

Abstract
Aristolochia manshuriensis Kom (AMK) is an herb used as a traditional medicine; however, it causes side effects such as nephrotoxicity and carcinogenicity. Nevertheless, AMK can be applied in specific ways medicinally, including via ingestion of low doses for short periods of time. Non-alcoholic steatohepatitis (NASH) induced the hepatocyte injury and inflammation. The protective effects of AMK against NASH are unclear; therefore, in this study, the protective effects of AMK ethyl acetate extract were investigated in a high-fat diet (HFD)-induced NASH model. We found decreased hepatic steatosis and inflammation, as well as increased levels of lipoproteins during AMK extract treatment. We also observed decreased hepatic lipid peroxidation and triglycerides, as well as suppressed hepatic expression of lipogenic genes in extract-treated livers. Treatment with extract decreased the activation of c-jun N-terminal kinase 1/2 (JNK1/2) and increased the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2). These results demonstrate that the protective effect of the extract against HFD-induced NASH occurred via reductions in reactive oxygen species production, inflammation suppression, and apoptosis related to the suppression of JNK1/2 activation and increased ERK1/2 phosphorylation. Taken together, these results indicate that that ethyl acetate extract of AMK has potential therapeutic effects in the HFD-induced NASH mouse model.
Keywords: Aristolochia manshuriensis Kom, c-jun N-terminal kinase 1/2, extracellular signal-regulated kinase 1/2, inflammation, nonalcoholic steatohepatitis
Introduction
Non-alcoholic fatty liver disease (NAFLD) includes non-alcoholic steatohepatitis (NASH), which is related to metabolic liver diseases involving steatosis, hepatocyte injury, and inflammation [3,8]. This disease is caused by increases in excess hepatic neutral fats [19]. Two hypotheses have been suggested for the pathogenesis of NASH; namely, early metabolic obstruction producing steatosis or pathogenic stimulation leading to increased production of reactive oxygen species (ROS), oxidative stress, and lipid peroxidation [4,8]. Therefore, oxidative stress plays a central role in the pathogenesis of NASH, and these stresses increase in response to inflammatory responses known to cause lipid peroxidation [21]. Accordingly, the inhibition of hepatic inflammation and fatty acid cytotoxicity are important to the treatment of NASH.
A strong relationship between hepatic apoptosis and oxidative stress has been described [31]. A previous study reported that ROS induce cell death and injuries via indirect activation of not fully characterized mitogen-activated protein kinases (MAPKs) pathways, including the extracellular signal-regulated kinase (ERK) pathway, and that it has induction and pro-apoptotic roles [7,18]. In contrast, the activation of several MAPKs, including c-jun N-terminal kinase (JNK) and p38, induced cellular death [18,32].
Medical plants have been used to treat diseases for thousands of years worldwide. Over the past few decades, scientific investigations of plant-derived herbal medicines (often imported from Asia) have increased in Western countries. Aristolochia manshuriensis Kom (AMK) is a member of the Aristolochiaceae family distributed in East Asia that is used in traditional Chinese medicines (TCMs) as an anti-inflammatory, analgesic, antitussive, anti-asthmatic, and antibacterial agent [34]. The main active constituent of AMK is aristolochic acid, which reportedly leads to nephrotoxicity and carcinogenicity [23]. Several studies and some TCM doctors have proposed that AMK can still be used as a medicine if it is applied in specific ways such as consuming low doses for short periods of time and exterior application as antibacterial and anti-inflammatory agents. We previously reported that the AMK extract suppressed adipogenesis in 3T3-L1 adipocytes and the high-fat diet (HFD)-induced obesity animal model without side effects [17]. However, the potential therapeutic effects of AMK in NASH are unclear. Therefore, this study was conducted to evaluate the role of the AMK ethyl acetate (EtOAc) extract in NASH prevention in C57BL/6 mice administered a HFD.
Materials and Methods
In vivo and investigational protocol
Male C57BL/6 mice obtained from Samtaco (Korea) weighing 17–18 g were housed in groups of five at 60 ± 5% relative humidity and 22 ± 2℃ under a 12 h light/dark cycle. The HFD, which induces NASH primarily by inhibiting phosphatidylcholine biosynthesis (Central Lab. Animal, Korea), was administered to the HFD group for 15 weeks, while the normal diet (ND) group received a normal diet. The animals were casually separated into four groups (n = 10). Specifically, the ND group received a normal diet, the HFD group received HFD alone, the butyl alcohol (BuOH) group received HFD plus BuOH extract of AMK (62.5 mg/kg/day, orally), and the EtOAc extract group received the HFD plus the AMK EtOAc extract (2.5 mg/kg/day). After 15 weeks, the mice were fasted for 6 h, then euthanized using ethyl ether anesthesia. Blood and liver tissue was subsequently harvested and stored at −70℃ until biochemical analysis. All protocols were designed in accordance with the Guidelines for the Care and Use of Experimental Animals and were approved in agreement with the regulations of the Ethics Committee of the Korea Institute of Oriental Medicine.
Preparation of EtOAc extract of AMK
AMK (300 g) was extracted overnight at a set temperature 4℃ in a mixture of water and ethanol. The AMK extract was powdered under pressure.
Biochemical analyses
The levels of various lipid proteins including low-density lipoprotein (LDL), high-density lipoprotein (HDL) cholesterol, total cholesterol (TC), and triglycerides (TG) were measured using enzymatic kits (Pars Azmoon, Iran). Hepatic cell injury was evaluated based on measurement of the activity of hepatic proteins such as aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (ALP) using commercial kits (Pars Azmoon). Hepatic antioxidant activation was assessed based on the glutathione (GSH) and glutathione reductase (GR) activities according to method described by Jollow et al. [12], while glutathione peroxidase (GPx) was assessed as described by Paglia and Valentine [20].
Superoxide dismutase (SOD) activity
SOD action in the reaction blend of NADH-phenazine-methosulphate (PMS)-NBT was assessed based on the suppression of amino blue tetrazolium formazan formation [13]. The blend contained 0.1 mL of supernatant, 0.3 mL of 300 mM NBT, 0.1 mL of 186 mM PMS, and 1.2 mL of 52 mM sodium pyrophosphate buffer (pH 8.3) and was initiated by the addition of 0.2 mL of 750 mM NADH solution. Glacial acetic acid (0.1 mL) was added after incubation at 30℃ for 90 sec to stop the reaction. Next, 4 mL n-butanol was added and the chromogen color intensity in the butanol was assessed spectrophotometrically at 560 nm.
Histopathological assay
The paraffin sections of mouse liver tissue (4 µm) were stained using hematoxylin and eosin, and graded for hepatic steatosis and inflammation [35] as follows: 0, no inflammatory foci; 1, mild; 2, moderate; and 3, severe. The percentage of hepatic steatosis was graded as 0, 0%; 1, 1% to 30%; 2, 31% to 55%; 3, 56% to 78%; and 4, 79%.
Oxidative stress measurement
This study used the oxidized fluorescent derivative 2,7-dichlorodihydrofluorescin (DCF) to measure the lipoperoxides related to thiobarbituric acid reactive substances (TBARS) using TBARS assay kit (Cayman Chemical, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR) of hepatic messenger RNA expression levels
We used TRIzol reagent (Invitrogen, USA) to extract total RNA from the liver samples and then reverse transcribed the RNA into complementary DNA using Accupower RT PreMix (Bioneer, Korea). Next, qRT-PCR was conducted using a SYBR green master mix (Thermo Scientific, USA). Ct values were normalized to β-actin. For PCR, the following primers were used: interleukin (IL)-6 (sense) ACAACCACGGCCTTC CCTACTT, (antisense) GTGTAATTAAGCCTCCGACT; IL-8 (sense) TTGGCTGGCTTATCTTC, (antisense) CCTTATCAA ATACGGAGTA; tumor necrosis factor (TNF)-α (sense) CGTGCTCCTCACCCACAC, (antisense) GGGTTCATAC CAGGGTTTGA; transforming growth factor (TGF)-1β (sense) CAACTTCTGTCTGGGACCCT, (antisense) TAGTA GACGATGGGCAGTGG; proliferator-activated receptor (PPAR)-α (sense) TGGGGATGAAGAGGGCTGAG, (antisense) GGGGACTGCCGTTGTCTGT; PPAR-γ (sense) CAGGCT TGCTGAACGTGAAG, (antisense) GGAGCACCTTGGCG AACA; cytochrome P450 (CYP) 2E1 (sense) AGTGTTC ACACTGCACCTGG, (antisense) CCTGGAACACAGGAA TGTCC; CYP4A10 (sense) ACACTGCTCCGCTTCGAACT, (antisense) CAAGTCGGGCTAAGGGCA; CYP4A14 (sense) GCCAGAATGGAGGATAGGAACA, (antisense) ATGAATG TGTCCACCTCTGCAC; stearoyl coenzyme A desaturase (SCD)-1 (sense) GTACCGCTGGCACATCAACTT, (antisense) GGCTAAGACAGTAGCCTTAG; fatty acid synthase (FAS, sense) TGGTGG-GTTTGGTGAATTGTC, (antisense) GCTT GTCCTGCTCTAACTGGAAGT; CCAAT/enhancer binding protein (C/EBP)-β (sense) GTTTCGGGAGTTGATGC-AATC, (antisense) AACAACCCCGCAGGAACAT; and β-actin (sense) TGTCCACCTTCCAGCAGATGT, (antisense) AGCT CAGTAACAGTCCGCCTAGA.
Western blotting
Ice cold Pro-Preptm buffer (iNtRON Biotechnology, Korea) was used to lyse hepatic tissue samples, after which they were centrifuged at 18,340 × g and 4℃ for 20 min to remove insoluble materials. Protein extracts were resolved by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and then transferred to membranes at 150 mA for 1 h. The membranes were subsequently blocked for 1 h at room temperature with PBS containing 5% bovine serum albumin (BSA) and 0.1% Tween 20, then incubated with 1:1,000 dilutions of primary antibodies (anti-JNK1/2, anti-pJNK1/2, anti-ERK1/2, and anti-pERK1/2; Cell Signaling Technology, USA) overnight at 4℃. Next, samples were incubated for 1 h at room temperature with peroxidase-conjugated anti-mouse and anti-rabbit secondary antibody (1:1,000; Cell Signaling Technology). The activity of peroxidase was then measured using an ECL kit (Thermo Fisher Scientific, USA).
Statistical analyses
All values are presented as the mean ± standard deviation (SD). Significant differences were identified using an unpaired Student's t-test and p values < 0.05 and < 0.01 were considered significant.
Results
Histopathological findings
The ND group showed no pathological changes and the liver tissue from the mice showed normal histology. Liver steatosis and inflammation were induced in both the HFD and BuOH groups. Following treatment with EtOAc extract, these factors decreased to grade 0 (Table 1). As shown in Table 1, fibrosis was not found in the HFD or BuOH-treated mice. We also did not observe any significant changes in liver morphology. In EtOAc extract-treated mice, body weight decreased significantly compared with the HFD group; however, no significant differences in food intake were observed relative to other groups (Table 2).
Table 1. Effects of Aristolochia manshuriensis Kom (AMK) ethyl acetate (EtOAc) extract on hepatic steatosis and necroinflammation scores.

Data are the means ± SD, n = 9. ap < 0.01 indicates statistically significant differences compared with ND mice. bp < 0.01 indicates statistically significant differences when compared with HFD mice. ND, normal diet; HFD, high fat diet; AMK-BuOH extract, AMK-butyl alcohol extract; AMK-EtOAc extract, AMK-ethyl acetate extract.
Table 2. Body weight and food intake in mice were treated with AMK-EtOAc extract.

Data are the means ± SD, n = 9. ap < 0.01 indicates statistically significant differences compared with ND mice. bp < 0.01 indicates statistically significant differences when compared with HFD mice.
Assay of the serum lipoprotein profile and hepatic enzymes
Serum TG and TC levels in the ND, HFD, and EtOAc groups are shown panel B in Fig. 1. The administration of the EtOAc extract (2.5 mg/kg) markedly increased the serum TC and TG levels (p < 0. 05) compared to HFD-fed mice. Fig. 2A shows the effects of EtOAc extract on the serum lipoprotein profiles of mice. The EtOAc extract of AMK dramatically reduced the LDL concentration (33%, p < 0.05) relative to that of the HFD-fed mice, and the serum levels of HDL and very low-density lipoprotein (VLDL) improved by about 20% and 33%, respectively, in the EtOAc extract-treated mice relative to the HFD-fed mice (panel A in Fig. 2). Additionally, serum of mice treated with extract of AMK EtOAc had ALP, AST, and ALT enzyme levels 54%, 52%, and 56% higher (p < 0.05) than those of HFD-fed mice, respectively, implying that AMK protected against HFD-induced liver injury (panel B in Fig. 2).
Fig. 1. Effects of EtOAc extract of AMK on non-alcoholic steatohepatitis (NASH) in mice. (A) Morphological evaluation of fatty liver. (B) Evaluation of triglyceride (TG) and cholesterol (TC) in NASH mice sera. The levels were measured in mice fed with ND, HFD, or HFD plus EtOAc extract of AMK (2.5 mg/kg) for 15 weeks. Each group consisted of 10 mice. Data are expressed as the means ± SD. *p < 0.05, ***p < 0.001 vs. HFD-fed mice.
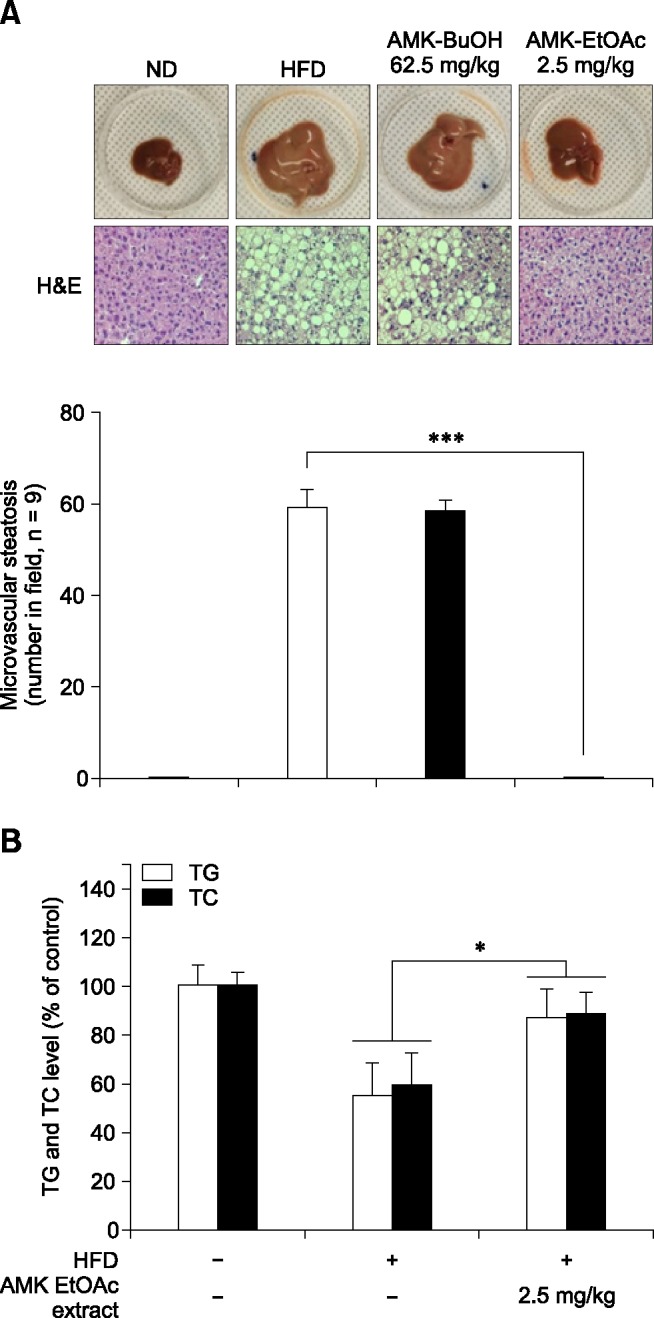
Fig. 2. Effects of EtOAc extract of AMK on NASH mice. (A) Serum levels of high-density lipoprotein (HDL), low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL) in NASH mice. (B) alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) in mice with NASH. The levels were measured in mice fed with ND, HFD, or HFD plus EtOAc extract of AMK (2.5 mg/kg) for 15 weeks. Each group consists of 10 mice. Data are expressed as the means ± SD. *p < 0.05 vs. HFD-fed mice.

Effect of EtOAc extract on hepatic TGs and lipid peroxidation
Consumption of HFD increased TG (panel A in Fig. 3) and TBARS (panel B in Fig. 3) levels compared with the ND mice. However, treatment with the EtOAc extract markedly inhibited these increases. In the hepatocytes of mice treated with EtOAc extract of AMK (2.5 mg/kg), the levels of both TG and TBARS was significantly decreased compared to mice fed with the HFD alone (Fig. 3).
Fig. 3. Effects of EtOAc extract of AMK on hepatic TG and lipoperoxide content in NASH mice. (A) Hepatic TG and (B) lipoperoxide content were assessed in mice fed ND, HFD, or HFD treated with EtOAc extract of AMK (2.5 mg/kg) for 12 weeks. Data are the means ± SD of 10 mice in each group. *p < 0.05 vs. HFD-fed mice. SOD, superoxide dismutase; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione; TBARS, thiobarbituric acid reactive substances.

Effect of EtOAc extract on liver oxidative status
The activities of SOD, GSH, GR, and GPx decreased significantly in HFD-fed mice (p < 0.05). The administration of EtOAc extract markedly amplified the activities of SOD (118%), GPx (88%), GR (68%) and GSH (80%) relative to HFD-fed mice (panel C in Fig. 3).
Effect of EtOAc extract on the expression of genes related to fatty acid regulation and peroxidation
In this study, the hepatic levels of lipogenic genes including PPAR-γ, FAS, SCD1, and C/EBPβ, as well as the lipolytic genes PPAR-α, CYP4A10, and CYP4A14, which are known to be PPAR-α downstream target molecules (panels A and B in Fig. 4), were measured to evaluate the hepatic TG-reducing effects of AMK. Administration of EtOAc extract greatly increased the expression of PPAR-α, which was reduced by HFD (panel A in Fig. 4). However, HFD-induced C/EBP-β was significantly decreased following treatment with the EtOAc extract (panel B in Fig. 4). The other lipogenic genes were altered compared to the HFD-fed mice, but these changes were not significant (panels A and B in Fig. 4). CYP2E1, which is a major mediator of lipid peroxidation, was promoted by HFD. The expression of CYPE21 mRNA was markedly increased by HFD, but was significantly reduced following treatment with the EtOAc extract (panel B in Fig. 4). The expression of CYPE21 protein was strongly increased by EtOAc extract (panel A in Fig. 5).
Fig. 4. Effects of HFD and treatment with EtOAc extract of AMK on hepatic mRNA expression. Expression of regulation genes such as (A) hepatic fatty acid, (B) adiopogenesis and (C) inflammation was measured in the hepatocytes of mice fed ND, HFD, or HFD plus EtOAc extract of AMK (2.5 mg/kg) for 15 weeks. Data are the means ± SD of 10 mice in each group. *p < 0.05, **p < 0.01 vs. HFD-fed mice.

Fig. 5. Effect of AMK-EtOAc extract on inflammatory and fatty acid regulation proteins. (A) Mitogen-activated protein kinases proteins including ERK1/2 and JNK1/2 in hepatocytes of mice with NASH. (B) TNF-α, CYP2E1, PPAR-α, Bax, caspase-3, ERK1/2, and JNK1/2 proteins in hepatocytes of mice with NASH. Data are the means ± SD of 10 mice in each group.

Effect of EtOAc extract on the expression of pro-inflammatory factors and inflammatory pathways
In mice fed the HFD, a noticeable elevation of mRNA expression of pro-inflammatory factors (TNF-α, IL-6, -8, and -1β) was observed relative to ND mice. Conversely, treatment of HFD-fed mice with EtOAc extract of AMK at 2.5 mg/kg significantly inhibited the mRNA expression of TNF-α and IL-6 (panel C in Fig. 4), and markedly reduced the expression of TNF-α protein compared with HFD-fed mice (panel B in Fig. 5).
Involvement of the MAPK pathway
The regulation of some MAPKs members in the oxidative stress process has been described. We sought to determine the possible involvement of MAPKs and elucidate the mechanism of their involvement based on the effects observed in mice fed HFD with or without EtOAc extract of AMK. HFD induced significant phosphorylation (p) of JNK1/2 (panel A in Fig. 5). However, pERK1/2 was increased in HFD-fed mice compared with the ND mice. Treatment with EtOAc extract of AMK markedly decreased pJNK1/2 and pERK1/2 relative to the HFD-fed mice (panel A in Fig. 5). The role of some pro-apoptotic proteins was regulated by JNK1/2. In HFD fed mice, the levels of pro-apoptotic proteins including Bcl-2-associated X (Bax) and caspase-3 were significantly increased. However, stimulation of the pro-apoptotic proteins was markedly inhibited in EtOAc extract-treated mice compared with HFD-fed mice (panel B in Fig. 5).
Discussion
NASH occurs as a result of the excessive accumulation of lipids, which leads to an imbalance between lipid degradation and formation in the hepatocytes [2]. In the hepatocytes of mice that received HFD, antioxidant capacity decreased; therefore, accrued lipids tend to be oxidized by elevated ROS. The increased level of ROS in the hepatocytes also damages the DNA [22]. Accordingly, oxidative stress is regarded as a central causal component during development of NASH. There have recently been several reports of the anti-oxidative activity of members of the Aristolochiaceae family [29,30]. The results of the present study show the anti-NASH effect of the EtOAc extract in mice.
In the present study, EtOAc extract significantly regulated the lipoprotein profile compared to HFD-fed mice. Specifically, LDL levels decreased dramatically (p < 0.05) relative to the HFD-fed mice, as did ALP, ALT, and AST. Histopathological evaluation of the liver tissue of the EtOAc extract-fed mice also led to marked improvement of the state of steatohepatitis compared with HFD-fed mice to levels similar to those observed in ND mice. These results suggest that the EtOAc extract reduced the extent of steatosis and inflammation in mice induced with NASH. We demonstrated that treatment with the EtOAc extract of AMK suppressed lipoperoxides and decreased TG accumulation. Furthermore, the EtOAc extract improved actions of SOD, GPx, and GR enzymes when compared with HFD-fed mice. Enzymes of the biological system related to the neutralization of free radicals are protected by these enzymes; therefore, these findings suggest that EtOAc extract of AMK potently increases the free radical reducing ability of biological systems. Our data showed that extract of AMK EtOAc markedly suppressed serum ALT and AST levels in mice. A previous study reported that NASH might increase the serum levels of ALP to the upper normal limit in affected individuals [6]. Therefore, our data are indicative of an improvement in the condition of NASH. Moreover, histopathological findings and biochemical assays revealed that the EtOAc extract did not cause any hepatotoxicity.
Steatohepatitis and steatosis without fibrosis developed via up-regulation of CYP2E1. The elevation of CYP2E1 inhibits lipid peroxidation, thereby contributing to oxidative stress damage, and upregulation of CYP2E1 has been reported in NASH [26]. We showed that the extract of AMK EtOAc reduced the expression of CYP2E1 by approximately 2- to 3-fold compared to the HFD-fed mice. Therefore, these findings indicate that EtOAc extract-mediated reduction of lipid peroxidation and prevention of steatohepatitis may, at least in part, be caused by the downregulation of CYP2E1 expression. The promoter of CYP2E1 has a binding site for C/EBP-β transcription factor [1]. One possible mechanism mediating the down-regulation of CYP2E1 by EtOAc extracts of AMK may be via suppression of the expression of C/EBP-β. A previous study reported that the height of both C/EBP-β and CYP2E1 is related to HFD-induced steatohepatitis, and that down-regulation of C/EBP-β inhibited the expression of CYP2E1 protein in mice fed HFD [24]. In addition, the induction of CYP2E1 was mediated by C/EBP-β [14]. In C/EBP gene deleted mice fed HFD, hepatic TG accumulation and steatohepatitis were markedly reduced [24]. Administration of the EtOAc extract clearly reduced C/EBP-β, making it a newly recognized hepatic lipogenesis controller. Overall, these findings indicate that C/EBP-β plays an essential role as a regulator of CYP2E1 in the HFD animal model. Furthermore, the EtOAc extracts of AMK down-regulated CYP2E1 by suppressing C/EBP-β.
Inflammatory factors including TNF-α and IL-6 are critically involved in the progression of NASH [16], and are imperative sources of associated oxidative stress, dramatically increasing ROS levels [25]. A previous study reported that continuous exposure to IL-6 impairs hepatic regeneration and sensitizes the liver to injury [15]. In this study, administration of EtOAc extract significantly reduced TNF-α and IL-6 gene expression. This inhibition of inflammation by EtOAc extract may be partially associated with the reduction of mRNA expression levels of these regulators and suppression of hepatic lipoperoxide. The inflammatory process was regulated by PPAR-α via the TNF-α signaling pathway. PPAR-α is a nuclear receptor that plays a central role in lipid metabolism. Moreover, PPAR-α agonists can inhibit hepatic steatosis in animal models of NAFLD [11]. The results of the present study indicated that the activation of PPAR-α in the hepatic steatosis of HFD-fed mice was lower than in ND mice and was upregulated by the EtOAc extract of AMK. In contrast, the TNF-α level was decreased by the EtOAc extract of AMK. These results suggest that the anti-lipogenesis of AMK EtOAc extract may be associated with the activation of PPAR-α, supporting the notion that hepatic steatosis is more susceptible to oxidative stress and the resulting lipid peroxidation, leading to inflammation. We also found that pJNK1/2 was highly active in HFD-fed mice, but significantly suppressed following administration of the EtOAc extract. JNK1/2 is a major mediator of the inflammatory response pathway that is potently activated by stress signals such as ROS [9]. It is well known that the JNK/c-Jun pathway induces liver damage via oxidative stress and lipoapoptosis [28]. Therefore, JNK has been implicated as the consequence and cause of increased inflammation [10]. In this respect, one of the prospective mechanisms of EtOAc extract-mediated actions on hepatic inflammation is the anti-oxidative amelioration of cellular ROS production via the suppression of JNK1/2 activation. This hypothesis was reinforced by the finding that the JNK1/2 pathway protected mice from HFD-induced steatohepatitis, and reduced hepatic TG, liver injury, and inflammation [27]. Therefore, suppression of JNK1/2 activation by EtOAc extract of AMK may also contribute to the reduction of lipid accumulation.
It has been reported that JNK1/2 induces cell death in HFD-induced NASH via regulation of the mitochondrial death mechanism [9]. The activation of JNK1/2 induces cellular apoptosis [32] or the regulation of pro-apoptotic proteins such as Bax [18]. A strong relationship between hepatocyte apoptosis and oxidative stress has been reported, and the identification of apoptosis as a main source of hepatic damage such as inflammation was described [31]. Caspase-3 is a major factor in apoptosis that is regarded as the central causative component in the initiation of hepatic wounds [33]. The EtOAc extract of AMK significantly inhibited cleaved caspase-3 and Bax levels in HFD-fed mice. Therefore, these findings suggest that the anti-apoptotic effects of EtOAc extract primarily occur via its strong antioxidant capability. Our results indicate that HFD-induced apoptosis is linked with JNK1/2 phosphorylation.
Several studies have reported the association between ERK1/2 activation and oxidative stress in cellular responses [7,18]. The activation of ERK1/2 weakened cell death in cells in vitro [5]. Our data indicated that ERK1/2 phosphorylation was related to HFD-induced NASH. However, the EtOAc extract of AMK decreased the activation of JNK1/2 and improved pERK1/2 in HFD-fed mice. In terms of antioxidant effects, it is speculated that the EtOAc extract of AMK-mediated attenuation of ROS generation inhibited JNK1/2 and ERK1/2. The results of this study confirm that the protective effects of the AMK EtOAc extract against HFD-induced NASH are regulated by inhibition of ROS, suppression of inflammation, and reduction of apoptosis. The anti-NASH effects of the AMK EtOAc extract are a result of the inhibition of JNK1/2 and increase in pERK1/2. These findings suggest that the EtOAc extract of AMK has a protective effect mediated by the regulation of JNK1/2 and ERK1/2 activation in HFD-induced NASH. Therefore, the present data support the notion that EtOAc extract of AMK has potential therapeutic effects. Further molecular investigations are currently being conducted in our laboratory to elucidate these effects.
Acknowledgments
This paper was supported by Wonkwang University in 2014.
Footnotes
There is no conflict of interest.
References
- 1.Abdel-Razzak Z, Garlatti M, Aggerbeck M, Barouki R. Determination of interleukin-4-responsive region in the human cytochrome P450 2E1 gene promoter. Biochem Pharmacol. 2004;68:1371–1381. doi: 10.1016/j.bcp.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17(Suppl):S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- 3.Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, McConnell JM, Byrne CD. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- 4.Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 5.Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–929. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 6.Cortez-Pinto H, Camilo ME. Non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH): diagnosis and clinical course. Best Pract Res Clin Gastroenterol. 2004;18:1089–1104. doi: 10.1016/j.bpg.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 7.Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- 8.Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 9.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 10.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54(Suppl 2):S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 11.Jin X, Zimmers TA, Perez EA, Pierce RH, Zhang Z, Koniaris LG. Paradoxical effects of short- and long-term interleukin-6 exposure on liver injury and repair. Hepatology. 2006;43:474–484. doi: 10.1002/hep.21087. [DOI] [PubMed] [Google Scholar]
- 12.Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR. Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology. 1974;11:151–169. doi: 10.1159/000136485. [DOI] [PubMed] [Google Scholar]
- 13.Kakkar P, Das B, Viswanathan PN. A modified spectrophotometric assay of superoxide dismutase. Indian J Biochem Biophys. 1984;21:130–132. [PubMed] [Google Scholar]
- 14.Kelicen P, Tindberg N. Lipopolysaccharide induces CYP2E1 in astrocytes through MAP kinase kinase-3 and C/EBPβ and -δ. J Biol Chem. 2004;279:15734–15742. doi: 10.1074/jbc.M311850200. [DOI] [PubMed] [Google Scholar]
- 15.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 16.Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- 17.Kwak DH, Lee JH, Kim T, Ahn HS, Cho WK, Ha H, Hwang YH, Ma JY. Aristolochia manshuriensis Kom inhibits adipocyte differentiation by regulation of ERK1/2 and Akt pathway. PLoS One. 2012;7:e49530. doi: 10.1371/journal.pone.0049530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 19.Pagano G, Pacini G, Musso G, Gambino R, Mecca F, Depetris N, Cassader M, David E, Cavallo-Perin P, Rizzetto M. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: further evidence for an etiologic association. Hepatology. 2002;35:367–372. doi: 10.1053/jhep.2002.30690. [DOI] [PubMed] [Google Scholar]
- 20.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–169. [PubMed] [Google Scholar]
- 21.Pathil A, Mueller J, Warth A, Chamulitrat W, Stremmel W. Ursodeoxycholyl lysophosphatidylethanolamide improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology. 2012;55:1369–1378. doi: 10.1002/hep.25531. [DOI] [PubMed] [Google Scholar]
- 22.Pessayre D, Fromenty B, Mansouri A. Mitochondrial injury in steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1095–1105. doi: 10.1097/00042737-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Pozdzik AA, Salmon IJ, Husson CP, Decaestecker C, Rogier E, Bourgeade MF, Deschodt-Lanckman MM, Vanherweghem JL, Nortier JL. Patterns of interstitial inflammation during the evolution of renal injury in experimental aristolochic acid nephropathy. Nephrol Dial Transplant. 2008;23:2480–2491. doi: 10.1093/ndt/gfn140. [DOI] [PubMed] [Google Scholar]
- 24.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE. CCAAT/enhancing binding protein β deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- 25.Sánchez-Alcázar JA, Schneider E, Martínez MA, Carmona P, Hernández-Muñoz I, Siles E, De La Torre P, Ruiz-Cabello J, García I, Solis-Herruzo JA. Tumor necrosis factor-α increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. J Biol Chem. 2000;275:13353–13361. doi: 10.1074/jbc.275.18.13353. [DOI] [PubMed] [Google Scholar]
- 26.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 27.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 28.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNF-α- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 29.Shi LS, Kuo PC, Tsai YL, Damu AG, Wu TS. The alkaloids and other constituents from the root and stem of Aristolochia elegans. Bioorg Med Chem. 2004;12:439–446. doi: 10.1016/j.bmc.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 30.Shirwaikar A, Somashekar AP, Udupa AL, Udupa SL, Somashekar S. Wound healing studies of Aristolochia bracteolata Lam. with supportive action of antioxidant enzymes. Phytomedicine. 2003;10:558–562. doi: 10.1078/094471103322331548. [DOI] [PubMed] [Google Scholar]
- 31.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22(Suppl 1):S45–S48. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]
- 32.Ura S, Masuyama N, Graves JD, Gotoh Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells. 2001;6:519–530. doi: 10.1046/j.1365-2443.2001.00439.x. [DOI] [PubMed] [Google Scholar]
- 33.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Li X, Wang H. Possible mechanisms explaining the tendency towards interstitial fibrosis in aristolochic acid-induced acute tubular necrosis. Nephrol Dial Transplant. 2007;22:445–456. doi: 10.1093/ndt/gfl556. [DOI] [PubMed] [Google Scholar]
- 35.Yu J, Ip E, Dela Peña A, Hou JY, Sesha J, Pera N, Hall P, Kirsch R, Leclercq I, Farrell GC. COX-2 induction in mice with experimental nutritional steatohepatitis: role as pro-inflammatory mediator. Hepatology. 2006;43:826–836. doi: 10.1002/hep.21108. [DOI] [PubMed] [Google Scholar]