± 0.06 | 7.42 ± 0.03 | 7.42 ± 0.04 |
| Control + NAC | 7.48 ± 0.12 | 7.42 ± 0.03 | 7.38 ± 0.02 | 7.32 ± 0.05* |
| LPS | 7.36 ± 0.03 | 7.27 ± 0.06** | 7.26 ± 0.07*** | 7.2 ± 0.07*** |
| LPS + HES-RA | 7.36 ± 0.03 | 7.26 ± 0.07*** | 7.31 ± 0.07* | 7.27 ± 0.05*** |
| LPS + HES-RA + NAC | 7.37 ± 0.04 | 7.26 ± 0.09*** | 7.27 ± 0.08*** | 7.20 ± 0.03*** |
| HCO3 − (mmol/L) | ||||
| Time control | 20.6 ± 0.9 | 21 ± 0.6 | 21.2 ± 1.3 | 21.6 ± 1.3 |
| Control + NAC | 18.8 ± 0.6 | 18.4 ± 1.5* | 19.3 ± 1.2 | 14.2 ± 3.8*** |
| LPS | 20.2 ± 0.7 | 15 ± 0.9*** | 15.3 ± 1.2*** | 12.8 ± 0.5*** |
| LPS + HES-RA | 21.6 ± 0.7 | 14.8 ± 1.5*** | 18.1 ± 0.9***, ++ | 16.7 ± 1.6***, +++ |
| LPS + HES-RA + NAC | 20.7 ± 0.8 | 14.7 ± 2.5*** | 16.6 ± 1.6*** | 14 ± 1.7***, ## |
| Base excess (mmol/L) | ||||
| Time control | −3.5 ± 1.2 | −2.7 ± 1.6 | −1.7 ± 1.3 | −1.8 ± 1.5 |
| Control + NAC | −3.5 ± 0.5 | −4.4 ± 1.7 | −4.5 ± 1.3 | −8.9 ± 2.4*** |
| LPS | −4 ± 1.3 | −10.8 ± 1.9*** | −10.5 ± 1.7*** | −14.1 ± 1.3*** |
| LPS + HES-RA | −2.9 ± 1.1 | −11.3 ± 2.7*** | −7 ± 2***, ++ | −8.9 ± 1.9***, +++ |
| LPS + HES-RA + NAC | −3.4 ± 1.5 | −11.2 ± 4.1*** | −9.1 ± 2.8*** | −12.5 ± 1.7***, ## |
| Anion gap K+ (mmol/L) | ||||
| Time control | 17.7 ± 0.8 | 18.0 ± 0.7 | 16.4 ± 0.5 | 16.8 ± 1.3 |
| Control + NAC | 18.9 ± 2.0 | 18.5 ± 2.0 | 17.0 ± 1.2 | 18.9 ± 4.8 |
| LPS | 18.9 ± 2.3 | 21.8 ± 1.1 | 21.6 ± 1.8 | 23.2 ± 1.6*** |
| LPS + HES-RA | 17.5 ± 1.3 | 22.7 ± 1.3 | 19.2 ± 1.0 | 20.9 ± 2.5*** |
| LPS + HES-RA + NAC | 18.4 ± 1.0 | 22.5 ± 2.5** | 20.6 ± 2.3* | 23.1 ± 1.9*** |
| Lactate (mmol/L) | ||||
| Time control | 2.32 ± 0.52 | 2.22 ± 0.34 | 2.08 ± 0.31 | 1.73 ± 0.27 |
| Control + NAC | 3.10 ± 0.64 | 2.92 ± 0.76 | 2.17 ± 0.36 | 2.10 ± 0.33 |
| LPS | 2.42 ± 0.32 | 3.08 ± 0.28 | 3.08 ± 0.5 | 4.32 ± 0.6*** |
| LPS + HES-RA | 1.92 ± 0.2 | 3.32 ± 0.55 | 2.75 ± 0.48 | 5.08 ± 1.83*** |
| LPS + HES-RA + NAC | 2.70 ± 0.21 | 3.83 ± 1.36** | 3.30 ± 1.17 * | 4.98 ± 1.21*** |
| Hct (%) | ||||
| Time control | 49.6 ± 1.3 | 41.6 ± 1.9 | 39 ± 2.9 | 33.3 ± 3 |
| Control + NAC | 48.8 ± 2.6 | 42.8 ± 4.4 | 41.1 ± 4.2 | 33.8 ± 2.6 |
| LPS | 49.3 ± 4.0 | 39.6 ± 3.0 | 35.1 ± 2.9 | 34.0 ± 3.5 |
| LPS + HES-RA | 49.3 ± 1.7 | 43.3 ± 2.5 | 30.1 ± 4.5***, + | 17.5 ± 3***, +++ |
| LPS + HES-RA + NAC | 48.6 ± 0.8 | 44.1 ± 3.2 + | 33.0 ± 2.6** | 18.0 ± 2.0***, +++ |
LPS lipopolysaccharide, HES-RA hydroxyethyl starch-ringer acetate, NAC N-acetylcysteine
Values are presented as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 versus control; ++ p < 0.05, +++ p < 0.001 versus LPS; ## p < 0.01 versus LPS + HES-RA
LPS induced a significant increase in ER% O2 at T1, T2, and T3 compared to the control group (p < 0.01, p < 0.01, and p < 0.001, respectively), and this value was only improved in LPS + HES-RA group at T2. In the control group receiving NAC, ER%O2 was also increased at T3 compared to control (p < 0.001).
Kidney function and biomarkers of kidney injury
The evolution of TNa+, VO2ren/TNa+, ClCrea, and EFNa+ at different time points is presented in Table 2. TNa+ levels were lower in the LPS, LPS + HES-RA, and LPS + HES + RA + NAC groups (p < 0.001) than the control group at T1, T2, and T3. Fluid resuscitation did not restore these values. The addition of NAC also decreased the TNa + in the control + NAC group compared to the control group (p < 0.01). VO2ren/TNa+ increased in LPS groups receiving HES-RA and HES + RA + NAC at T3 compared to the control group (p < 0.05 and p < 0.001, respectively) and the LPS alone group (p < 0.05 and p < 0.001, respectively). Compared to the control and LPS alone groups, EFNa+ values were increased in the LPS groups treated with HES-RA with and without NAC at T2 and T3. NAC administration in the control group tended to decrease TNa+ and EFNa+ without reaching a significant level. Fluid resuscitation improved urine output regardless of the addition of NAC compared to the LPS group (p < 0.001). No effect of NAC on urine output was noted in the control + NAC group compared to the control group, but a significant decrease in Clcreat was observed (p < 0.001).
Table 2.
Parameters of renal function and excretion at different time points of the experiment
| T0 (baseline) | T1 (shock) | T2 (30 min) | T3 (120 min) | |
|---|---|---|---|---|
| TNa+ (mmol/min−1) | ||||
| Time control | 15.3 ± 9.6 | 14.1 ± 7.0 | 18.8 ± 6.8 | 19.7 ± 4.7 |
| Control + NAC | 15.0 ± 6.4 | 13.2 ± 6.5 | 15.9 ± 6.5 | 9.5 ± 4.3** |
| LPS | 12.3 ± 5.5 | 0.00 ± 0.00*** | 0.00 ± 0.00*** | 0.00 ± 0.00*** |
| LPS + HES-RA | 12.0 ± 8.3 | 0.00 ± 0.00*** | 5.0 ± 1.12*** | 2.2 ± 1.3*** |
| LPS + HES-RA + NAC | 11.9 ± 5.7 | 0.00 ± 0.00*** | 2.8 ± 2.8*** | 2.2 ± 1.8*** |
| VO2/TNa+ | ||||
| Time control | 1.77 ± 1.6 | 2.19 ± 1.3 | 1.59 ± 0.6 | 1.79 ± 0.7 |
| Control + NAC | 1.36 ± 0.4 | 2.19 ± 0.9 | 1.95 ± 1.18 | 6.32 ± 3.9 |
| LPS | 2.5 ± 1.2 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| LPS + HES-RA | 2.24 ± 1.1 | 0.00 ± 0.00 | 6.06 ± 1.7 | 17.17 ± 7.1*, + |
| LPS + HES-RA + NAC | 1.97 ± 1.3 | 0.00 ± 0.0* | 23.36 ± 22.2 | 28.53 ± 28***, +++ |
| ClCrea (ml/min) | ||||
| Time control | 0.11 ± 0.6 | 0.10 ± 0.04 | 0.14 ± 0.04 | 0.14 ± 0.03 |
| Control + NAC | 0.11 ± 0.04 | 0.09 ± 0.04 | 0.11 ± 0.04 | 0.06 ± 0.03*** |
| LPS | 0.09 ± 0.04 | 0.00 ± 0.00*** | 0.00 ± 0.00*** | 0.00 ± 0.00*** |
| LPS + HES-RA | 0.09 ± 0.06 | 0.00 ± 0.00*** | 0.05 ± 0.01***, + | 0.02 ± 0.01*** |
| LPS + HES-RA + NAC | 0.09 ± 0.04 | 0.00 ± 0.00*** | 0.04 ± 0.01*** | 0.02 ± 0.01*** |
| EFNa+ | ||||
| Time control | 4.8 ± 4.7 | 13.2 ± 5.7 | 11.9 ± 4.9 | 10.4 ± 2.4 |
| Control + NAC | 2.3 ± 0.8 | 3.22 ± 1.7 | 2.9 ± 1.7 | 6.9 ± 6.6 |
| LPS | 3.7 ± 1.4 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| LPS + HES-RA | 3.5 ± 2.8 | 0.00 ± 0.00 | 35.3 ± 7.3**, +++ | 17.1 ± 8.1 |
| LPS + HES-RA + NAC | 4.4 ± 3.9 | 0.00 ± 0.00 | 45.8 ± 36.7***, +++ | 31.4 ± 28.4***, +++ |
| Urine volume (ml) | ||||
| Time control | 0.39 ± 0.14 | 0.53 ± 0.13 | 0.45 ± 0.13 | 0.19 ± 0.03 |
| Control + NAC | 0.25 ± 0.09 | 0.34 ± 0.17 | 0.2 ± 0.04 | 0.16 ± 0.06 |
| LPS | 0.31 ± 0.13 | 0.06 ± 0.09*** | 0.04 ± 0.05*** | 0.02 ± 0.05*** |
| LPS + HES-RA | 0.37 ± 0.3 | 0.12 ± 0.19*** | 0.6 ± 0.23 +++ | 0.16 ± 0.08 +++ |
| LPS + HES-RA + NAC | 0.33 ± 0.26 | 0.04 ± 0.09*** | 0.65 ± 0.47 +++ | 0.2 ± 0.13 +++ |
Values are presented as mean ± SD
TNa+ tubular sodium reabsorption, Cl creat clearance of creatinine, LPS lipopolysaccharide, HES-RA hydroxyethyl starch-ringer acetate, NAC N-acetylcysteine
*p < 0.05, **p < 0.01, and ***p < 0.001 versus control; + p < 0.05 and +++ p < 0.001 versus LPS
Biomarkers of AKI (NGAL and L-FABP) were significantly increased in the kidney after LPS infusion (p < 0.05 and p < 0.001, respectively, versus the control group) (Fig. 3). The resuscitation fluid combined with NAC significantly decreased these biomarkers compared to the LPS alone group (p < 0.001 and <0.05, respectively). Moreover, L-FABP was lower in the group of septic rats resuscitated with HES-RA + NAC than with HES-RA alone (p < 0.001).
Fig. 3.

Immunostaining intensity (HSCORE) of NGAL (a) and L-FABP (b) in kidney cortex of all groups. *p < 0.05, ***p < 0.001 versus control group; +p < 0.05, +++p < 0.001 versus LPS group, ###p < 0.001 versus LPS + HES-RA group
Plasma electrolytes and acid-base status
Bicarbonate and plasma lactate levels, pH, base excess, and anion gap with K+ are shown in Table 3. LPS infusion significantly increased the plasma lactate level and anion gap, which could not be corrected by the administration of HES-RA either with or without NAC (p < 0.001) compared to the control group. Base excess, pH, and bicarbonate levels were similarly decreased after LPS infusion and were partially corrected by fluid resuscitation. NAC infusion alone in the control group resulted in a significant decrease in pH, base excess, and bicarbonate levels compared to the control group (p < 0.01). NAC administration worsened the acid-base status in the LPS resuscitated and control groups. The level of bicarbonate and base excess were significantly lower in rats resuscitated with fluid plus NAC than with fluid alone (p < 0.01).
Oxidative stress and inflammatory cytokines
The levels of biomarkers of oxidative stress, pro-inflammatory cytokines, and products of glycocalyx degradation are represented in Fig. 4. The levels of TNF-α (3A) and IL-6 (3B) in kidney homogenates from the LPS group were significantly increased compared to the control group (528.1 ± 143.9 pg/mg protein versus 291.8 ± 99.1 pg/mg protein, p < 0.05; and 1246 ± 441 pg/mg protein versus 753.8 ± 122 pg/mg protein, p < 0.05, respectively). The same results were observed regarding hyaluronic acid (HA) (3C), nitric oxide (3D), and MDA (3E) after LPS infusion (p < 0.05). The addition of NAC to HES-RA during fluid resuscitation resulted in a significant lower level of HA and nitric oxide compared to the LPS group (p < 0.01). Infusion of HES-RA alone decreased the levels of MDA compared to LPS alone (p < 0.01) (3E). Protein carbonyl levels were not altered (3E).
Fig. 4.
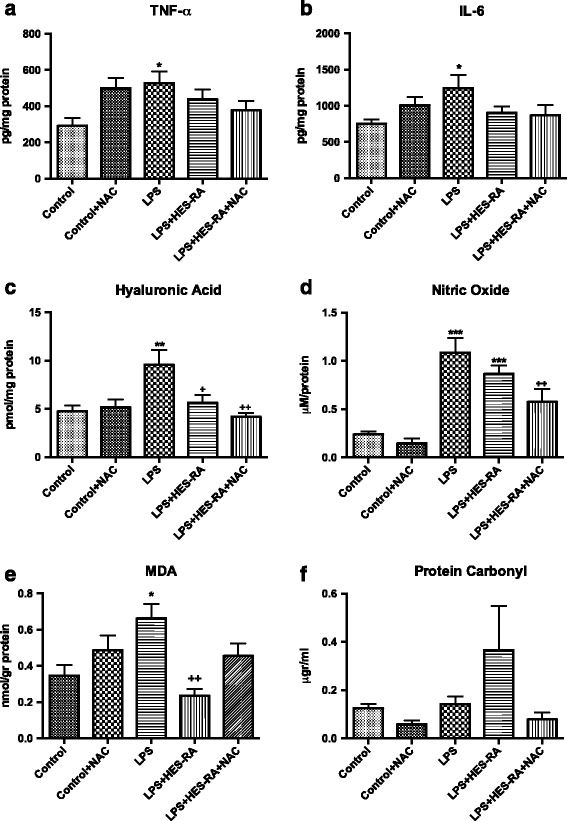
Levels of biomarkers of oxidative stress and pro-inflammatory cytokines in renal tissue. Renal tissue TNF-α (a), IL-6 (b), hyaluronic acid (c), nitric oxide (d), MDA (e), and protein carbonyl (f). *p < 0.05, **p < 0.01, ***p < 0.001 versus control; +p < 0.05, ++p < 0.01 versus LPS group
Discussion
In the present study, we found that fluid supplemented with NAC improved cortical renal oxygenation, oxygen delivery, and oxygen consumption compared to the LPS group. Fluid resuscitation alone was partially effective in correcting kidney hypoxia but did not reach a significant level compared to the LPS group. The addition of NAC to the resuscitation fluid did not further improve systemic or renal hemodynamics compared to HES-RA alone. It has been suggested that a specific effect of NAC on microvascular oxygenation exists independent of renal macrovascular perfusion. In an experimental study, Heyman et al. showed that NAC induced vasodilation in a pre-constricted renal microvasculature rat model [18]. The vasculature may be similarly constricted after LPS infusion leading to microcirculation heterogeneity [25]. Although creatinine levels did not differ between septic rats receiving fluids either with or without NAC, the fluid resuscitation combined with NAC decreased the levels of renal NO, hyaluronic acid, and early markers of acute kidney injury such as NGAL or L-FABP. Previous studies demonstrated a significant decrease in inflammatory biomarkers in specific organs such as the lungs and kidneys in models of sepsis [14–16, 26–29]. However, none of these studies reported the beneficial effects of NAC on tissue oxygenation during sepsis by decreasing oxidative stress and inflammation.
Several studies using microcirculatory techniques have now questioned the significance of arterial RBF and have focused on the renal microcirculation as the hemodynamic culprit in the pathophysiology of septic AKI [6, 25, 30]. Microcirculatory dysfunction may contribute to renal hypoxia even in the absence of overt renal hypoperfusion. The microcirculation of the renal cortex has been shown to be severely injured in animal models of sepsis. After LPS infusion in rats, Legrand et al. showed that fluid resuscitation could not fully restore renal microcirculatory dysfunction [6]. In dogs, endotoxemia was found to be associated with renal hypoperfusion and hypoxia in the renal cortex but was concomitant with increased renal venous PO2, supporting the concept that convective shunting of oxygen may contribute to the development of tissue hypoxia [31]. In our study, the LPS-induced renal microvascular heterogeneity and hypoxia appeared to be corrected with the NAC-supplemented fluid.
We also observed negative effects of NAC infusion. Delayed hypotension occurred in the control + NAC group, highlighting the vasodilatory effects of this compound as previously described [12, 32]. The mechanism involved in this lowering effect might be mediated by the interaction of sulfhydryl groups with enzymes such as the guanylate cyclase, which is the primary receptor for NO [32]. This drop in the MAP may contribute to tissue hypoperfusion and hypoxia as well as the lower pH observed in the control + NAC group compared to the control group. However, no significant change was observed in renal blood flow measured in the renal artery. Conflicting data exist between experimental and human clinical studies regarding the effects of NAC on regional blood flow and cardiac output [19, 33–37]. First, NAC administration was shown to improve survival in experimental models of peritonitis-induced sepsis [38, 39]. In mongrel dogs subjected to endotoxemia, Zhang et al. demonstrated the myocardial protective effects of NAC pretreatment (150 mg/kg) with enhanced oxygen delivery but lower systemic and pulmonary pressures [37]. In contrast, in a study involving 20 patients with septic shock, NAC infusion (150 mg/kg for 15 min followed by continuous infusion) that was initiated within 24 h after the onset of septic shock resulted in a decrease in left ventricular stroke work—revealing myocardial depression—without a significant impact on MAP at 48 h after treatment initiation [33]. In a similar population, Rank et al. demonstrated the exact opposite effect with an improvement in liver blood flow, oxygen delivery, and oxygen consumption related to an increase in cardiac index [34]. However, the infusion of NAC lasted less than 2 h in this latter study. Agustí et al. reported an increase in the cardiac index associated with vasodilatation but without improvements in splanchnic microcirculation after NAC infusion in patients presenting with septic shock and multiple organ failure [35]. Clinical studies yielded controversial results with the use of NAC in sepsis [11]. NAC treatment during the first hours of sepsis or septic shock may decrease peroxidative stress [40], improve hepatic function [34], and enhance tissue oxygenation and cardiac function [41], whereas delayed administration adversely affected the outcomes of critically ill patients with multiple organ failure [33, 42].
Most published studies have examined the effects of NAC when given as a pretreatment, e.g., before the insult. Here, we evaluated the ability of NAC when administered during the resuscitation process to correct tissue hypoxia and inflammation already present. It seems that the beneficial effects on tissue oxygenation, if any, are not similar if NAC is administered before or after the insult. The effects also depend on the time elapsed since the insult. We showed that NAC-supplemented fluid did not provide additional benefits on the acid status and renal function compared to fluid alone. Only kidney oxygenation was significantly higher in the group receiving fluid supplemented with NAC compared to the LPS alone group, whereas fluid alone did not reach a level of significance. Pretreatment with NAC appeared to be more efficient than post-injury treatment in protecting tissues against oxidative stress and inflammation in models of sepsis and ischemia/reperfusion injury. Due to the mechanisms of action of NAC, it is conceivable that it would be easier to prevent certain pathways from being activated rather than to modulate already highly activated signals with redundant or alternative pathways.
The effects of NAC on renal oxygenation could be mediated by different mechanisms. First, tissue NO levels were significantly increased after LPS infusion. By decreasing NO levels in the cortex, NAC combined with fluid administration may improve microvascular dysfunction, microvascular delivery of oxygen, and cortical oxygenation. Similarly, our group previously demonstrated that the prostaglandin analog iloprost restored kidney function in a rat model of endotoxemia and prevented the occurrence of hypoxic regions [43]. The improvement of renal microvascular oxygenation was mediated in part by the inhibition of inducible nitric oxide synthase expression in the kidney. Another major player involved in endothelial dysfunction in sepsis-induced AKI is the widespread damage to the endothelial glycocalyx, which may contribute to microvascular dysfunction via impaired flow-dependent vasodilatation. Hyaluronic acid levels reflect the disruption of the glycocalyx [44]. In the present study, hyaluronic acid levels were significantly increased after LPS infusion. Fluid resuscitation either with or without the addition of NAC significantly dampened these levels. However, the decrease was more prevalent with NAC treatments, but all of the significant benefits were due to the fluids as opposed to NAC. NAC may still further improve microvascular oxygenation by decreasing endothelial glycocalyx damage in addition to fluid resuscitation. In contrast, a clinical study monitored volume loading with HES during elective surgery (20 ml/kg) and observed increased serum glycocalyx biomarkers with HES alone [45]. In our study, fluid resuscitation with HES seemed to be beneficial with regard to these biomarkers levels. One of the main pathways of glycocalyx disruption is thought to be the formation of ROS such as peroxynitrite. As NAC is a well-known scavenger of ROS, we measured the tissue levels of malondialdehyde as a marker of lipid peroxidation. We did not observe a significant decrease when using NAC-supplemented fluid compared to fluid alone. This result can be explained because of the high dose of NAC in our study. Some authors have previously suggested that high doses of NAC may increase MDA levels, whereas lower doses may decrease MDA levels [46]. Additionally, a lack of effect of NAC on lipid peroxidation in cases of established endotoxemia has been shown [47].
Limitations
In the light of the ongoing debate about the deleterious effects of HES on the kidney, a possible limitation of our study is our use of HES as a resuscitation fluid. However, the present study is a mechanistic study wherein we investigated whether we could ameliorate the inflammatory effects of fluid administration in a sepsis model. In our experience, all fluids cause inflammation [48], and it is the fluid volume that determined the extent of injury. It could be argued that we should have chosen a balanced crystalloid solution instead of a colloid-based solution. However, crystalloid solutions have issues as well. For example, Ringer’s lactate causes even more inflammation than HES [49, 50], and we would also have had to administer a larger volume to maintain blood pressure causing more hemodilution. We chose a colloid solution to keep the amount of fluid required to correct blood pressure to a minimum. We could have used albumin, but even albumin can promote renal failure, as has been shown in a recent study in cardiac surgery [51]. In conclusion, arguably, all fluids have deleterious effects on the kidney to a greater or lesser extent. However, in this proof of concept study, we hope to have introduced the idea that controlling the inflammatory component imposed by fluids such as HES can be implemented by co-administration of an anti-inflammatory drug such as NAC. Our results suggest that such an approach could be used for other fluids, but this approach would require testing in subsequent studies.
Conclusions
In conclusion, the addition of NAC to fluid resuscitation may improve renal oxygenation and attenuate microvascular dysfunction and AKI. Decreases of renal NO levels and hyaluronic acid levels may be involved in this beneficial effect. A therapeutic strategy combining the macrovascular effects of fluids and the microvascular effects of NAC may be critical to preventing sepsis-induced AKI. This study sets forth a new concept for changing the procedure of fluid resuscitation by the addition of antioxidant therapy during the initial phase of resuscitation.
Acknowledgements
We are grateful to Fresenius Kabi for providing the Volulyte®. We would like to thank Albert van Wijk, technician at the Laboratory of Experimental Surgery, Academic Medical Centre, Amsterdam, The Netherlands, for his excellent and skillful technical assistance.
Funding
This was supported by the Department of Translational Physiology funds solely.
Authors’ contributions
BE, study design, carried out experiment, data analaysis, writing the first draft, and review of manuscript; PG, data analaysis, writing the final version and review of manuscript; LZ, data analysis and writing the first draft of manuscript; FN, study design and review of manuscript; AK, conducted immunohistochemical studies; EG, conducted immunohistochemical studies; CDT, review manuscript; CI, study design, review and writing the final version of manuscript. All authors read and approved the final manuscript.
Competing interests
Prof. Ince has received grants and consultant fees from Fresenius Kabi, Baxter Healthcare, BBraun, and AM Pharma. Dr. Philippe Guerci is supported by a grant from the Société Française d’Anesthésie-Réanimation (SFAR), France. Dr Frank Nocken is currently working for Fresenius Kabi Deutschland GmbH and provided the HES 130/0.4 (Volulyte®) in this study. The remaining authors declare no conflicts of interest.
Ethics approval
All experiments in this study were approved by the institutional Animal Experimentation Committee of the Academic Medical Center of the University of Amsterdam (DFL102538). The care and handling of the animals were in accordance with the guidelines for Institutional Animal Care and Use Committees. The study was conducted in accordance with the Declaration of Helsinki.
Abbreviations
- AKI
Acute kidney injury
- CaO2
Arterial oxygen content
- ClCrea
Creatinine clearance
- CvO2
Renal venous oxygen content
- CμPO2
Microvascular renal cortical PO2
- DO2ren
Renal oxygen delivery
- EFNa+
Fractional excretion of sodium
- ER
Extraction ratio
- HES
Hydroxyethyl starch
- L-FABP
Liver-type fatty acid-binding protein
- LPS
Lipopolysaccharide
- MAP
Mean arterial pressure
- MDA
Malondialdehyde
- MμPO2
Microvascular renal medullar PO2
- NAC
N-Acetylcysteine
- NGAL
Neutrophil gelatinase-associated lipocalin
- NO
Nitric oxide
- RA
Ringer’s acetate
- RBF
Renal blood flow
- TNa+
Total amount of sodium reabsorbed
- TNF-α
Tumor necrosis factor-α
- VO2ren
Renal oxygen consumption
References
- 1.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 2.Ait-Oufella H, Maury E, Lehoux S, Guidet B, Offenstadt G. The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010;36:1286–1298. doi: 10.1007/s00134-010-1893-6. [DOI] [PubMed] [Google Scholar]
- 3.Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, Hernandez G, Murray P, De Backer D, Workgroup ADQIXIV. The endothelium in sepsis. Shock. 2016;45:259–270. doi: 10.1097/SHK.0000000000000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ince C. The microcirculation is the motor of sepsis. Crit Care. 2005;9(Suppl 4):S13–9. doi: 10.1186/cc3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Legrand M, Mik EG, Johannes T, Payen D, Ince C. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol Med. 2008;14:502–516. doi: 10.2119/2008-00006.Legrand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Legrand M, Bezemer R, Kandil A, Demirci C, Payen D, Ince C. The role of renal hypoperfusion in development of renal microcirculatory dysfunction in endotoxemic rats. Intensive Care Med. 2011;37:1534–1542. doi: 10.1007/s00134-011-2267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oudemans-van Straaten HM, Spoelstra-de Man AM, de Waard MC. Vitamin C revisited. Crit Care. 2014;18:460. doi: 10.1186/s13054-014-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galley HF, DiMatteo MA, Webster NR. Immunomodulation by anaesthetic, sedative and analgesic agents: does it matter? Intensive Care Med. 2000;26:267–274. doi: 10.1007/s001340051149. [DOI] [PubMed] [Google Scholar]
- 9.Berger MM, Chioléro RL. Antioxidant supplementation in sepsis and systemic inflammatory response syndrome. Crit Care Med. 2007;35:S584–90. doi: 10.1097/01.CCM.0000279189.81529.C4. [DOI] [PubMed] [Google Scholar]
- 10.Angstwurm MWA, Engelmann L, Zimmermann T, Lehmann C, Spes CH, Abel P, Strauss R, Meier-Hellmann A, Insel R, Radke J, Schüttler J, Gärtner R. Selenium in intensive care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med. 2007;35:118–126. doi: 10.1097/01.CCM.0000251124.83436.0E. [DOI] [PubMed] [Google Scholar]
- 11.Szakmany T, Hauser B, Radermacher P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst Rev. 2012;9:CD006616. doi: 10.1002/14651858.CD006616.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sehirli AO, Sener G, Satiroglu H, Ayanoğlu-Dülger G. Protective effect of N-acetylcysteine on renal ischemia/reperfusion injury in the rat. J Nephrol. 2003;16:75–80. [PubMed] [Google Scholar]
- 14.Ozdulger A, Cinel I, Koksel O, Cinel L, Avlan D, Unlu A, Okcu H, Dikmengil M, Oral U. The protective effect of N-acetylcysteine on apoptotic lung injury in cecal ligation and puncture-induced sepsis model. Shock. 2003;19:366–372. doi: 10.1097/00024382-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Hsu B-G, Lee R-P, Yang F-L, Harn H-J, Chen HI. Post-treatment with N-acetylcysteine ameliorates endotoxin shock-induced organ damage in conscious rats. Life Sci. 2006;79:2010–2016. doi: 10.1016/j.lfs.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 16.Carbonell LF, Díaz J, Hernández I, Cuevas S, Valero F, Quesada T, Fenoy F, Salom MG. N-acetylcysteine exerts protective effects and prevents lung redox imbalance and peroxynitrite generation in endotoxemic rats. Med Chem. 2007;3:29–34. doi: 10.2174/157340607779317580. [DOI] [PubMed] [Google Scholar]
- 17.Huet O, Cherreau C, Nicco C, Dupic L, Conti M, Borderie D, Pène F, Vicaut E, Benhamou D, Mira J-P, Duranteau J, Batteux F. Pivotal role of glutathione depletion in plasma-induced endothelial oxidative stress during sepsis. Crit Care Med. 2008;36:2328–2334. doi: 10.1097/CCM.0b013e3181800387. [DOI] [PubMed] [Google Scholar]
- 18.Heyman SN, Goldfarb M, Shina A, Karmeli F, Rosen S. N-acetylcysteine ameliorates renal microcirculation: studies in rats. Kidney Int. 2003;63:634–641. doi: 10.1046/j.1523-1755.2003.00783.x. [DOI] [PubMed] [Google Scholar]
- 19.Schaller G, Pleiner J, Mittermayer F, Posch M, Kapiotis S, Wolzt M. Effects of N-acetylcysteine against systemic and renal hemodynamic effects of endotoxin in healthy humans. Crit Care Med. 2007;35:1869–1875. doi: 10.1097/01.CCM.0000275385.45557.25. [DOI] [PubMed] [Google Scholar]
- 20.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci U S A. 2013;110:18351–18352. doi: 10.1073/pnas.1318345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haase VH. Inflammation and hypoxia in the kidney: friends or foes? Kidney Int. 2015;88:213–215. doi: 10.1038/ki.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johannes T, Mik EG, Ince C. Dual-wavelength phosphorimetry for determination of cortical and subcortical microvascular oxygenation in rat kidney. J Appl Physiol. 2006;100:1301–1310. doi: 10.1152/japplphysiol.01315.2005. [DOI] [PubMed] [Google Scholar]
- 24.Mik EG, Johannes T, Ince C. Monitoring of renal venous PO2 and kidney oxygen consumption in rats by a near-infrared phosphorescence lifetime technique. Am J Physiol Renal Physiol. 2008;294:F676–81. doi: 10.1152/ajprenal.00569.2007. [DOI] [PubMed] [Google Scholar]
- 25.Ergin B, Kapucu A, Demirci-Tansel C, Ince C. The renal microcirculation in sepsis. Nephrol Dial Transplant. 2015;30:169–177. doi: 10.1093/ndt/gfu105. [DOI] [PubMed] [Google Scholar]
- 26.Hsu B-G, Yang F-L, Lee R-P, Peng TC, Harn H-J, Chen HI. N-acetylcysteine ameliorates lipopolysaccharide-induced organ damage in conscious rats. J Biomed Sci. 2004;11:152–162. doi: 10.1007/BF02256558. [DOI] [PubMed] [Google Scholar]
- 27.Andrades M, Ritter C, de Oliveira MR, Streck EL, Fonseca Moreira JC, Dal-Pizzol F. Antioxidant treatment reverses organ failure in rat model of sepsis: role of antioxidant enzymes imbalance, neutrophil infiltration, and oxidative stress. J Surg Res. 2011;167:e307–13. doi: 10.1016/j.jss.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Campos R, Shimizu MHM, Volpini RA, de Bragança AC, Andrade L, Lopes FDTQDS, Olivo C, Canale D, Seguro AC. N-acetylcysteine prevents pulmonary edema and acute kidney injury in rats with sepsis submitted to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2012;302:L640–50. doi: 10.1152/ajplung.00097.2011. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Jo YH, Kim K, Lee JH, Rim KP, Kwon WY, Suh GJ, Rhee JE. Effect of N-acetylcysteine (NAC) on acute lung injury and acute kidney injury in hemorrhagic shock. Resuscitation. 2013;84:121–127. doi: 10.1016/j.resuscitation.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Zafrani L, Payen D, Azoulay E, Ince C. The microcirculation of the septic kidney. Semin Nephrol. 2015;35:75–84. doi: 10.1016/j.semnephrol.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Gullichsen E, Nelimarkka O, Halkola L, Niinikoski J. Renal oxygenation in endotoxin shock in dogs. Crit Care Med. 1989;17:547–550. doi: 10.1097/00003246-198906000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Girouard H, Chulak C, Wu L, Lejossec M, de Champlain J. N-acetylcysteine improves nitric oxide and alpha-adrenergic pathways in mesenteric beds of spontaneously hypertensive rats. Am J Hypertens. 2003;16:577–584. doi: 10.1016/S0895-7061(03)00863-X. [DOI] [PubMed] [Google Scholar]
- 33.Peake SL, Moran JL, Leppard PI. N-acetyl-L-cysteine depresses cardiac performance in patients with septic shock. Crit Care Med. 1996;24:1302–1310. doi: 10.1097/00003246-199608000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Rank N, Michel C, Haertel C, Lenhart A, Welte M, Meier-Hellmann A, Spies C. N-acetylcysteine increases liver blood flow and improves liver function in septic shock patients: results of a prospective, randomized, double-blind study. Crit Care Med. 2000;28:3799–3807. doi: 10.1097/00003246-200012000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Agustí AGN, Togores B, Ibañez J, Raurich JM, Maimó A, Bergada J, Marse P, Jorda R. Effects of N-acetylcysteine on tissue oxygenation in patients with multiple organ failure and evidence of tissue hypoxia. Eur Respir J. 1997;10:1962–1966. doi: 10.1183/09031936.97.10091962. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, Spapen H, Nguyen DN, Rogiers P, Bakker J, Vincent JL. Effects of N-acetyl-L-cysteine on regional blood flow during endotoxic shock. Eur Surg Res. 1995;27:292–300. doi: 10.1159/000129412. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Spapen H, Nguyen DN, Benlabed M, Buurman WA, Vincent JL. Protective effects of N-acetyl-L-cysteine in endotoxemia. Am J Physiol. 1994;266:H1746–54. doi: 10.1152/ajpheart.1994.266.5.H1746. [DOI] [PubMed] [Google Scholar]
- 38.Ritter C, Andrades ME, Reinke A, Menna-Barreto S, Moreira JCF, Dal-Pizzol F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit Care Med. 2004;32:342–349. doi: 10.1097/01.CCM.0000109454.13145.CA. [DOI] [PubMed] [Google Scholar]
- 39.de Mello RO, Lunardelli A, Caberlon E, de Moraes CMB, Christ Vianna Santos R, da Costa VL, da Silva GV, da Silva SP, Buaes LEC, da Silva Melo DA, Donadio MVF, Nunes FB, de Oliveira JR. Effect of N-acetylcysteine and fructose-1,6-bisphosphate in the treatment of experimental sepsis. Inflammation. 2011;34:539–550. doi: 10.1007/s10753-010-9261-9. [DOI] [PubMed] [Google Scholar]
- 40.Ortolani O, Conti A, De Gaudio AR, Moraldi E, Cantini Q, Novelli G. The effect of glutathione and N-acetylcysteine on lipoperoxidative damage in patients with early septic shock. Am J Respir Crit Care Med. 2000;161:1907–1911. doi: 10.1164/ajrccm.161.6.9903043. [DOI] [PubMed] [Google Scholar]
- 41.Spies CD, Reinhart K, Witt I, Meier-Hellmann A, Hannemann L, Bredle DL, Schaffartzik W. Influence of N-acetylcysteine on indirect indicators of tissue oxygenation in septic shock patients: results from a prospective, randomized, double-blind study. Crit Care Med. 1994;22:1738–1746. doi: 10.1097/00003246-199422110-00007. [DOI] [PubMed] [Google Scholar]
- 42.Molnár Z, Shearer E, Lowe D. N-Acetylcysteine treatment to prevent the progression of multisystem organ failure: a prospective, randomized, placebo-controlled study. Crit Care Med. 1999;27:1100–1104. doi: 10.1097/00003246-199906000-00028. [DOI] [PubMed] [Google Scholar]
- 43.Johannes T, Ince C, Klingel K, Unertl KE, Mik EG. Iloprost preserves renal oxygenation and restores kidney function in endotoxemia-related acute renal failure in the rat. Crit Care Med. 2009;37:1423–1432. doi: 10.1097/CCM.0b013e31819b5f4e. [DOI] [PubMed] [Google Scholar]
- 44.Dane MJC, van den Berg BM, Lee DH, Boels MGS, Tiemeier GL, Avramut MC, van Zonneveld AJ, van der Vlag J, Vink H, Rabelink TJ. A microscopic view on the renal endothelial glycocalyx. Am J Physiol Renal Physiol. 2015;308:F956–66. doi: 10.1152/ajprenal.00532.2014. [DOI] [PubMed] [Google Scholar]
- 45.Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, Conzen P, Becker BF, Rehm M. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. 2014;18:538. doi: 10.1186/s13054-014-0538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitri LE, Sardjono TW, Simamora D, Sumarno RP, Setyawati SK. High dose of N-acetylcysteine increase H2O2 and MDA levels and decrease GSH level of HUVECs exposed with malaria serum. Trop Biomed. 2011;28:7–15. [PubMed] [Google Scholar]
- 47.Caglikulekci M, Dirlik M, Pata C, Plasse M, Tamer L, Ogetman Z, Ercan B. Effect of N-acetylcysteine on blood and tissue lipid peroxidation in lipopolysaccharide-induced obstructive jaundice. J Invest Surg. 2006;19:175–184. doi: 10.1080/08941930600674702. [DOI] [PubMed] [Google Scholar]
- 48.Aksu U, Bezemer R, Yavuz B, Kandil A, Demirci C, Ince C. Balanced vs unbalanced crystalloid resuscitation in a near-fatal model of hemorrhagic shock and the effects on renal oxygenation, oxidative stress, and inflammation. Resuscitation. 2012;83:767–773. doi: 10.1016/j.resuscitation.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 49.Hussmann B, Lendemans S, de Groot H, Rohrig R. Volume replacement with Ringer-lactate is detrimental in severe hemorrhagic shock but protective in moderate hemorrhagic shock: studies in a rat model. Crit Care. 2014;18:R5. doi: 10.1186/cc13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohrig R, Rönn T, Lendemans S, Feldkamp T, de Groot H, Petrat F. Adverse effects of resuscitation with lactated ringer compared with ringer solution after severe hemorrhagic shock in rats. Shock. 2012;38:137–145. doi: 10.1097/SHK.0b013e31825b4ed9. [DOI] [PubMed] [Google Scholar]
- 51.Frenette AJ, Bouchard J, Bernier P, Charbonneau A, Nguyen LT, Rioux J-P, Troyanov S, Williamson DR. Albumin administration is associated with acute kidney injury in cardiac surgery: a propensity score analysis. Crit Care. 2014;18:602. doi: 10.1186/s13054-014-0602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]