

Abstract
The all-ferrous Rieske cluster, [2Fe-2S]0, has been produced in solution and characterized by protein-film voltammetry and UV–visible, EPR, and Mössbauer spectroscopies. The [2Fe-2S]0 cluster, in the overexpressed soluble domain of the Rieske protein from the bovine cytochrome bc1 complex, is formed at –0.73 V at pH 7. Therefore, at pH 7, the [2Fe-2S]1+/0 couple is 1.0 V below the [2Fe-2S]2+/1+ couple. The two cluster-bound ferrous irons are both high spin (S = 2), and they are coupled antiferromagnetically (–J ≥ 30 cm–1, H =–2JS1·S2) to give a diamagnetic (S = 0) ground state. The ability of the Rieske cluster to exist in three oxidation states (2+, 1+, and 0) without an accompanying coupled reaction, such as a conformational change or protonation, is highly unusual. However, uncoupled reduction to the [2Fe-2S]0 state occurs at pH > 9.8 only, and at high pH the intact cluster persists in solution for <1 min. At pH < 9.8, the all-ferrous cluster is stabilized significantly by protonation. A combination of experimental data and calculations based on density functional theory suggests strongly that the proton binds to one of the cluster μ2-sulfides, consistent with observations that reduced [3Fe-4S] clusters are protonated also. The implications for our understanding of coupled reactions at iron–sulfur clusters and of the factors that determine the relative stabilities of their different oxidation states are discussed.
Iron–sulfur (FeS) clusters are essential to all forms of life. Most frequently, they are simple electron carriers, but they also constitute catalytic centers, structural scaffolds, and sensors, and they undergo oxidative degradation (1, 2). The rhombic [2Fe-2S], cuboidal [3Fe-4S], and cubane [4Fe-4S] clusters are the most common, but elaboration of these basic modules has produced clusters that contain heterometals and up to eight iron centers. For example, the catalytic clusters in acetyl-CoA synthase contain nickel, and the P-cluster and iron–molybdenum cofactor of nitrogenase can be considered to be two cuboidal subclusters joined by sulfurs. Assembly, disassembly, and interconversion of the simpler clusters are exploited in oxygen sensing and in the control of intracellular iron levels, and they also constitute enzyme active sites, such as in aconitase, chloroplast ferredoxin:thioredoxin reductase, and biotin synthase. In contrast, uncontrolled cluster disassembly (during oxidative stress) accelerates the production of reactive oxygen species and therefore is potentially very damaging.
Formally, each iron center in a cluster can be ferric or ferrous, so [2Fe-2S], [3Fe-4S], and [4Fe-4S] clusters have three, four, and five possible oxidation states, respectively. In proteins, the oxidation states of [3Fe-4S] clusters cover the widest range because many have been observed in the 1+ (all-ferric), 0, and 2–(all-ferrous) states (3). Formation of the [3Fe-4S]0 state is associated with protonation (4), and reduction to the 2–state occurs only upon the uptake of a total of three protons (3). Therefore, the overall charge is conserved, and it is likely that [3Fe-4S] clusters exist in more than two oxidation states because they can be protonated. The protons probably bind on the three μ2-sulfides, consistent with the ability of [3Fe-4S]0 clusters to coordinate a fourth metal ion (5). In contrast, most [4Fe-4S] clusters in proteins are confined to only two oxidation states, either the 2+ and 1+ states or the 3+ and 2+ states in high-potential iron proteins. The [4Fe-4S] cluster in the Fe-protein of nitrogenase (“the Fe-protein”) is unique because it can exist in the all-ferrous state and is stable in more than two oxidation states (2+,1+, and 0) (6). This versatility may be because of its unusually high solvent accessibility (7), although during turnover it undergoes significant conformational changes coupled to nucleotide binding (8), and protonation of the all-ferrous state has not been excluded. The nitrogenase [8Fe-7S] P-cluster (“the P cluster”), the only high-nuclearity cluster not involved directly in small molecule activation, also exists in three oxidation states, but interconversions between them are coupled to changes in cluster structure and ligation and to protonation; the most reduced state may be all-ferrous (9, 10). There is no confirmed example of an FeS cluster in a protein undergoing sequential uncoupled redox transformations.
Under physiological conditions, [2Fe-2S] clusters have been observed in the 2+ and 1+ states only. The all-ferrous state may not have been observed because the potential for its formation is too negative, the all-ferrous cluster is unstable, or two-electron reduction is precluded in the absence of a charge-compensation mechanism. In contrast, in anaerobic nonaqueous solvents, synthetic [2Fe-2S] analogues can be reduced sequentially by two electrons (11); the reduction potentials are reported to be only ≈0.25 V apart, but the proposed all-ferrous nature of the fully reduced state has not been demonstrated. A [2Fe-2S]0 cluster was generated artificially in spinach ferredoxin by irreversible complexation of the protein to a chromium reductant, increasing the reduction potential significantly and adding extra positive charge (12). Voltammetric signals from the soluble Rieske domain from the bovine heart cytochrome bc1 complex (–0.84 V at pH 7, potential reported to be pH independent) were attributed to formation of the all-ferrous cluster, but no characterization was attempted (13). Here, we describe the reversible formation of a stable, unmodified [2Fe-2S]0 cluster-containing protein and its extensive characterization. The [2Fe-2S]0 cluster is a Rieske cluster, so its formation is facilitated by the neutral, electronegative histidine ligands. Our results provide insight into the potentials, stabilities, and reactivities of the different oxidation states of the simplest FeS clusters and are also relevant to understanding the structures and functions of higher nuclearity clusters.
Materials and Methods
Protein Preparation. The design, cloning, overexpression, and purification of the truncated Rieske protein from the bovine cytochrome bc1 complex (BtRp) and its S163A, Y165F, and C144A + C160A (AA) mutants are described in ref. 14. BtRp lacks residues 1–69 of the mature protein and has a six-histidine tag on its N terminus. Its spectroscopic properties (UV–visible and EPR) and reduction potential ([2Fe-2S]2+/1+) are very similar to those of its structurally characterized native equivalent, resolved from the complex by proteolysis (14–16).
Protein-Film Voltammetry. Protein-film voltammetry was carried out as described in refs. 17 and 18 under anaerobic conditions (O2 < 2 ppm). Typically, the potential was prepoised at the low-potential limit for 20 s, then scanned at 10 mV·s–1. Reduction potentials are relative to the standard hydrogen electrode and were independent of scan rate, and poise potential and time.
Generation of the All-Ferrous Rieske Protein in Solution. All-ferrous BtRp was generated in solution by using EuII-diethylenetriamine-N,N,N′,N″,N′″-pentaacetate (EuII-DTPA) (19). All procedures were performed under strictly anaerobic conditions (O2 ≤ 1 ppm). A stock of 50 mM Eu2O3 (Sigma) in 0.5 M HCl was diluted to 2 mM in 50 mM Hepes/0.3 M NaCl (pH 8.0), final pH ≈ 7, and electrolyzed exhaustively at –0.6 V by using a graphite “pot” electrode (3) to reduce the EuIII. The EuII concentration was verified spectroscopically. The EuII and protein solutions were brought to the experimental pH and mixed to give a molar excess of EuII of 1.4–2.0. Then, upon the addition of DTPA to the same molar excess (Sigma, 50 mM DTPA in 0.2 M NaOH), BtRp was reduced stoichiometrically to the all-ferrous form.
Mössbauer Spectroscopy. Cells expressing BtRp were grown on M9 minimal medium, supplemented with 5 mg·ml–1 (NH4)257Fe(SO4)2·6H2O. They were incubated at 37°C until induction at OD600 = 0.2 with 0.5 mM isopropyl β-d-thiogalactopyranoside and then for 18 h at 25°C. Purified 57Fe-BtRp was concentrated to 1.5–2.0 mM for preparation of the all-ferrous Rieske cluster at 300–600 μM. Mössbauer spectra were recorded on an alternating constant-acceleration spectrometer, with the sample temperature maintained by either an Oxford Instruments Variox or Mössbauer-Spectromag cryostat. The 57Co/Rh source (1.8 GBq) was positioned at a zero-field position at room temperature. Isomer shifts are relative to iron metal at 300 K.
Density Functional Theory Calculations. The pKs of the cysteine ligands and the cluster sulfides in the all-ferrous state were calculated as described in ref. 20 by using a combination of continuum electrostatic and density functional theory calculations, and based on the structure of the soluble Rieske protein domain from bovine cytochrome bc1 (Protein Data Bank ID code 1RIE, www.pdb.org) (15). The change in vibrational energy upon deprotonation of a typical sulfur center was estimated from normal mode analyses of H3S+ and H2S (–27.37 kJ·mol–1) and 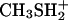 and CH3SH (–27.12 kJ·mol–1). The results from different conformations and tautomers were averaged thermodynamically to obtain macroscopic pK values.
and CH3SH (–27.12 kJ·mol–1). The results from different conformations and tautomers were averaged thermodynamically to obtain macroscopic pK values.
Results
Reversible Formation of the All-Ferrous [2Fe-2S]0 Rieske Cluster. Fig. 1A shows a film voltammogram of BtRp adsorbed on a freshly polished graphite-edge electrode, with clearly resolved, reversible oxidation and reduction peaks at –0.813 and –0.851 V, respectively (average –0.832 V). Interconversion of the oxidized, [2Fe-2S]2+, and normal-reduced, [2Fe-2S]1+, states occurs at much higher potential (+0.305 Vat pH 7 for BtRp) and is described in detail in refs. 16 and 18. The reduction potential measured with BtRp diffusing (not adsorbed) to the electrode (–0.806 V, 0°C, pH 8, 0.3 M NaCl) was close to the value recorded by protein-film voltammetry under identical conditions (–0.791 V), so adsorption does not perturb the cluster environment. The low-potential couple is due to formation of the all-ferrous cluster and not to a reduction of the disulfide bond (17), which bridges over the cluster between the two coordinating loops (15), for the following reasons. (i) The charges passed for the high- and low-potential couples (using the same film) are within 15% of one another, and the peak half-height widths are very similar (90–100 mV). Therefore, the low-potential couple represents a one-electron transition. (ii) The low-potential couple is observed in disulfide-free mutants of BtRp. Film voltammograms from mutant AA (C144A + C160A) clearly show the low-potential transition, occurring at a potential similar to that exhibited by WT BtRp (Fig. 1B). The [2Fe-2S]0 cluster was observed also in the high-potential Rieske proteins from Rhodobacter sphaeroides (RsRp) and Thermus thermophilus (TtRp). As predicted from their [2Fe-2S]2+/1+ potentials (18), the [2Fe-2S]1+/0 couples in BtRp and RsRp occur at very similar potential (–0.730 and –0.747 V, respectively, at pH 7), but in TtRp the potential is considerably lower (–0.918 V at pH 7).
Fig. 1.

Cyclic voltammograms showing the reversible interconversion of the all-ferrous, [2Fe-2S]0, and normal-reduced, [2Fe-2S]1+, Rieske clusters and background-subtracted signals (expanded). (A)WT BtRp (pH 8.82). (B) Mutant AA (which lacks the disulfide) (pH 7.90). The potential was held at the low-potential limit for 20 s before scanning (10 mV·s–1). The peak shapes are close to the ideal (Nernstian) shape, with half-height widths of 90–100 mV. Conditions: 10 mM sodium acetate, Hepes, Mes, and N-[tris(hydroxymethyl) methyl]-3-aminopropanesulfonic acid (TAPS) buffers, 2 M NaCl, 20°C.
Reduction to the All-Ferrous State Is Accompanied by Protonation. Protein-film voltammetry measurements showed that the [2Fe-2S]1+/0 reduction potential is strongly pH dependent (Fig. 2). At pH < 9, the gradient of the curve of E vs. pH is –58 mV per decade; therefore, one proton is taken up by the [2Fe-2S]0 state. This proton is distinct from the two protons that can be dissociated from the two histidine ligands. In the [2Fe-2S]1+ state, they are fully bound at pH < 11 (14, 18), and they remain bound at all observable pH values in the [2Fe-2S]0 state. The data shown in Fig. 2A are recorded in 2 M NaCl, confirming that the pH dependence is not because of nonspecific changes in protein charge (21); similar behavior was observed in 0.1 and 0.01 M NaCl also. At pH > 10, the reduction potential becomes pH independent (at –0.89 V), defining the pK of the [2Fe-2S]0 cluster, pK = 9.77, according to the modeled lines shown in Fig. 2, calculated by using Eq. 1 and Fig. 3. The pH-independent region is present unambiguously in D2O (Fig. 2B) because the pK decreases to 8.87.
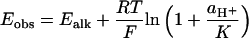 |
[1] |
Fig. 2.

The pH dependence of the reduction potential of the [2Fe-2S]1+/0 transition of the Rieske cluster in BtRp in H2O(A) and in D2O(B). Fitted lines are from Eq. 1, and experimental conditions are as for Fig. 1.
Fig. 3.

Thermodynamic scheme relating the [2Fe-2S]1+ cluster to the [2Fe-2S]0 cluster and showing that the [2Fe-2S]0 cluster may be protonated or deprotonated. No evidence has been observed for the [2Fe-2S]1+-H+ cluster, which would formally complete the square, so it has been omitted.
Generation of the All-Ferrous Cluster in Solution. For spectroscopy, the all-ferrous cluster was produced stoichiometrically by using EuII-DTPA (E = –1.14 V at pH 8.0) (19). UV–visible and EPR spectroscopies, after reoxidation by [Fe(CN)6]3–, and voltammetry demonstrated that formation of the all-ferrous cluster was reversible and confirmed that the properties of the cluster were unaltered. Direct reduction of the [2Fe-2S]1+-containing protein by electrolysis in a graphite pot electrode (3) was also possible at –0.9V(0°C, pH 8) but was marred by rapid reoxidation, even under strictly anaerobic conditions (O2 < 1 ppm). TiIII(citrate) and CrII(EDTA) were used previously to reduce the Fe-protein from Azotobacter vinelandii (6, 22), but the BtRp [2Fe-2S]1+ cluster was not reduced by TiIII(citrate), and the protein precipitated rapidly upon reduction by CrII(EDTA).
Fig. 4 shows the UV–visible spectra of the [2Fe-2S]0 and [2Fe-2S]1+ clusters. As expected, the all-ferrous state is pale yellow/green, whereas the more oxidized states are red/brown. Its spectrum is similar to the spectra of the [2Fe-2S]0 state of spinach ferredoxin, generated by complexation to a chromium reductant (12), and the [3Fe-4S]2–-3H+ cluster in Sulfolobus acidocaldarius ferredoxin (3). Interestingly, the all-ferrous Fe-protein is distinct because it is pink-red, due to an absorption band at ≈520 nm (19, 22). The all-ferrous cluster of BtRp is EPR silent, in both perpendicular and parallel mode, indicating that it has zero, or integer, spin.
Fig. 4.

UV–visible spectra showing the reduction of the [2Fe-2S]1+ cluster to the all-ferrous state, [2Fe-2S]0-H+ (pH 7). The cluster remained in the [2Fe-2S]1+ state in the presence of EuII, then addition of DTPA reduced the cluster rapidly to the [2Fe-2S]0-H+ state. Experimental details are described in Materials and Methods.
Mössbauer Spectroscopy. Fig. 5A shows the zero-field Mössbauer spectrum of the all-ferrous cluster at pH 7, 4.2 K. Its asymmetric form results from two symmetric quadrupole doublets, with Lorentzian lineshapes and equal intensity (subspectra a and b), and a third doublet with ≈16% intensity (subspectrum c). The high isomer shifts (δ) and large quadrupole splittings (ΔEQ) indicate that all three doublets are due to high-spin (S = 2) ferrous iron (23). Subspectrum c is caused by adventitious iron because of its high δ (>1.2 mm·s–1), which exceeds the range for tetrahedral FeIIS2X2 sites and is typical of six-coordinate Fe2+. Subspectra a and b are ascribed to the two tetrahedral cluster irons and demonstrate complete conversion from [2Fe-2S]1+ to [2Fe-2S]0. They are clearly distinguishable because of the asymmetric cluster coordination and display remarkably narrow linewidths (Γ= 0.25 mm·s–1), confirming that the sites are highly homogeneous. The absence of magnetic hyperfine splitting is consistent with a diamagnetic (S = 0) or integer spin ground state. Zero-field spectra were recorded also at 160 and 80 K for direct comparison with the paramagnetic [2Fe-2S]1+ Rieske cluster and were essentially the same as at 4.2 K. The spectrum of the [2Fe-2S]1+ state of BtRp at 160 K was very similar to that of TtRp, described in detail in ref. 24.
Fig. 5.

Mössbauer spectra of the [2Fe-2S]0-H+ Rieske cluster (pH 7). (A) B = 0, 4.2 K. (B) B = 7 T perpendicular to the γ-beam, 25 K. A was modeled by using Lorentzian doublets (see Table 1), and the lines shown in B are spin-Hamiltonian simulations (see text). The “nested” configuration of doublets is shown, and subspectra a and b represent the cysteine- and histidine-coordinated centers, respectively. Subspectrum c represents contamination from cluster degradation (16%).
The two quadrupole doublets (a and b) are of equal intensity, so “nested” and “staggered” configurations yield equivalent data fits. Analyses of magnetically perturbed spectra (see below) did not favor either, so the nested configuration was adopted for the following reasons: (i) in the staggered configuration the difference in δ is unreasonably large (Table 1) and would exceed the difference between FeII in reduced ferredoxins (FeIIS4) and Rieske centers (FeIIS2N2); and (ii) δ and ΔEQ values from the nested configuration agree more closely with those of related proteins and model compounds (24–26). The doublet with higher δ (Table 1) is from the histidine-coordinated iron (FeH), because the nitrogen ligands are more electronegative and the Fe–N bond is less covalent (23). Therefore, the cysteine-coordinated iron (FeC) has δ = 0.70 mm·s–1 and ΔEQ = 2.76 mm·s–1 at 4.2 K, consistent with values from tetrahedral FeIIS4 sites in the [2Fe-2S]0 cluster in Aquifex aeolicus ferredoxin (27) (δ = 0.71 mm·s–1, ΔEQ = 2.75 mm·s–1 at 4.2 K) and rubredoxin (δ = 0.7 ± 0.02 mm·s–1 at 4.2 K) (28). FeH has δ = 0.78 mm·s–1 and ΔEQ = 2.24 mm·s–1 (160 K) and should match the ferrous ion in the [2Fe-2S]1+ clusters of BtRp (δ = 0.69 mm·s–1, ΔEQ = 2.87 mm·s–1, 160 K) and TtRp (δ = 0.65 mm·s–1, ΔEQ = 2.81 mm·s–1, 230 K) (24). Increases in δ for one cluster iron, upon the reduction of another, have been observed for Fe3+ in [2Fe-2S]2+,1+ and [3Fe-4S]1+,0 clusters and also for the Fe2.5+ pair in [4Fe-4S]3+,2+,1+ clusters (26). For BtRp, the increased δ of FeH upon the reduction of FeC suggests that reduction weakens the FeH–sulfide bonds. The reason may be that the cluster dimensions increase, hydrogen bonds between cluster and protein strengthen, or perhaps a bridging sulfide is protonated (see below).
Table 1. Mössbauer parameters for Fe2+ in the [2Fe-2S]1+ cluster and the [2Fe-2S]0 cluster at high and low pH.
| Cluster | Center | T, K | δ, mm·s-1 | ΔEQ, mm·s-1 | Γ, mm·s-1 |
|---|---|---|---|---|---|
| [2Fe-2S]+ | FeII-His | 160 | 0.69 | 2.87 | 0.28 |
| [2Fe-2S]0 (low pH) | FeII-Cys (n) | 160 | 0.66 | 2.71 | 0.25 |
| FeII-His (n) | 160 | 0.78 | 2.24 | 0.25 | |
| FeII-Cys (n) | 4.2 | 0.70 | 2.76 | 0.26 | |
| FeII-His (n) | 4.2 | 0.81 | 2.32 | 0.26 | |
| FeII-Cys (s) | 4.2 | 0.65 | 2.65 | 0.26 | |
| FeII-His (s) | 4.2 | 0.87 | 2.43 | 0.26 | |
| [2Fe-2S]0 (high pH) | FeII-Cys (n) | 160 | 0.65 | 2.94 | 0.40 |
| FeII-His (n) | 160 | 0.79 | 2.72 | 0.40 | |
| FeII-Cys (n) | 4.2 | 0.69 | 3.06 | 0.40 | |
| FeII-His (n) | 4.2 | 0.80 | 2.79 | 0.40 |
(n) and (s) refer to fits using the nested and staggered configurations. Typical errors are ± 0.02 mm·s-1 for both δ and ΔEQ.
Magnetically perturbed Mössbauer spectra (Fig. 5B) were recorded to explore spin coupling in the [2Fe-2S]0 cluster and to establish a lower limit for the spin coupling constant. The magnetic splittings are typical of a diamagnetic system (S = 0), demonstrating that the two S = 2 ferrous ions are strongly antiferromagnetically coupled and could be simulated satisfactorily by using the values given in Table 1. No internal fields, arising from the thermal population of excited paramagnetic states, could be detected at up to 80 K (7 T applied field). A quantitative analysis was derived from spin-Hamiltonian simulations by using the usual nuclear Hamiltonian (25) and an explicit exchange coupling term –2 JS1·S2. The local axial and rhombic zero-field parameters were Di = 7 cm–1, E/Di = 0, and the hyperfine parameters were taken from Table 1, with positive ΔEQ, asymmetry parameters η = 0.8 (a) and 0.6 (b), and Ai/gNβN = –20 T. For the all-ferrous cluster, any simulation with –J ≥ 30 cm–1 is in accord with the experimental spectra, consistent with the estimated value from the [2Fe-2S]0 cluster in Aquifex aeolicus ferredoxin, –J > 40 cm–1, derived by using similar conditions (27). Therefore, the [2Fe-2S]0 cluster, like [2Fe-2S]1+ and [2Fe-2S]2+ clusters, is an antiferromagnetically coupled spin pair. J is expected to decrease upon reduction because of the weaker coordination of FeII and an increase in the Fe–Fe distance, as observed in, for example, the [2Fe-2S]2+ and [2Fe-2S]1+ clusters in spinach ferredoxin (–J = 183 and –J > 100 cm–1, respectively) (29). The Rieske [2Fe-2S]1+ clusters in TtRp and benzene dioxygenase have –J = 100 and 190 cm–1, respectively (30). Therefore, although a meaningful evaluation is precluded without an exact measurement for the [2Fe-2S]0 cluster, the reported value falls within the expected range.
To explore the protonation of the [2Fe-2S]0 cluster, Mössbauer samples were prepared also at high pH (pH > pK). Samples at pH 10, pD 9, and pD 10 all showed considerable cluster deterioration [as evidenced by the high relative intensity (65–75%) of subspectrum c]. Note that very little degradation of the [2Fe-2S]1+ cluster is observed under equivalent alkaline conditions. Therefore, the unprotonated [2Fe-2S]0 cluster is considerably less stable than its protonated counterpart. This finding is consistent with the lack of voltammetric signals at high pH {in contrast, the [2Fe-2S]2+/1+ couple can be observed even at pH 14 (18)}. Similar behavior was reported for the [3Fe-4S]2– cluster (3). The high-pH spectra (Fig. 6) could be fitted by using essentially the same subspectra as the low-pH spectra (Fig. 5A), with equal intensities for subspectra a and b, and values are reported in Table 1. The δ values at high pH are within 0.01 cm–1 of their corresponding low pH values, but all of the ΔEQ values have increased, suggesting small differences in the coordination symmetry (23). The increased linewidths probably reflect the mixture of states present at pH ≈ pK. No differences in δ or ΔEQ were observed between protonated and deprotonated states of the [3Fe-4S]0 cluster of Azotobacter vinelandii ferredoxin I, where the proton is thought to bind to a μ2-sulfide (31).
Fig. 6.

Zero-field Mössbauer spectrum of the (deprotonated) [2Fe-2S]0 Rieske cluster at pH 10, 4.2 K. The data were modeled by using Lorentzian doublets and the nested configuration, and subspectra a and b represent the cysteine- and histidine-coordinated sites, respectively. Subspectrum c is the product of cluster degradation.
The Site of Protonation. The reduction potential of the AA mutant is pH dependent at all observable values (pH 4.5–9, –58 mV per decade), and at pH 7 the reduction potential is equal to that of WT BtRp (–0.729 V vs. –0.730 V). Therefore, the proton does not bind to the disulfide. The behavior of mutants S163A and Y165F, however, is distinct from that of WT BtRp. S163 hydrogen bonds to the cluster μ2-sulfide S1, and Y165 hydrogen bonds to Sγ of the cluster ligand C139 (15). S163A and Y165F both have pH-dependent reduction potentials (–58 mV per decade) and at pH 7, where the [2Fe-2S]0 state is protonated, their reduction potentials are close to that of WT BtRp, –0.730 V and –0.770 V, respectively. However, the pK values of the [2Fe-2S]0 states are altered significantly, to outside the experimental range (pK ≥ 11, Ealk ≤ –1.0 V). This finding is consistent with the electrostatic influence of the hydrogen-bonding dipole, which increases the electron affinity of the cluster and decreases its proton affinity. At pH 7, reduction is electroneutral, and the two effects cancel. Importantly, this observation suggests strongly that the sites for reduction and protonation are spatially close together and inaccessible to solvent, indicating that the proton-binding site is on (or closely associated with) the cluster. Similar behavior was described in ref. 32 for the [3Fe-4S]1+/0 cluster in Azotobacter vinelandii ferredoxin I. The behavior of the BtRp [2Fe-2S]2+/1+ couple is strikingly different, because cluster reduction is coupled to deprotonation of the solvent-accessible and distant histidine ligands, so the S163A and Y165F mutations affect reduction potential much more than they affect pK (14).
Therefore, possible protonation sites are the cluster μ2-sulfides, as proposed for [3Fe-4S]0 and [3Fe-4S]2– clusters (3, 4), and the Sγ groups of the two cysteine ligands. Coordination of a ferrous heme iron by neutral cysteine has recently been demonstrated (33). Calculations of the protonation probabilities of all other titratable residues in the [2Fe-2S]1+ and [2Fe-2S]0 states (assuming no significant structural changes) revealed no significant dependence on redox state for any residue (20). This finding supports the proposal that the proton binds to the cluster core. Density functional theory calculations indicated that the geometries of the unprotonated [2Fe-2S]0 cluster, and each protonated cluster, were very similar to the geometry of the [2Fe-2S]1+ cluster (15).
Comparison of the calculated macroscopic pK values for each protonation site with the experimental results (Table 2) indicates clearly that the proton binds to a cluster μ2-sulfide, not to a cysteine Sγ. The calculated value for WT BtRp differs from the experimental value by only 1 pH unit, and, consistent with observed trends, calculated pK values for S163A and Y165F are higher. Consideration of the pK values in S163A and Y165F (to rule out the influence of the side-chain hydrogen bonds) shows that, as expected, the two sulfides and two cysteine Sγs are differentiated by the local environment. In WT BtRp, the side-chain hydrogen bonds from S163 and Y165 strongly disfavor protonation on S1 and C139, respectively, so calculations were performed for both the native conformation and the conformations with the hydrogen bonds broken. The free energy costs of breaking the hydrogen bonds were calculated to be equivalent to 2.1 (S163) and 5.6 (Y165) pK units (in the deprotonated state). Thus, in the absence of the hydrogen bond from S163, the free energy for protonation of S1 corresponds to a pK of 10.9, close to the value in S163A. Interestingly, some of the energy required to break the hydrogen bonds is offset by the formation of new interactions between the protons bound at S1 and C139 and the Oδ– atoms of S163 and Y165, respectively. The net result of the local environment and the hydrogen bonding is that the protonation of S1 is favored slightly over the protonation of S2. However, this difference is small compared with the clear preference for protonation on one of the sulfides, rather than on one of the cysteine ligands, which is maintained consistently.
Table 2. Calculated microscopic pK values for the addition of a single proton to the μ2-sulfides of the cluster and Sγ of the cysteine ligands and comparison of calculated macroscopic pK values with the experimental results.
| Calculated microscopic pK values
|
||||||
|---|---|---|---|---|---|---|
| Cysteine ligands
|
μ2-sulfides
|
Macroscopic pK values
|
||||
| Protein | C139 | C158 | S1 | S2 | Calculated | Experimental |
| WT | 2.1 | 0.5 | 8.8 | 7.5 | 8.9 | 9.77 |
| S163A | 2.0 | 1.3 | 10.5 | 8.3 | 10.5 | >11 |
| Y165F | 4.1 | 1.2 | 9.3 | 8.2 | 9.3 | >11 |
Discussion
The Reduction Potential of the [2Fe-2S]1+/0 Couple. In BtRp, the [2Fe-2S]1+/0 reduction potential is –0.73 V at pH 7, very close to the [3Fe-4S]0/2– potential, typically –0.70 V at pH 7 (3). It is also comparable to the lowest reported [4Fe-4S]2+/1+ potential (–0.72 V) (34) and to the potentials of cluster FX in type I reaction centers (approximately –0.70 V) (35) and the [4Fe-4S]1+/0 couple in the Fe-protein of nitrogenase (–0.79 V at pH 8) (6). However, the neutral and electronegative histidine ligands, the hydrogen bonds, and the control of solvent accessibility all combine to raise the [2Fe-2S]2+/1+ potentials of high-potential Rieske clusters (18), and therefore they influence the [2Fe-2S]1+/0 potential also. In an all-cysteine-ligated cluster, the [2Fe-2S]1+/0 potential is likely to be significantly lower. In BtRp, the pH-independent 2+/1+ and 1+/0 potentials (histidines always neutral, cluster always deprotonated) are separated by 1.2 V. In all-cysteine-ligated clusters the 2+/1+ couple occurs typically at –0.2 V, giving an estimated value of –1.4 V for the 1+/0 couple.
The BtRp cluster is unique, so far, in being able to adopt three oxidation states without using a coupled protonation or conformational change to compensate for the extra charge (see below). Therefore, it is not surprising that the separation between the two redox couples is so large. For the [3Fe-4S] cluster in S. acidocaldarius ferredoxin, the 1+/0 and 0/2–couples are separated by only 0.45 V at pH 7 (3), but the separation varies with pH, and the pK values of the 2–state are not known. Therefore, the free energy of protonation cannot be deconvoluted from that of electron transfer. In the Fe-protein, the most recent value for the [4Fe-4S]1+/0 potential (–0.79 V at pH 8) is only 0.48 V away from the 2+/1+ potential (6), but the Fe-protein is known to undergo significant conformational changes (8). The high-potential iron protein ([4Fe-4S]3+/2+) of Rhodopila globiformis can be reduced to the [4Fe-4S]1+ state, but the 2+/1+ potential is ill-defined because equilibrium is never attained, and reduction seems gated by an undefined conformational change (36). Finally, in the [8Fe-7S] P-cluster, the P2+/1+ and P1+/N potentials actually cross at pH ≈ 7.4 (10), but the cluster undergoes large redox-state-dependent structural changes (9), as well as protonation. Therefore, in FeS clusters, adjacent redox couples appear to become closer as the cluster nuclearity increases because of the more sophisticated methods of charge compensation that are adopted. Finally, note that the aquo-ions of first-row transition metals, such as Cr and Mn, exist in a wide range of formal oxidation states but that despite the polarity and high dielectric of water, no more than two oxidation states ever adopt the same coordination environment. Consequently, the 0.25-V separation between the proposed 2+/1+ and 1+/0 couples in model [2Fe-2S] compounds (11) is likely to include an unidentified coupled reaction (such as protonation or ion pairing) or to be because of ligand, not iron, reduction.
The Stability of the [2Fe-2S]0 Cluster. Despite significant contemporary interest, the factors that influence the stability of FeS clusters in their various oxidation states, and the reasons they degrade during oxidative stress, are not well understood. One may question the existence of all-ferrous clusters on the basis of Fe2+ being low in the Irving–Williams series. The Irving–Williams series suggests that the FeII–S2– bonds in [2Fe-2S]0 are weaker than the FeIII–S2– bonds in [2Fe-2S]2+, consistent with the weaker antiferromagnetic coupling. However, in aqueous solution, the relative stabilities are determined by the difference in free energy between [2Fe-2S]2+ and 2[Fe(H2O)6]3+, and [2Fe-2S]0 and 2[Fe(H2O)6]2+. Fe3+ is a “hard” acid, with a preference for O rather than S ligands, but Fe2+ is “softer” and more compatible with S. Therefore, all-ferrous clusters may be even less susceptible to degradation than all-ferric clusters. Indeed, stable all-ferrous [3Fe-4S], [4Fe-4S], [7Fe-8S] (P), and [2Fe-2S] clusters have been reported, and the highly oxidized [4Fe-4S]3+ clusters in high-potential iron proteins are stable only if protected from solvent (37). Note, however, that these arguments pertain only to the thermodynamic driving force for cluster degradation, not to its rate. Little is known about the fluxionality of FeS clusters, but chalcogenide exchange has been observed in several cases (1), suggesting that, to some extent at least, exchange occurs between the cluster and solution.
In BtRp, the unprotonated [2Fe-2S]0 cluster does not persist in solution, but it is stabilized significantly by protonation, probably on one of the μ2-sulfides. It is unlikely that bonding interactions within the [2Fe-2S]0 core are strengthened by protonation (for example, antiferromagnetic coupling in [2Fe-2(SR)] is weaker than in [2Fe-2S]) (25). Instead, it is likely that the surrounding protein structure is destabilized by the addition of two electrons to the cluster, without charge compensation, but that two electrons and a proton can be accommodated. For example, the hydrogen-bonding network may be disrupted, the orientation of backbone dipoles altered, or the solvent structure changed. Note that [4Fe-4S]2+/1+ clusters disassemble when oxidized to the 3+ (or 4+) level but that a stable [3Fe-4S]1+ cluster of equal charge may be formed also, by expulsion of positive charge as FeII (38).
Protonation of the [2Fe-2S]0 Cluster. The evidence presented here suggests strongly that the [2Fe-2S]0 cluster is protonated on one of its μ2-sulfides, not on a cysteine Sγ. Therefore, the μ2-sulfides of [2Fe-2S] and [3Fe-4S] clusters distinguish them from [4Fe-4S] clusters, which contain only μ3-sulfides, by allowing them to compensate for reduction by direct protonation of the cluster core. Cluster basicity increases upon reduction, so that [3Fe-4S]1+ clusters are not protonated, but one and three protons are bound in the 0 and 2–states, respectively (3, 4); the basicity of the [3Fe-4S] core may be enhanced by the tridentate “crown” of sulfide ligands. In [2Fe-2S] clusters, the sulfide lone pairs point away from one another, so the cluster must be reduced to the all-ferrous state to exhibit a quantifiable proton affinity.
Acknowledgments
We thank Prof. F. A. Armstrong and Dr. K. A. Vincent, Oxford University (Oxford), for advice on EuII-DTPA and Bernd Mienert for technical help with the Mössbauer spectroscopy. This work was supported by the Medical Research Council. E.B. was supported by the Max-Planck-Gesellschaft, G.M.U. was supported by Deutsche Forschungsgemeinschaft Grant 174/2-1,2, and T.E. was supported by the Klaus-Tschira Foundation. Computations were carried out on the Heidelberg Linux Cluster System, HELICS (Interdisziplinäres Zentrum für Wissenschaftliches Rechnen, Heidelberg).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BtRp, overexpressed soluble Rieske protein from the bovine cytochrome bc1 complex; DTPA, diethylenetriamine-N,N,N′,N″,N′″-pentaacetate; FeS, iron–sulfur.
References
- 1.Beinert, H., Holm, R. H. & Münck, E. (1997) Science 277, 653–659. [DOI] [PubMed] [Google Scholar]
- 2.Rees, D. C. & Howard, J. B. (2003) Science 300, 929–931. [DOI] [PubMed] [Google Scholar]
- 3.Duff, J. L. C., Breton, J. L. J., Butt, J. N., Armstrong, F. A. & Thomson, A. J. (1996) J. Am. Chem. Soc. 118, 8593–8603. [Google Scholar]
- 4.Shen, B., Martin, L. L., Butt, J. N., Armstrong, F. A., Stout, C. D., Jensen, G. M., Stephens, P. J., La Mar, G. N., Gorst, C. M. & Burgess, B. K. (1993) J. Biol. Chem. 268, 25928–25939. [PubMed] [Google Scholar]
- 5.Butt, J. N., Armstrong, F. A., Breton, J., George, S. J., Thomson, A. J. & Hatchikian, E. C. (1991) J. Am. Chem. Soc. 113, 6663–6670. [Google Scholar]
- 6.Guo, M., Sulc, F., Ribbe, M. W., Farmer, P. J. & Burgess, B. K. (2002) J. Am. Chem. Soc. 124, 12100–12101. [DOI] [PubMed] [Google Scholar]
- 7.Strop, P., Takahara, P. M., Chiu, H.-J., Angove, H. C., Burgess, B. K. & Rees, D. C. (2001) Biochemistry 40, 651–656. [DOI] [PubMed] [Google Scholar]
- 8.Burgess, B. K. & Lowe, D. J. (1996) Chem. Rev. 96, 2983–3011. [DOI] [PubMed] [Google Scholar]
- 9.Peters, J. W., Stowell, M. H. B., Soltis, S. M., Finnegan, M. G., Johnson, M. K. & Rees, D. C. (1997) Biochemistry 36, 1181–1187. [DOI] [PubMed] [Google Scholar]
- 10.Lanzilotta, W. N., Christiansen, J., Dean, D. R. & Seefeldt, L. C. (1998) Biochemistry 37, 11376–11384. [DOI] [PubMed] [Google Scholar]
- 11.Mayerle, J. J., Frankel, R. B., Holm, R. H., Ibers, J. A., Phillips, W. D. & Weiher, J. F. (1973) Proc. Natl. Acad. Sci. USA 70, 2429–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Im, S.-C., Kohzuma, T., McFarlane, W., Gaillard, J. & Sykes, A. G. (1997) Inorg. Chem. 36, 1388–1396. [DOI] [PubMed] [Google Scholar]
- 13.Verhagen, M. F. J. M., Link, T. A. & Hagen, W. R. (1995) FEBS Lett. 361, 75–78. [DOI] [PubMed] [Google Scholar]
- 14.Leggate, E. J. (2003) Ph.D. thesis (Cambridge University, Cambridge, U.K.).
- 15.Iwata, S., Saynovits, M., Link, T. A. & Michel, H. (1996) Structure 4, 567–579. [DOI] [PubMed] [Google Scholar]
- 16.Link, T. A. (1999) Adv. Inorg. Chem. 47, 83–157. [Google Scholar]
- 17.Zu, Y., Fee, J. A. & Hirst, J. (2002) Biochemistry 41, 14054–14065. [DOI] [PubMed] [Google Scholar]
- 18.Zu, Y., Couture, M. M.-J., Kolling, D. R. J., Crofts, A. R., Eltis, L. D., Fee, J. A. & Hirst, J. (2003) Biochemistry 42, 12400–12408. [DOI] [PubMed] [Google Scholar]
- 19.Vincent, K. A., Tilley, G. J., Quammie, N. C., Streeter, I., Burgess, B. K., Cheesman, M. R. & Armstrong, F. A. (2003) Chem. Commun. 20, 2590–2591. [DOI] [PubMed] [Google Scholar]
- 20.Ullmann, G. M., Noodleman, L. & Case, D. A. (2002) J. Biol. Inorg. Chem. 7, 632–639. [DOI] [PubMed] [Google Scholar]
- 21.Zu, Y., di Bernardo, S., Yagi, T. & Hirst, J. (2002) Biochemistry 41, 10056–10069. [DOI] [PubMed] [Google Scholar]
- 22.Angove, H. C., Yoo, S. J., Burgess, B. K. & Münck, E. (1997) J. Am. Chem. Soc. 119, 8730–8731. [Google Scholar]
- 23.Gütlich, P. (1975) in Mössbauer Spectroscopy, ed. Gonser, U. (Springer, Berlin), Vol. 5, pp 53–96. [Google Scholar]
- 24.Fee, J. A., Findling, K. L., Yoshida, T., Hille, R., Tarr, G. E., Hearshen, D. O., Dunham, W. R., Day, E. P., Kent, T. A. & Münck, E. (1984) J. Biol. Chem. 259, 124–133. [PubMed] [Google Scholar]
- 25.Trautwein, A. X., Bill, E., Bominaar, E. L. & Winkler, H. (1991) Struct. Bonding 78, 1–95. [Google Scholar]
- 26.Mouesca, J.-M. & Lamotte, B. (1998) Coord. Chem. Rev. 178–180, 1573–1614. [Google Scholar]
- 27.Yoo, S. J., Meyer, J. & Münck, E. (1999) J. Am. Chem. Soc. 121, 10450–10451. [Google Scholar]
- 28.Schulz, C. & Debrunner, P. G. (1976) J. Phys. Colloque 12, 153–158. [Google Scholar]
- 29.Palmer, G., Dunham, W. R., Fee, J. A., Sands, R. H., Iizuka, T. & Yonetani, T. (1971) Biochim. Biophys. Acta 245, 201–207. [DOI] [PubMed] [Google Scholar]
- 30.Bertrand, P., Gayda, J.-P., Fee, J. A., Kuila, D. & Cammack, R. (1987) Biochim. Biophys. Acta 916, 24–28. [DOI] [PubMed] [Google Scholar]
- 31.Hu, Z., Jollie, D., Burgess, B. K., Stephens, P. J. & Münck, E. (1994) Biochemistry 33, 14475–14485. [DOI] [PubMed] [Google Scholar]
- 32.Camba, R., Jung, Y.-S., Hunsicker-Wang, L. M., Burgess, B. K., Stout, C. D., Hirst, J. & Armstrong, F. A. (2003) Biochemistry 42, 10589–10599. [DOI] [PubMed] [Google Scholar]
- 33.Perera, R., Sono, M., Sigman, J. A., Pfister, T. D., Lu, Y. & Dawson, J. H. (2003) Proc. Natl. Acad. Sci. USA 100, 3641–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macedo, A. L., Besson, S., Moreno, C., Fauque, G., Moura, J. J. G. & Moura, I. (1996) Biochem. Biophys. Res. Commun. 229, 524–530. [DOI] [PubMed] [Google Scholar]
- 35.Vassiliev, I. R., Antonkine, M. L. & Golbeck, J. H. (2001) Biochim. Biophys. Acta 1507, 139–160. [DOI] [PubMed] [Google Scholar]
- 36.Heering, H. A., Bulsink, Y. B. M., Hagen, W. R. & Meyer, T. E. (1995) Eur. J. Biochem. 232, 811–817. [PubMed] [Google Scholar]
- 37.Agarwal, A., Li, D. & Cowan, J. A. (1995) Proc. Natl. Acad. Sci. USA 92, 9440–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camba, R. & Armstrong, F. A. (2000) Biochemistry 39, 10587–10598. [DOI] [PubMed] [Google Scholar]