

Abstract
15N-1H NMR spectroscopy has been used to probe the dynamic properties of uniformly 15N labeled Escherichia coli ribosomes. Despite the high molecular weight of the complex (≈2.3 MDa), [1H-15N] heteronuclear single-quantum correlation spectra contain ≈100 well resolved resonances, the majority of which arise from two of the four C-terminal domains of the stalk proteins, L7/L12. Heteronuclear pulse-field gradient NMR experiments show that the resonances arise from species with a translational diffusion constant consistent with that of the intact ribosome. Longitudinal relaxation time (T1) and T1ρ 15N-spin relaxation measurements show that the observable domains tumble anisotropically, with an apparent rotational correlation time significantly longer than that expected for a free L7/L12 domain but much shorter than expected for a protein rigidly incorporated within the ribosomal particle. The relaxation data allow the ribosomally bound C-terminal domains to be oriented relative to the rotational diffusion tensor. Binding of elongation factor G to the ribosome results in the disappearance of the resonances of the L7/L12 domains, indicating a dramatic reduction in their mobility. This result is in agreement with cryoelectron microscopy studies showing that the ribosomal stalk assumes a single rigid orientation upon elongation factor G binding. As well as providing information about the dynamical properties of L7/L12, these results demonstrate the utility of heteronuclear NMR in the study of mobile regions of large biological complexes and form the basis for further NMR studies of functional ribosomal complexes in the context of protein synthesis.
Protein synthesis in living systems takes place on the ribosome, a complex macromolecular assembly whose structural and functional properties are rapidly emerging from a powerful combination of electron microscopy (EM) and x-ray crystallography (1, 2). In Escherichia coli, the ribosome is composed of 54 different proteins and three RNA molecules (23S, 16S, and 5S rRNA) This 2.3-MDa complex is termed the 70S ribosome and is made up of two components, the 30S and 50S subunits. The translation of genetic information into functional proteins involves a number of auxiliary factors, many of which are GTPases, including IF2, EF-Tu, elongation factor G (EF-G), and RF3 (2). These molecules bind to overlapping sites on the 50S subunit and regulate the transition of the ribosome through various states on the translational pathway. The binding sites are collectively known as the GTPase-associated region, due to the role of this region in stimulating the GTPase activity of the auxiliary factors.
The GTPase-associated region (GAR) includes helices 42–44 and 95 (the α-sarcin loop) of 23S RNA, and the proteins L10, L11, and L7/L12 (1). The latter (L7/L12) is located on the ribosomal stalk and is unique among the ribosomal proteins, because it is the only protein present in multiple copies (four proteins per ribosomal particle). Although atomic details of much of the GAR have been revealed in the crystal structures of the 50S subunit (3, 4), regions such as the L7/L12 stalk give only fragmented or weak density in cryo-EM (5) and x-ray diffraction maps. In the 5.5-Å 70S structure (6) (the most complete structure available for the stalk region), for example, the density for L10 is absent; only two of the four copies of L7/L12 have been identified in this structure (6) and are tentatively placed at the base of the stalk in a compact conformation. The remaining two copies of L7/L12 are believed to form the stalk; their absence in high-resolution structures may result from the fact that they do not occupy well defined positions within the individual ribosomes.
The L7/L12 molecules have no apparent contact with rRNA and are known to undergo conformational changes at different steps of the translational cycle (7, 8). 1H NMR spectroscopy on isolated 50S and 70S ribosomes has provided evidence for regions of local motional averaging, revealing several narrow resonances that have been attributed to L7/L12 (9, 10). 15N NMR spectroscopy of isolated L7/L12 proteins shows two apparently independent structured domains linked by a flexible hinge, with the nuclear Overhauser effect patterns indicating an N-terminal domain (NTD) dimer independent of the respective C-terminal domain (CTD) (11). Similar studies of a pentameric complex (the four copies of L7/L12 along with L10) show that the binding of L10 to the NTD of L7/L12 causes the disappearance of the resonances of the latter (12). The implied flexibility of L7/L12 and possible significance of this dynamic behavior for ribosomal function make detailed study of this region of the complex of interest.
We describe here a heteronuclear NMR investigation of intact ribosomal particles that permits the analysis of the structure and dynamics of bound L7/L12 proteins and points the way to future NMR studies of ribosomal structure and function.
Materials and Methods
Preparation of E. coli Ribosomes. Ribosomal particles were prepared from E. coli MRE600 cells (CDN, Tallinn, Estonia) grown in minimal medium containing 15NH4Cl. Further preparation details, quality controls, and concentration measurements of ribosomal particles are described in Supporting Text, which is published as supporting information on the PNAS web site, as is the preparation of EF-G.
Sample Preparation and NMR Spectroscopy. Characterization of NMR samples. The concentration of 70S ribosomes in samples used for the NMR experiments was 15–25 μM, that of the 50S subunit was 25–40 μM, and that of the 30S was 15–35 μM. Unless otherwise stated, samples were made up in buffer solutions containing 10 mM KHPO4, 100 mM KCl, and 10 mM magnesium acetate, pH 7.5, with 10% D2O. Samples were generally used for no more than 12 h, because some deterioration in spectral quality was evident after 16–24 h. Control heteronuclear single-quantum correlation spectra (HSQCs) were recorded before and after long accumulations to ascertain sample integrity.
NMR experiments used to measure diffusion rates, as well as 15N-spin relaxation experiments and fast-HSQC (13) experiments, were performed at 298 K on a 600 MHz Bruker (Karlsruhe, Germany) DRX spectrometer equipped with a (1H/13C/15N) cryoprobe with a single-axis pulsed-field gradient capability. [1H-15N] HSQC experiments were additionally recorded at 750 and 700 MHz (Bruker Avance) by using conventional TXI probes. [1H, 15N] correlated spectra were collected with 2,048 (t2) and 80–100 (t1) complex points with 16–40 scans per t1 increment unless otherwise stated.
Pulsed-field gradient-diffusion measurements. These experiments (14) were performed by using a modification of the published heteronuclear multiple quantum correlation-type pulse sequence (15). Pulse lengths and delays were kept constant, and z-gradient pulses were incremented in each series of experiments from 5% to 90% of their maximum value (the maximum z-gradient was 50 G/cm) in tailored steps. Further details are available in Supporting Text.
Experiments were carried out with 15N labeled 70S and 50S ribosomes and with selectively 15N labeled low-molecular weight proteins, carbonic anhydrase (CA) (≈30 kDa) and the regulatory domains of calcium-dependent protein kinase (≈20 kDa), for calibration of the relationship of the diffusion coefficient, D, with the molecular weight, M. 2D diffusion spectra were typically collected with 120–160 scans per t1 increment.
15N relaxation experiments. Longitudinal 15N spin relaxation rates (R1) were measured by using a standard pulse sequence (16), and transverse relaxation rates (R2) were extracted from R1ρ (17, 18) experiments modified to incorporate a WATERGATE (19). Experimental details are described in Supporting Text.
Data sets were processed by using xwinnmr (Bruker), felix2000 (Accelrys, San Diego), nmrpipe, and nmrdraw (20), and peak analysis was performed by using nmrview (21), matlab (Math-Works, Natick, MA), and kaleidagraph (Synergy Software, Reading, PA).
Results
HSQC Spectra of the Ribosome and Ribosomal Subunits. The [1H-15N] HSQC spectrum of 15N labeled E. coli 70S ribosomes is shown in Fig. 1A. Approximately 100 well resolved amide crosspeaks are observed, despite the fact that the overall rotational correlation time of an intact complex of the size of the ribosome is expected to be ≈1,000 ns. Such a value of the correlation time would give rise to NMR linewidths in excess of 1,000 Hz and hence to unresolvable resonances. Because the ribosome contains ≈7,500-aa residues, the appearance of a set of narrow well resolved signals indicates that they stem from a small region or regions of the structure with substantial motion independent of that of the ribosome itself.
Fig. 1.

[1H-15N] HSQC spectra of uniformly 15N labeled E. coli ribosomes. Spectra of (A) intact 70S; (B) 50S; (C) 30S ribosomes; and (D) overlay of spectra of the 50S and 30S subunits, acquired at 750 MHz in 10 mM KCl/10 mM MgCl2/10 mM KHPO4, pH 7.2, 303 K. Peaks in B are labeled according to the assignments of isolated L7/L12 protein, because the chemical shift values are essentially identical [the differences are less than ± 0.02 (1H) and ±0.1 (15N) ppm (11)]. Crosspeaks likely to originate from A39, V40, and A41 in the interdomain hinge are labeled with an asterisk. The deviations of the chemical shifts of these signals from those of isolated L7/L12 are greater than for other labeled peaks, and multiple resonances are assigned for these residues in the isolated protein.
Similar HSQC experiments carried out on the 50S and 30S subunits (Fig. 1 B and C) show that the majority (≈80) of the crosspeaks in the 70S ribosome spectrum overlay closely with signals from the 50S subunit, whereas the remaining (≈20) crosspeaks correlate closely with resonances in the spectrum of the 30S subunit (Fig. 1 C and D). The crosspeaks that are attributable to residues in the 30S subunit have limited dispersion and are largely located in the central region of the amide spectrum in a manner characteristic of disordered regions of proteins (Fig. 1C); by contrast, the crosspeaks from the 50S subunit are typical of those of a highly structured protein. Examination of the HSQC spectra in more detail indicates that the large majority of the observable resonances from the 50S correspond to those reported for the interdomain hinge and the CTD (residues 43–120) of free L7/L12 (11) (Fig. 1B). This finding is consistent with the appearance of several narrow resonances that were attributed to these proteins in the 1D 1H NMR spectrum of the ribosome (9) and indicates that at least one of the four copies of L7/L12 has substantial motional freedom within the ribosomal complex.
To ensure that the resonances observed in the HSQC spectrum originate from L7/L12 proteins incorporated into the ribosomal structure and not from any fragmented or dissociated species, the ribosomes were purified by using a sucrose gradient before and after recording the NMR spectra. The sucrose gradient profile indicated that no detectable changes in the composition of the particles occurred during the time used to record the NMR spectra. Furthermore, 2D gel electrophoresis confirmed that L7/L12 cosedimented with the ribosome, showing that they were tightly bound. 1D gel electrophoresis of the sample supernatants after centrifugation to pellet the ribosomal particles showed no detectable quantities of free L7/L12 either before or after recording the spectra. These experiments indicate that the maximum free concentration of L7/L12 present in the NMR sample must have been <0.2 μM (4 pmol/20 μl), which would not be detectable in the HSQC experiments. As an additional control, the activity of the 70S ribosomes sedimented after the experiment was measured by using a Poly(Phe) synthesis assay; <10% of their activity was lost over the course (1–12 h) of the NMR experiments (see Supporting Text). Because Poly(Phe) synthesis depends on the presence on the ribosome of L7/L12, the large majority of the ribosomes must retain their complete integrity during the experiments.
Analysis of the Resonances Observed for L7/L12. The linewidths of the L7/L12 resonances are inconsistent with the correlation times expected for the overall tumbling of these particles. To probe the origin of the narrow lines observed in HSQC spectra of 50S and 70S ribosomes, the translational diffusion properties of the species giving rise to the observed resonances were examined by using pulsed-field gradient experiments as described in Materials and Methods. Approximately 25 well resolved resonances of L7/L12 in the 50S and 70S ribosomal particles were analyzed from data sets (collected for two independent samples in each case) with four gradient strengths between 2.5 and 45 G/cm. To obtain absolute values for molecular masses, 15N labeled samples of proteins known to be monomeric in solution, CA and calcium-dependent protein kinase were used to calibrate the gradient strength by using the same pulse sequence. Unlabeled bovine pancreatic tryptic inhibitor and BSA were also used as calibrants by using a homonuclear version of the same pulse sequence. To eliminate the effects of the increased viscosity of the solutions containing ribosomes on the translational diffusion coefficient, DT, the diffusion properties of CA (25 μM) added to a sample of the 50S ribosome were measured.
The relative diffusion constants obtained from these experiments are shown in Fig. 2. It is clear that L7/L12 in the ribosomal particles diffuses much more slowly than either of the isolated monomeric proteins. Although the errors in the diffusion constants are large because of the small diffusion constants, the diffusion constants are clearly indicative of particles in excess of 1 MDa. The resonances observed in the HSQC spectra therefore arise from L7/L12 molecules attached to the ribosomes.
Fig. 2.

Pulsed-field gradient–NMR characterization of the intact ribosome. Relative diffusion coefficients of the 70S and 50S ribosomes and of the small monomeric proteins calcium-dependent protein kinase and CA. The data for CA-2 show the reduced diffusion resulting from CA being added to a solution containing the 50S subunit (CA-1 corresponds to measurements with the isolated protein). Assuming constant densities and spherical particles, then M1/M2 = (D2/D1)3 (where M1 and M2 are the molecular masses, and D1 and D2 are the diffusion coefficients of proteins 1 and 2, respectively). These values provide an estimate of the apparent molecular mass for the ribosomal particles in excess of 1 MDa.
The peaks observed in the HSQC spectra of the 70S particle and the 50S subunit overlay well with those reported for the isolated L7/L12 proteins, and the chemical shift and nuclear Overhauser effect pattern analysis of the latter (11) can be extended to residues 43–120 of the ribosomally bound proteins. The highly structured region, residues 54–120, is consistent with the compact structure of three β-strands and three α-helices reported previously (22). Residues 42–53 appear to adopt highly disordered conformations consistent with their location in the linker region between the NTD and the CTD. No resonances of the N-terminal residues 1–38 are detectable, indicating that this region of the protein is not undergoing fast motional averaging.
Fast HSQC experiments were used to investigate the number of copies of the L7/L12 domain. These experiments yield H,N crosspeaks that can be integrated readily because nonuniform signal losses that result from saturation transfer from water are reduced (see also Supporting Text). Initially, these experiments were performed on a 50 μM sample of a small soluble protein 15N labeled titin-I27. Approximately 40 well resolved peaks were integrated and showed remarkably uniform integrals (the standard deviation was 10%). The experiment was then carried out on a sample of the 50S subunit (19.1 μM) containing 15N labeled titin (41.4 μM), and repeated an additional three times. Analysis of the L7/L12 resonances of the hinge region (residues 43–51) in the fast HSQC experiment indicates that the intensities of the visible L7/L12 peak correspond to 1.9 ± 0.5 molecules per ribosome particle. Comparison of the integrals of the resonances of the structured part of L7/L12 (residues 52–120) with the signals of titin indicates, however, that the visible intensity corresponds to 1.1 ± 0.3 L7/L12 molecules per ribosomal particle. We conclude that a minimum of one and maximum of two of the four copies of the L7/L12 CTD experience the fast dynamical averaging necessary to give narrow resonances in the HSQC spectrum of the ribosome.
Although the relaxation rates (described below) of the hinge and the structured regions of the observable L7/L12 proteins differ significantly (average T2 of 80 ms vs. 40 ms, respectively) as a result of their different dynamical properties, this phenomenon is unlikely to account for the differing peak integrals (see Supporting Text). It is possible that one of the CTD regions experiences an exchange contribution to its T2 relaxation, for example from interactions with other regions of the ribosome, giving rise to broadening of one of the sets of resonances of the CTD but not of those from the associated more flexible hinge region (see below). Regardless of the origins of the differing intensities, however, we can conclude that at least two copies of L7/L12 are constrained within the main body of the ribosome, a result that is in line with the electron densities for two L7/L12 copies in the x-ray structure of the 70S (6), but that significant regions of at least two copies of the CTD are undergoing extensive independent motion.
Interaction of the Ribosome with EF-G. The region of the ribosome in which the L7/L12 proteins are located undergoes substantial conformational changes during the various stages of the elongation cycle (23–26). We therefore investigated the effects on the NMR spectra of the presence of added EF-G. A series of spectra were obtained with differing molecular equivalent (mol. eq.) of EF-G added to the 70S. The spectra show that increases in the concentration of EF-G give rise to a progressive reduction in the intensities of the L7/L12 peaks. At EF-G levels above 0.1 mol. eq. only the intense resonances of the amino acids in the hinge region (res. 45–50) of L7/L12 are visible. At the highest concentrations of EF-G (4 mol. eq.) no L7/L12 signals could be observed (in both the presence and absence of fusidic acid). This reduction in the intensities of residues in the structured region of the CTD at substoichiometric concentrations of EF-G may result from further line-broadening of the type described in the previous paragraph that results from exchange between the mobile state of the L7/L12 molecules and an immobilized state as a result of the interaction of the CTD with the ribosomally associated EF-G. That the linker and CTD disappear at different stoichiometric ratios is consistent with the differing dynamical behavior of the linker and the structured regions of the CTD that is evident in the NMR spectra of the ribosome in the absence of EF-G (see above). These results are consistent with the previous 1H NMR study where narrow signals attributed to the side chains of residues in L7/L12 disappeared when EF-G was added (10). The experiments therefore suggest that the interaction of the ribosome with EF-G causes a loss of the rapid independent motion of L7/L12 that is present in the absence of EF-G.
Dynamics of the L7/L12 Proteins on the Ribosome. 15N spin relaxation parameters of proteins contain information about the overall rotational correlation time (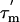 ) and the amplitude of internal motions (S2) of N-HN vectors. In addition, if tumbling is anisotropic, the relaxation data also contain information about the orientation of each N-HN vector relative to the long axis of the diffusion tensor. To explore the motional behavior of L7/L12, R1 and R1ρ 15N-spin relaxation rates were measured for the L7/L12 resonances observed in the spectra of 50S subunits. The relatively sharp peaks in the [1H-15N] HSQC spectrum of the 50S subunit (Fig. 1B) suggest that the CTD of L7/L12 tumbles almost independently of the ribosome. The relaxation data were analyzed assuming that the CTD of L7/L12 diffuses with its own apparent rotational correlation time (
) and the amplitude of internal motions (S2) of N-HN vectors. In addition, if tumbling is anisotropic, the relaxation data also contain information about the orientation of each N-HN vector relative to the long axis of the diffusion tensor. To explore the motional behavior of L7/L12, R1 and R1ρ 15N-spin relaxation rates were measured for the L7/L12 resonances observed in the spectra of 50S subunits. The relatively sharp peaks in the [1H-15N] HSQC spectrum of the 50S subunit (Fig. 1B) suggest that the CTD of L7/L12 tumbles almost independently of the ribosome. The relaxation data were analyzed assuming that the CTD of L7/L12 diffuses with its own apparent rotational correlation time (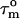 ). R2 rates were extracted from the R1ρ measurements (as described in Supporting Text). The measured longitudinal relaxation time (T1) and transverse relaxation time (T2) values for 71 L7/L12 residues are shown in Figs. 3 A and B.
). R2 rates were extracted from the R1ρ measurements (as described in Supporting Text). The measured longitudinal relaxation time (T1) and transverse relaxation time (T2) values for 71 L7/L12 residues are shown in Figs. 3 A and B.
Fig. 3.

15N-spin relaxation properties of ribosomal L7/L12. (A) T1 and (B) T2 relaxation times of L7/L12 incorporated in the 50S subunit. T2 values are derived from the R1ρ experiments, as detailed in Supporting Text. Average errors, 8% and 12%, for the T1 and T2 relaxation times, respectively, are relatively large through low S/N ratios resulting from the need to minimize accumulation times to avoid sample degradation (see text). Residues removed by the R1/R2 filter (see Results) are marked x at the bottom. (C) Values of the apparent rotational correlation time 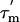 measured from the relaxation times. Errors in
measured from the relaxation times. Errors in 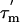 values are ≈±1.4 ns, based on the average errors in the T1 and T2 values given above; error bars for
values are ≈±1.4 ns, based on the average errors in the T1 and T2 values given above; error bars for 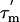 have, however, been omitted for clarity. Average
have, however, been omitted for clarity. Average 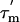 values for the α-helices are indicated with thick lines and their standard deviations as thin lines above and below each helical region. (D) Order parameters, S2 (based on an average
values for the α-helices are indicated with thick lines and their standard deviations as thin lines above and below each helical region. (D) Order parameters, S2 (based on an average 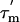 of 13.6 ns). Values for residues 90, 100, 106, and 107 are omitted, because they are affected by conformational exchange.
of 13.6 ns). Values for residues 90, 100, 106, and 107 are omitted, because they are affected by conformational exchange.
A reliable estimate of the overall rotational correlation time, 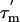 , is required for the further analysis of the relaxation data; we estimated the apparent overall rotational correlation time,
, is required for the further analysis of the relaxation data; we estimated the apparent overall rotational correlation time, 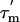 , from the ratio of the R2 and R1 relaxation rates (16, 27). Because this estimate is only valid if the internal motion is not coupled to the overall tumbling, it is necessary to identify any residues that are affected by internal motions, slower than ≈200 ps, and remove them from the analysis. A filtering method was used to exclude L7/L12 residues experiencing such slow motions by selecting residues with R1 and/or R2 values that deviate significantly from the average values (28); data for 16 residues were excluded on this basis (Fig. 3). The apparent rotational correlation times,
, from the ratio of the R2 and R1 relaxation rates (16, 27). Because this estimate is only valid if the internal motion is not coupled to the overall tumbling, it is necessary to identify any residues that are affected by internal motions, slower than ≈200 ps, and remove them from the analysis. A filtering method was used to exclude L7/L12 residues experiencing such slow motions by selecting residues with R1 and/or R2 values that deviate significantly from the average values (28); data for 16 residues were excluded on this basis (Fig. 3). The apparent rotational correlation times, 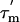 , for the 57 remaining residues, calculated from their R1 and R2 rates, are shown in Fig. 3C.
, for the 57 remaining residues, calculated from their R1 and R2 rates, are shown in Fig. 3C.
When the overall rotational correlation time, 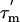 is longer than ≈6 ns, as is the case here, the R2 relaxation rate is to a first approximation independent of the internal rotational correlation time τi. The apparent R2 relaxation rate thus depends primarily on
is longer than ≈6 ns, as is the case here, the R2 relaxation rate is to a first approximation independent of the internal rotational correlation time τi. The apparent R2 relaxation rate thus depends primarily on 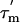 , the order parameter S2, and the conformational exchange rate, Rex (27, 29). When Rex = 0, R2 is proportional to the product of S2 and
, the order parameter S2, and the conformational exchange rate, Rex (27, 29). When Rex = 0, R2 is proportional to the product of S2 and 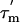 (27); the order parameter can then be estimated from the
(27); the order parameter can then be estimated from the 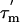 and R2 data alone. In Fig. 3D, the S2 values for L7/L12 calculated in this way, using a
and R2 data alone. In Fig. 3D, the S2 values for L7/L12 calculated in this way, using a 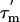 of 13.6 ns (the average of the values shown in Fig. 3C), are shown for all residues. Residues in the linker region (43–51) display low S2 values, (average 0.35 ± 0.04), implying a high degree of internal flexibility. Residues 52–120, however, have an average S2 of 0.75 ± 0.1, a value characteristic of highly structured protein domains (30).
of 13.6 ns (the average of the values shown in Fig. 3C), are shown for all residues. Residues in the linker region (43–51) display low S2 values, (average 0.35 ± 0.04), implying a high degree of internal flexibility. Residues 52–120, however, have an average S2 of 0.75 ± 0.1, a value characteristic of highly structured protein domains (30).
The 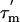 values (Fig. 3C) show significant variation along the protein sequence, suggesting that the tumbling of L7/L12 is anisotropic. From the maximum and minimum values of
values (Fig. 3C) show significant variation along the protein sequence, suggesting that the tumbling of L7/L12 is anisotropic. From the maximum and minimum values of 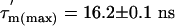 ,
, 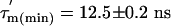 and
and 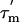 (the average and the standard deviation of the five residues with the longest and shortest
(the average and the standard deviation of the five residues with the longest and shortest 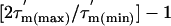 values, respectively), the apparent rotational anisotropy (
values, respectively), the apparent rotational anisotropy (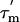 ) was calculated (27) to be 1.6 ± 0.1. This can be compared with the rotational anisotropy of 1.4 estimated (31), from the x-ray structure of an isolated CTD (res 52–120) of L7/L12 (22).
) was calculated (27) to be 1.6 ± 0.1. This can be compared with the rotational anisotropy of 1.4 estimated (31), from the x-ray structure of an isolated CTD (res 52–120) of L7/L12 (22).
The 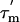 value for a given N-HN vector in a molecule with a small degree of anisotropy depends on the angle φ between the relative orientation of the N-HN vector and the principal axis of the diffusion tensor (27) (see Supporting Text). Further analysis can be carried out by recognizing also that all N-HN vectors in an α-helix are parallel (within ±15°) to the helical axis. Therefore, the N-HN vectors in a rigid α-helix will have similar
value for a given N-HN vector in a molecule with a small degree of anisotropy depends on the angle φ between the relative orientation of the N-HN vector and the principal axis of the diffusion tensor (27) (see Supporting Text). Further analysis can be carried out by recognizing also that all N-HN vectors in an α-helix are parallel (within ±15°) to the helical axis. Therefore, the N-HN vectors in a rigid α-helix will have similar 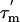 values, and the average value can be used to define the angle φ that each α-helix makes relative to the principal axis of the diffusion tensor. The average values of φ (and in parentheses,
values, and the average value can be used to define the angle φ that each α-helix makes relative to the principal axis of the diffusion tensor. The average values of φ (and in parentheses, 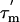 ) for each α-helix in L7/L12 calculated in this way are: helix I, 53°±14° (13.6 ± 0.7 ns); helix II, 53°±10° (13.6 ± 0.6 ns); and helix III, 23°±23° (15.4 ± 0.8 ns). To examine whether these angles define a unique preferred orientation of L7/L12 relative to the ribosome a grid search method was used; a database of angles defining each orientation of the L7/L12 CTD and the angles of the α-helices relative to the diffusion tensor was created, and systematically analyzed (further details in Supporting Text). A single unique value of the orientation tensor was found, where the helical angles are 66°,63°, and 32° for helix I, II, and III, respectively (Fig. 4A). These values are within the error margins of the helical angles derived from experiment and are therefore consistent with the average orientation of the CTD of L7/L12 bound to the ribosome.
) for each α-helix in L7/L12 calculated in this way are: helix I, 53°±14° (13.6 ± 0.7 ns); helix II, 53°±10° (13.6 ± 0.6 ns); and helix III, 23°±23° (15.4 ± 0.8 ns). To examine whether these angles define a unique preferred orientation of L7/L12 relative to the ribosome a grid search method was used; a database of angles defining each orientation of the L7/L12 CTD and the angles of the α-helices relative to the diffusion tensor was created, and systematically analyzed (further details in Supporting Text). A single unique value of the orientation tensor was found, where the helical angles are 66°,63°, and 32° for helix I, II, and III, respectively (Fig. 4A). These values are within the error margins of the helical angles derived from experiment and are therefore consistent with the average orientation of the CTD of L7/L12 bound to the ribosome.
Fig. 4.

Representation of the orientation of L7/L12 on the ribosome. (A) Orientation of the CTD of an L7/L12 protein derived from rotational anisotropy analysis of relaxation data. L7/L12 is positioned within the cone in an extended conformation [modified version of Protein Data Bank (PDB) 1ctf, with the hinge residues (43–51) modeled as a random coil]. Helical angle orientations are 66°,63°, and 32° for helices I, II, and III, respectively, relative to the y axis and the long axis of the diffusion tensor. (B) The hinge and CTD regions of L7/L12 (residues 43–120), modeled into the structure of the 50S subunit from T. thermophilus [(6) PDB 1giy] with L11 (in orange indicating the base of the stalk), rRNA (cyan), and the remaining ribosomal proteins (dark blue). L7/L12 is drawn in various colors and positioned in six orientations representing a full rotation around the principal axis (gray line) as seen in A. In this model, the cone representing the possible range of orientations of L7/L12 is placed in an arbitrary position similar to that of the ribosomal stalk, although the precise orientation of the CTD of L7/L12 relative to the ribosomal body is unknown.
For a free molecule of known structure, the inertia tensor is a good estimate of the diffusion tensor, and the orientations of the helices relative to the diffusion tensor can therefore be estimated. For the isolated CTD of L7/L12, the helical angles relative to the diffusion tensor are 59° (helix I), 70° (helix II), and 39° (helix III). Comparing these orientations with the helical orientations of the ribosome-bound CTD, we find that the diffusion tensor of the isolated CTD has to be rotated by 18°, 30°, and 44° (around the x, y, and z axes, respectively) to align it with the diffusion tensor of the ribosomally bound CTD. That the orientation of the diffusion tensor differs from that of the free molecule implies that the ribosome imposes a preferred orientation on the CTD of L7/L12. The relative orientations of helices I–III can therefore be used to model possible locations of the CTD of L7/L12 into the structure of the 50S from Thermus thermophilus (6) (Fig. 4B).
Discussion
The results we describe in this paper demonstrate the ability of NMR spectroscopy to provide detailed insights into the structure and dynamics of mobile regions of very large macromolecular systems. Of the 100 resonances observable in the HSQC spectrum of the 70S ribosome, ≈20 are from residues in the 30S subunit and are likely to originate from unstructured regions of the S1 protein (9). The remaining 80 or so crosspeaks observed from the 50S subunit have been shown to arise from residues 43–120 of L7/L12 as they have chemical shifts that are essentially identical to those reported for the isolated protein (11). The pulsed-field gradient experiments confirm that, despite the high degree of motional freedom that must be present to generate narrow NMR signals, the resonances observed from L7/L12 correspond to molecules attached to the main body of the ribosome particle.
15N relaxation experiments have yielded a 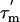 of 13.6 ns for the CTD of the protein, a value significantly longer than the correlation time of 4 ns predicted by hydrodynamic calculations for an isolated L7/L12 CTD but much shorter than the 1,000 ns anticipated for the intact ribosomal particle. This indicates that L7/L12 tumbles largely, but not completely, independently of the ribosomal body. The L7/L12 proteins are crucial for accurate translation of mRNA (32), and many biophysical studies of L7/L12 have been carried out to probe the mode of action of these molecules (33). The CTD of L7/L12 has been shown by fluorescence studies (34, 35) to be flexible; such motion is thought to enable L7/L12 to interact with the diverse set of auxiliary translation factors that bind at the base of the stalk and guide the ribosome through the translational cycle (36). Indeed, very recent evidence strongly suggests that the CTD of L7/L12 is responsible for elongation-factor Tu (EF-Tu) function (37). The NTD (residues 1–36) of L7/L12 mediates the dimer interaction and anchors the protein, via L10, to the slow-tumbling ribosomal body (38). In accord with this conclusion, no resonances of the NTD domain can be observed in the NMR spectra.
of 13.6 ns for the CTD of the protein, a value significantly longer than the correlation time of 4 ns predicted by hydrodynamic calculations for an isolated L7/L12 CTD but much shorter than the 1,000 ns anticipated for the intact ribosomal particle. This indicates that L7/L12 tumbles largely, but not completely, independently of the ribosomal body. The L7/L12 proteins are crucial for accurate translation of mRNA (32), and many biophysical studies of L7/L12 have been carried out to probe the mode of action of these molecules (33). The CTD of L7/L12 has been shown by fluorescence studies (34, 35) to be flexible; such motion is thought to enable L7/L12 to interact with the diverse set of auxiliary translation factors that bind at the base of the stalk and guide the ribosome through the translational cycle (36). Indeed, very recent evidence strongly suggests that the CTD of L7/L12 is responsible for elongation-factor Tu (EF-Tu) function (37). The NTD (residues 1–36) of L7/L12 mediates the dimer interaction and anchors the protein, via L10, to the slow-tumbling ribosomal body (38). In accord with this conclusion, no resonances of the NTD domain can be observed in the NMR spectra.
Two of the four L7/L12 molecules have been tentatively placed by model building [using the structure of isolated L7/L12 (39)] in the base of the stalk of the ribosome in a compact conformation (6), in agreement with a range of biophysical data, most notably that from immuno-EM (40) and cryo-EM experiments (7). This positioning is in line with our intensity analysis, which indicates that resonances of two of the copies of L7/L12 are not observable, consistent with their incorporation into the ribosomal structure. Moreover, ribosomes reconstituted with only one L7/L12 dimer appeared stalkless (41), and fluorescence anisotropy measurements have indicated that two copies of the CTD are independently mobile on the ribosome (34); the extended and mobile conformation of these domains, constituting the ribosomal stalk, is the likely form of L7/L12 from which we observe resonances. Our intensity measurements suggest it is possible that resonances of one of the two mobile CTDs (but not its linker region) are broadened relative to the other, a result that suggests that at least one CTD interacts, at least transiently, with the body of the ribosome. Alternatively, relaxation effects, including, for example, exchange between one or more sites with restricted motion, could reduce the observable intensities of all but the resonances of the more flexible linker region. This possibility is consistent with EM studies where, in conjunction with nanogold labeling, the two flexible CTDs of L7/L12 have been shown to reside in three further locations on empty ribosomes (7).
Consistent with cryo-EM studies that show that the ribosomal stalk assumes an elongated and well defined conformation upon EF-G binding (24, 25), the L7/L12 resonances disappear upon addition of EF-G, indicating that the high degree of mobility in the unbound state has been lost. It has been shown (25) that EF-G binds to regions close to the L7/L12 stalk [the binding positions overlap well with those for EF-Tu (42)] and in the pretranslocation GTP-bound state, EF-G causes a splitting of the stalk into two subdomains. In the GDP-bound state and in the tRNA-bound form (43), the L7/L12 stalk is observed in a well extended conformation. The absence of an extended stalk in cryo-EM reconstructions of empty ribosomes is likely to be a result of its high flexibility. Thus, in structural studies of the stalk, highly complementary information can be obtained from cryo-EM and NMR.
The relaxation analysis described in this paper enables the anisotropy of the tumbling of the mobile CTD to be defined and hence allows us to define its average orientation relative to the long axis of the ribosome, as shown in Fig. 4A. The anisotropy is revealed by the variations in the apparent 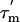 values for different regions of the CTD and further shows that the diffusion tensor of ribosomally bound L7/L12 differs from that of free L7/L12. This, in turn, indicates that the CTD has, on average, a preferred orientation relative to the ribosomal body, where the terminal α-helix and the three β-strands are parallel to the hinge that attaches the CTD to the NTD and hence to the body of the ribosome (Fig. 4A). The 15N relaxation data also yield order parameters (S2) for fast-timescale (picosecond) motions of the CTDs. The hinge between the two L7/L12 domains, residues 43–51, has very low-order parameters (S2 ≃ 0.35, i.e., a very high degree of internal flexibility), whereas residues 52–120, resulting from the CTD of the protein, have an average S2 of 0.75, characteristic of a highly structured protein. This result confirms that the hinge region (residues 43–51) serves as the flexible linker that allows the CTD to sample various positions on the ribosome, thereby potentially propagating conformational changes that occur at the base of the stalk to distant regions of the ribosome. The cryo-EM studies described above have shown the interactions of EF-G with the L7/L12 stalk (24–26); it is likely that the flexibility of the CTD of L7/L12 allows it to bind to EF-G as well as to other proteins at the base of the stalk, L10 (44), and L11 (44, 45), which are also thought to interact with the translation factors.
values for different regions of the CTD and further shows that the diffusion tensor of ribosomally bound L7/L12 differs from that of free L7/L12. This, in turn, indicates that the CTD has, on average, a preferred orientation relative to the ribosomal body, where the terminal α-helix and the three β-strands are parallel to the hinge that attaches the CTD to the NTD and hence to the body of the ribosome (Fig. 4A). The 15N relaxation data also yield order parameters (S2) for fast-timescale (picosecond) motions of the CTDs. The hinge between the two L7/L12 domains, residues 43–51, has very low-order parameters (S2 ≃ 0.35, i.e., a very high degree of internal flexibility), whereas residues 52–120, resulting from the CTD of the protein, have an average S2 of 0.75, characteristic of a highly structured protein. This result confirms that the hinge region (residues 43–51) serves as the flexible linker that allows the CTD to sample various positions on the ribosome, thereby potentially propagating conformational changes that occur at the base of the stalk to distant regions of the ribosome. The cryo-EM studies described above have shown the interactions of EF-G with the L7/L12 stalk (24–26); it is likely that the flexibility of the CTD of L7/L12 allows it to bind to EF-G as well as to other proteins at the base of the stalk, L10 (44), and L11 (44, 45), which are also thought to interact with the translation factors.
The ability to observe and characterize mobile regions of proteins is one of the major strengths of NMR spectroscopy. We have recently used cryo-EM to observe stalled ribosomes in which nascent chains are visible at least in outline, both within the tunnel and on the outside of the ribosomal particle (46). The success of the present study in characterizing the mobility of ribosomally bound L7/L12 suggests that the dynamic nature of nascent chains could, in principle, be accessible to NMR studies. Such observations are likely to contribute very significantly to an understanding of the nature of the various steps involved in cotranslational folding.
Supplementary Material
Acknowledgments
We thank Kresten Lindorff-Larsen (Cambridge University) and Janusz Sdunek (Umeå University) for assistance with the analysis of the dynamics and also Lewis Kay (University of Toronto) for helpful discussions. J.C. was supported by a Wellcome Trust International Prize Traveling Research Fellowship and G.L., by the Wenner–Gren Foundation. This study was supported by the Biotechnology and Biological Sciences Research Council and also a Wellcome Trust Programme Grant. We acknowledge the use of the 750 MHz spectrometer at the Oxford Centre for Molecular Sciences and the Engineering and Physical Sciences Research Council-funded Biomolecular NMR facility in the Chemistry Department at Cambridge University.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: EM, electron microscopy; HSQC, heteronuclear single-quantum correlation; CTD, C-terminal domain; NTD, N-terminal domain; EF-G, elongation factor G; T1, longitudinal relaxation time (R1=1/T1); T2, transverse relaxation time (R2=1/T2); 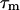 , apparent rotational correlation time; CA, carbonic anhydrase.
, apparent rotational correlation time; CA, carbonic anhydrase.
References
- 1.Moore, P. B. & Steitz, T. A. (2003) Annu. Rev. Biochem. 72, 813–850. [DOI] [PubMed] [Google Scholar]
- 2.Wilson, D. N. & Nierhaus, K. H. (2003) Angew. Chem. Int. Ed. Engl. 42, 3464–3486. [DOI] [PubMed] [Google Scholar]
- 3.Ban, N., Nissen, P., Hansen, J., Moore, P. B. & Steitz, T. A. (2000) Science 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 4.Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F. & Yonath, A. (2001) Cell 107, 679–688. [DOI] [PubMed] [Google Scholar]
- 5.Frank, J. (2000) Chem. Biol. 7, R133–R141. [DOI] [PubMed] [Google Scholar]
- 6.Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H. & Noller, H. F. (2001) Science 292, 883–896. [DOI] [PubMed] [Google Scholar]
- 7.Montesano-Roditis, L., Glitz, D. G., Traut, R. R. & Stewart, P. L. (2001) J. Biol. Chem. 276, 14117–14123. [DOI] [PubMed] [Google Scholar]
- 8.Gudkov, A. T. & Gongadze, G. M. (1984) FEBS Lett. 176, 32–36. [DOI] [PubMed] [Google Scholar]
- 9.Cowgill, C. A., Nichols, B. G., Kenny, J. W., Butler, P. D., Bradbury, E. M. & Traut, R. R. (1984) J. Biol. Chem. 259, 15257–15263. [PubMed] [Google Scholar]
- 10.Bushuev, V. N. & Gudkov, A. T. (1988) Methods Enzymol. 164, 148–158. [DOI] [PubMed] [Google Scholar]
- 11.Bocharov, E. V., Gudkov, A. T. & Arseniev, A. S. (1996) FEBS Lett. 379, 291–294. [DOI] [PubMed] [Google Scholar]
- 12.Bocharov, E. V., Gudkov, A. T., Budovskaya, E. V. & Arseniev, A.S. (1998) FEBS Lett. 423, 347–350. [DOI] [PubMed] [Google Scholar]
- 13.Mori, S., Abeygunawardana, C., Johnson, M. O., Berg, J. & van Zijl, P. C. M. (1995) J. Magn. Reson. Ser. B 108, 94–98. [DOI] [PubMed] [Google Scholar]
- 14.Price, W. S. (1997) Concepts Magn. Reson. 9, 299–336. [Google Scholar]
- 15.Dingley, A. J., Mackay, J. P., Shaw, G. L., Hambly, B. D. & King, G. F. (1997) J. Biomol. NMR 10, 1–8. [DOI] [PubMed] [Google Scholar]
- 16.Farrow, N. A., Muhandiram, D. R., Singer, A. U., Pascal, S. M., Kay, C. M., Gish, G., Shoelson, S. E., Pawson, T., Forman-Kay, J. D. & Kay, L. E. (1994) Biochemistry 33, 5984–6003. [DOI] [PubMed] [Google Scholar]
- 17.Szyperski, P., Luginbühl, P., Otting, G., Güntert, P. & Wüthrich, K. (1993) J. Biomol. NMR 3, 151–164. [DOI] [PubMed] [Google Scholar]
- 18.Evenäs, J., Malmendal, A. & Akke, M. (2001) Structure (London) 9, 185–195. [DOI] [PubMed] [Google Scholar]
- 19.Piotto, M., Saudek, V. & Sklenar, V. (1992) J. Biomol. NMR 6, 661–665. [DOI] [PubMed] [Google Scholar]
- 20.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 21.Johnson, B.A. & Blevins, R. A. (1994) J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 22.Leijonmarck, M. & Liljas, A. (1987) J. Mol. Biol. 195, 555–580. [DOI] [PubMed] [Google Scholar]
- 23.Gudkov, A. T. (1997) FEBS Lett. 407, 253–256. [DOI] [PubMed] [Google Scholar]
- 24.Agrawal, R. K., Penczek, P., Grassucci, R. A. & Frank, J. (1998) Proc. Natl. Acad. Sci. USA 95, 6134–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agrawal, R. K., Heagle, A. B., Penczek, P., Grassucci, R. A. & Frank, J. (1999) Nat. Struct. Biol. 7, 643–647. [DOI] [PubMed] [Google Scholar]
- 26.Stark, H., Rodnina, M. W., Wieden, H. J., van Heel, M. & Wintermeyer, W. (2000) Cell 100, 301–309. [DOI] [PubMed] [Google Scholar]
- 27.Larsson, G., Martinez, G., Schleucher, J., Wijmenga, S. S. (2003) J. Biomol. NMR 27, 291–312. [DOI] [PubMed] [Google Scholar]
- 28.Tjandra, N., Wingfield, P., Stahl, S. & Bax, A. (1996) J. Biomol. NMR 8, 273–284. [DOI] [PubMed] [Google Scholar]
- 29.Jin, D., Andrec, M., Montelione, G. T. & Levy, R. M. (1998) J. Biomol. NMR 12, 471–492. [DOI] [PubMed] [Google Scholar]
- 30.Bruschweiler, R. (2003) Curr. Opin. Struct. Biol. 13, 175–183. [DOI] [PubMed] [Google Scholar]
- 31.Orekhov, V., Yu., Nolde, D. E., Golovanov, A. P., Korzhnev, D. M. & Arseniev, A. S. (1995) Appl. Magn. Reson. 9, 581–588. [Google Scholar]
- 32.Pettersson, I. & Kurland, C. G. (1980) Proc. Natl. Acad. Sci. USA 77, 4007–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandra-Sanyal, S. & Liljas, A. (2000) Curr. Opin. Struct. Biol. 6, 633–636. [DOI] [PubMed] [Google Scholar]
- 34.Hamman, B. D., Oleinikov, A. V., Jokhadze, G. G., Traut, R. R. & Jameson, D. M. (1996) Biochemistry 35, 16672–16679. [DOI] [PubMed] [Google Scholar]
- 35.Hamman, B. D., Oleinikov, A. V., Jokhadze, G. G., Traut, R. R. & Jameson, D. M. (1996) Biochemistry 35, 16680–16686. [DOI] [PubMed] [Google Scholar]
- 36.Oleinikov, A. V., Jokhadze, G. G. & Traut, R. R. (1998) Proc. Natl. Acad. Sci. USA 95, 4215–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terasaki, M., Suzuki, T., Hanada, T. & Watanabe, K. (2004) J. Mol. Biol. 336, 331–342. [DOI] [PubMed] [Google Scholar]
- 38.Gudkov, A. T. & Behlke, J. (1978) Eur. J. Biochem. 90, 309–312. [DOI] [PubMed] [Google Scholar]
- 39.Wahl, M. C., Bourenkov, G. P., Bartunik, H. D. & Huber, R. (2000) EMBO. J. 19, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson, H. M., Sommer, A., Tewari D. S., Traut, R. R. & Glitz, D. G. (1986) J. Biol. Chem. 261, 6924–6932. [PubMed] [Google Scholar]
- 41.Tewari, D. S., Sommer, A. & Traut, R. R. (1986) J. Biol. Chem. 261, 6919–6923. [PubMed] [Google Scholar]
- 42.Stark, H., Rodnina, M., Rinke-Appel, J., Brimacombe, R., Wintermeyer, W. & van Heel, M. (1997) Nature 389, 403–406. [DOI] [PubMed] [Google Scholar]
- 43.Malhotra, A., Penczek, P., Agrawal, R .K., Gabashvili, I .S., Grassucci, R .A., Junemann, R., Burkhardt, N., Nierhaus, K. H. & Frank, J. (1998) J. Mol. Biol. 280, 103–116. [DOI] [PubMed] [Google Scholar]
- 44.Dey, D., Bochkariov, D. E., Jokhadze, G. G. & Traut, R. R. (1998) J. Biol. Chem. 273, 1670–1676. [DOI] [PubMed] [Google Scholar]
- 45.Traut, R. R., Dey, D., Bochkariov, D. E., Oleinikov, A.V., Jokhadze, G. G., Hamman, B. & Jameson, D. (1995) Biochem. Cell Biol. 73, 949–958. [DOI] [PubMed] [Google Scholar]
- 46.Gilbert, R. J. C., Fucini, P., Connell, S., Fuller, S. D., Nierhaus, K. H., Robinson, C. V., Dobson, C. M. & Stuart, D. I. (2004) Mol. Cell 14, 1–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.