

Abstract
Specific changes in immune repertoires at genetic level responding to the lethal H7N9 virus are still poorly understood. We performed deep sequencing on the T and B cells from patients recently infected with H7N9 to explore the correlation between clinical outcomes and immune repertoire alterations. T and B cell repertoires display highly dynamic yet distinct clonotype alterations. During infection, T cell beta chain repertoire continues to contract while the diversity of immunoglobulin heavy chain repertoire recovers. Patient recovery is correlated to the diversity of T cell and B cell repertoires in different ways – higher B cell diversity and lower T cell diversity are found in survivors. The sequences clonally related to known antibodies with binding affinity to H7 hemagglutinin could be identified from survivors. These findings suggest that utilizing deep sequencing may improve prognostication during influenza infection and could help in development of antibody discovery methodologies for the treatment of virus infection.
Influenza A (H7N9) is an emerging virus of avian origin that has caused three waves of infections since February 2013. As of February 2015, a total of 571 laboratory-confirmed cases have been reported to WHO, including 212 deaths1. New cases were notified in 2016 from China. H7N9 infection induced lethal complications like severe pneumonia and acute respiratory distress syndrome, and currently no specific treatment is available for this highly contagious viral infection2,3,4.
Effective humoral and cellular immune responses in influenza infection are critical for patient recovery from H7N9 infection5,6,7,8. Quantitative and qualitative analysis of antiviral immunity may aid in understanding the state of immune system and guide the therapeutics. Immune repertoire analysis based on next-generation sequencing (NGS) is a novel approach to analyze alterations during the antiviral immune response9,10,11. Many studies have reported the overwhelmingly diverse and dynamic property of immune repertoire changes in response to the antigen stimuli such as vaccination or allergy12,13,14. However, the changes of immune repertoire in clinical infections caused by lethal pathogens and their influence on patient recovery remain unknown.
In this study we explored alterations in the human immune repertoires after H7N9 infection. Specifically, we compared the dynamic behavior of T cell and B cell repertoires and analyzed signatures of these highly convergent immune repertoires. Also we identified antibody sequences from these NGS data. These results provide direct insights into the immune response, especially distinct features of T cell and B cell repertoire behaviors, after human influenza A (H7N9) infection and suggest potential implications in antibody development and prognostication.
Results
Next-generation sequencing results
Our study utilized peripheral blood samples collected at multiple time points from patients infected with H7N9 virus in 2013. Among these patients, 10 of 15 (66.7%) recovered from infection. From a total of 35 samples, deep sequencing based on Illimina MiSeq platform produced 150 base pairs surrounding the CDR3 of each T cell receptor β chain (TRB), and 250 base pairs covering CDR1 ~ 3 and beginning of C region of each Ig chain. Sequencing depth was comparable among samples (Supplementary Table S3). Because of the limited quantity of extracted RNA from blood samples, no reads of IGL or IGK were obtained from a non-survivor (patient J) and a survivor (patient C). These two samples were excluded from all analysis.
Ig heavy chain repertoire showed highly dynamic changes over time in a survived individual
An important feature of the immune response to foreign pathogens is clonal expansion of specific T and B cells and their subsequent contraction. To track the longitudinal immune repertoire dynamics in response to H7N9, we tried to collect sequential samples from these patients. For one of these patients, a total of four samples at different time points (11, 18, 25, and 42 days, respectively) were collected, while three or less were collected from others. During the disease progression, IGH repertoire of patient H showed typical alterations that are also found in other patients. The IGH sequences exceptionally high variation between time points (Fig. 1a), which shared sequences consisted only 0.6 ~ 20% of the whole repertoire. In addition, the dominant clones of IGH repertoires changes overtime (Fig. 1b), indicating that the consistent part of the IGH repertoire was very limited. In contrast, the TRB repertoires presented a more stable pattern, with dominant clones conserved and constant part during the infection made up 23 ~ 61% of the whole repertoires (Fig. 1c,d). These results reveal the overwhelming variation as a property of IGH repertoire after H7N9 virus infection that differs from TRB repertoire.
Figure 1. Dynamic change of IGH and TRB repertoires in an influenza A (H7N9) virus infected patient.

(a,c) shows the overlap of IGH and TRB CDR3 sequences at different time points—11, 18, 25 and 42 days after symptom onset in patient H (survivor). (b,d) shows the dynamic changes of fractions of dominant IGH and TRB clones. Each CDR3 used a unique color. Width of each CDR3 clones stands for the fraction of this clone at each time point. CDR3 clones ranked top 5 in IGH repertories or top 20 in TRB were selected as representative dominant clones.
Diversity of Ig heavy chain repertoire increases and is positively related to patient prognosis
Variation in immune response efficiency results in different infection severity and outcome. A proper diversity of the immune repertoire is crucial in order to mount an efficient adaptive immune response15. The overall D50 value of TRB and IGH in patients ranges from 0 ~ 5, indicating the repertoires are highly skewed (Fig. 2). For IGH repertoire, interestingly, the D50 values after 15 days were higher than the previous sample in all but one individual (Fig. 2a), suggesting an increasing tendency in IGH diversity. This is also the time point of neutralizing antibody (Nab) titers increasing in serologic measurements16 (Supplementary Figure S2). In contrast, the diversity of TRB repertoires did not increase over time. To simplify data analysis, we used the last sample of each patient, and the disease phase of these samples were comparable between groups (15–40 days in non-survival group and 15–46 days in survival group after onset). Importantly, D50 values of IGH repertoires were significantly lower in non-survivors than in survivors (P = 0.02, Student’s t test) (Fig. 2b), as were the Nab titers (Mann-Whitney U test, P = 0.04). On the contrary, survivors had lower D50 values of TRB repertoire than non-survivors (P = 0.02, Student’s t test) (Fig. 2b). As for IGL and IGK, the D50 values were comparable between the two groups (data not shown). For a more intuitive comparison of V-J rearrangement diversity of these repertoires, we performed 3D mapping of V-J pairing of two patients with different outcomes as examples (Fig. 3, Supplementary Figure S3). It is evident that the IGH repertoire of the non-survivor was significantly more convergent to several particular IGHV-IGHJ gene pairs than the survivor. On the contrary, the diversity of TRBV-TRBJ gene pairs in non-survivors was higher than that in the survivors. V-J pairs in IGK repertoire also skewed in the non-survivor (Supplementary Figure S3). These results, along with the finding that the IGH repertoire changed constantly over time in a single individual, indicate that as compared to other repertoires, the IGH repertoire diversity might be more associated with the patients’ recovery and the lower TCR diversity is related to the better outcome.
Figure 2. D50 values of IGH and TRB repertoires in H7N9 infected patients.
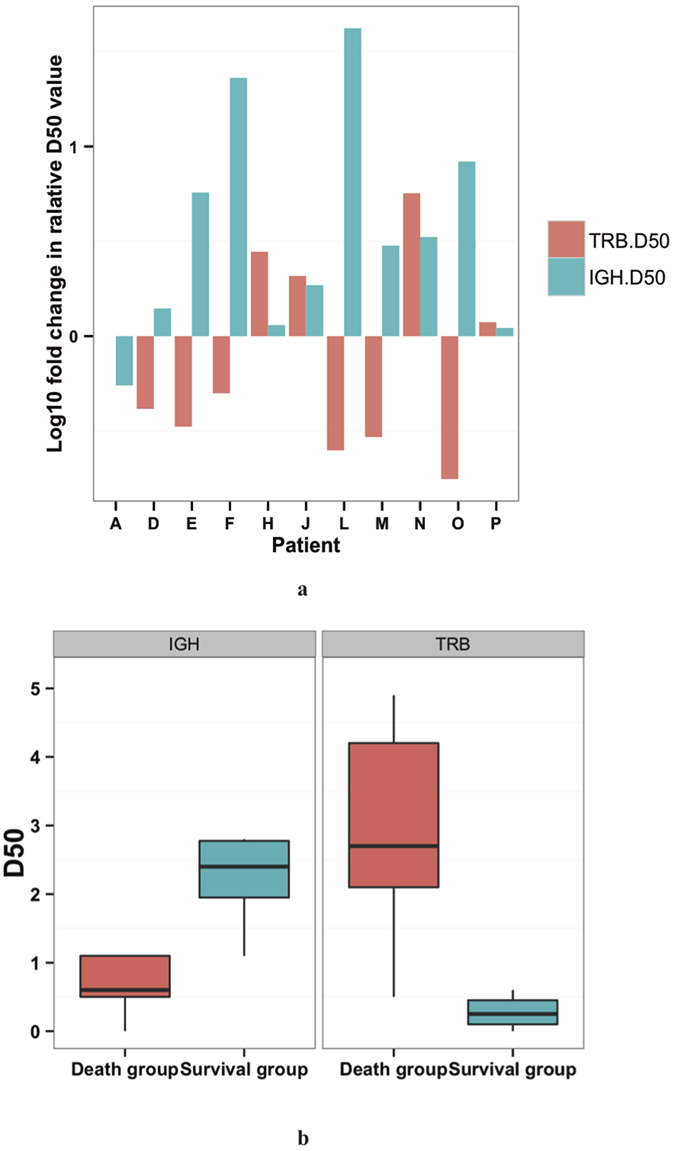
(a) shows the logged relative ratios of IGH (green) and TRB (red) D50 values of samples collected from each patient. The D50 values >15 days were divided by D50 value <15 days and the logged result were drown as vertical axis values. Every pair of green and red bars represented an individual. (b) is the boxplot of the D50 value of IGH and TRB repertoires in 15–42 days after onset by group.
Figure 3. Comparison of overall diversity of V-J gene pairing in IGH and TRB repertoires between.

(A,B) were representative IGH repertoires from a non-survivor and a survivor, respectively. (C,D) were TRB repertoires from the same patient as A and B at the same time point. Diversity of IGH repertoires was higher in survivor, while that of TRB repertoires was lower in survivor (Supplementary Figure S3).
Recombination patterns of V and J gene segments showed conserved patterns among patients
To further investigate H7N9-specific alterations of immune repertoire during infection, we questioned if there was distinct V or J gene or V-J pairing bias in survivors. For IGH repertoires, most of the IGHV and IGHJ genes and pairs were comparable across different patients (Fig. 4a, Supplementary Figure S4a). There were some IGHV and IGHJ genes of low frequency identified more in non-survivors than in survivors (Fig. 4b). These signatures are similar with previously reported antibody repertoires after vaccination17. In IGH repertoires, the V and J gene usage distribution pattern was similar in each unique CDR3 and all CDR3 clones (r = 9.59, p < 0.001) (Supplementary Figure S4b). This suggests that the biased usage of V and J genes was not only a result of the high expression level of some clones but also the expansion of particular cell clones in IGH repertoire. Frequency of TRBV and TRBJ genes are also not significant between survivors and non-survivors (Fig. 4c,d), indicating that V-J gene usage is biased in patients but not related to their prognosis.
Figure 4. V and J gene usage in H7N9 infected patients by outcome.

(a) shows frequency of IGHV gene usage in patients from non-survival group (red) and survival group (green). IGHV3-23 (9.73%), IGHV4-59 (8.76%) and IGHV3-30 (8.33%) were the three most frequent IGHV genes. (b) shows IGHJ gene usage frequency among patients. Expressing of IGHJ4 composed 44 ~ 64% of the repertoires. (c,d) show frequencies of TRBV and TRBJ genes usage. TRBV28 (14.28%), TRBV27 (12.69%) and TRBV20-1 (10.77%) accompanied by TRBJ1-1 (18.23%), TRBJ2-1 (16.31%) and TRBJ 2-3 (15.67%), were the three most frequent TRBV and TRBJ genes. Data are represented as mean ± SEM (See also Supplementary Figure S2).
Immune repertoires showed less CDR3 overlap in Ig repertoire across different individuals
Trimming and addition during combination effects the length of CDR3s, because the length of CDR3 loop influences its shape and ability to fold both on itself and in the company of others loops such as the CDR1 and CDR218,19. We found that in some patients, amino acid lengths of unique IGH CDR3s using IGHV1-69 gene were skewed to a particular length, resulting in a significant perturbation of overall distribution (Supplementary Figure S5).
We compared the sequence identity of IGH CDR3s in response to H7N9 across different individuals. Overlap between different patients was very limited, even in intergroup pairs (data not shown). In addition, we failed in identifying any IGH sequences differently expressed in two groups. However, we found some TRB, IGL and IGK CDR3 sequences shared among most of the patients. Interestingly, survivors shared many TRB CDR3 sequences that found to be absent or expressed at a much lower level in non-survivors (p < 0.05) (Fig. 5). These shared sequences in TRB were predominately not dominant clones but of high similarity, which indicates that these sequences may have similar function and are related to recovery from infection. For IGK and IGL, patients with different prognosis expressed comparably high level of same or similar CDR3 sequences, including dominant clones (Supplementary Figure S6).
Figure 5. Shared TRB CDR3 sequences expressed at different levels between survivors and non-survivors.

Comparison of the expression level of the shared sequences was performed using Wilcoxon Test, and the representative sequences expressed significantly different between survivors and non-survivors were drawn (p < 0.05). Each Colum is for one patient and the annotation bar represents the outcome of the patient (green for non-survival group, purple for survival group). Color of each rectangle stands for logged reads of the clone noted at right sided of the panel (blue as lowest, red as highest).
Identifying broadly-neutralizing and antigen-specific antibody sequences from Ig repertoires
Although our results suggest that the diversity of IGH repertoire was associated with prognosis in H7N9 patients, it remains a significant challenge to understand the immunological basis of this observation. It has been reported that the human antibody repertoire becomes highly skewed in response to influenza vaccination with some B cell clones expanding in the setting of decreased diversity20, which may suggest that the immune system was primed to produce specific influenza-specific antibodies. However, we found that the diversity of IGH repertoire presented a rising trend during H7N9 infection, accompanied by the increase of Nab titer (Supplementary Figure S2). In addition to this, the survivors present the higher diversity and Nab titers compared to non-survivors, suggesting that the extensive B cell clonal expansion is not necessarily correlated with improved outcomes, and that human immune system follows a different pattern in response to natural infection as compared to vaccination.
To examine whether the highly diverse IGH repertoires in some H7N9 patients correlate with the presence of neutralizing antibodies, we firstly compared our deep sequencing data to the reported influenza bnAbs sequences. Surprisingly, we found a panel of clones that have almost identical IGH CDR3 with a previously reported pan-influenza A neutralizing antibody, FI39021. As shown in Fig. 6a, one of these clones, N2_3050, shared ~92% identity with FI390, with a difference of only seven residues in all three CDR regions. This convergence is remarkable considering that IGH repertoires are very diverse among different people, and not a single shared IGH CDR3 sequence can be found in the H7N9 patients investigated here. All these clones were from the samples of two survivors, collected during the convalescent phase (P2, N2). These results suggest that bnAbs may have been elicited in some patients.
Figure 6. Identification of broadly neutralizing antibody (bnAb) and H7N9 specific antibody in Ig repertoires of H7N9 infected patients.

(a) Alignment of previously reported bnAb sequence (FI390) and the antibody sequence found in H7N9 patient. FI390 was as a bnAb against influenza virus reported by Pappas L. N2_3050 was the IGH sequence found in two recovered H7N9 patients with identical CDR3 amino acid sequence as FI390. Alignment was performed using Clustal W2. (b) Binding kinetics of scFvs N2_3050, o1b1 and l1b1 to H7N9 HA antigen using Surface Plasmon Resonance assays. Different concentrations (10 uM, 2 uM, 400 nM, 200 nM, 100 nM and 50 nM for N2_3050 and c1b1; 1 uM, 500 nM, 250 nM, 125 nM, 62.5 nM and 31.5 nM for l1b1, respectively) is used in the test. Calculated KD values are shown in the table presenting binding affinity of each scFvs.
We next sought to identify H7N9-specifc lineage members from IGH repertoires. We recently identified a human neutralizing antibody (Supplementary Table S5) from a non-immune human antibody library, which specifically neutralizes H7N9 influenza virus. We searched our deep sequencing database on the basis of in the last blood sample from all patients for sequences that could be clonally related to this antibody as they share the same IGHV1-69 and IGHJ1 germline families with a 14 amino acid long HCDR3. Interestingly, a number of clonally related sequences have been successfully identified in 8 of 10 H7N9 survivors, but have not been found in any of the non-surviving patients. Similarly to bnAb-related clones identified in patients P and N (survival group, 0.002–0.008% of the B-cell repertoires), these possible H7N9-specific lineage members represent a minor population (0.0001–0.01%) within the B-cell repertoire of each patient.
To further validate the functions of identified sequences in vitro, we synthesized some clonally related sequences, expressed the antibodies in scFv format, and measured their binding affinities to H7N9 HA (Table 1). 5 VHs that have similar sequences to our previous identified H7N9 antibody, 3 VHs that have similar sequences to another recently reported H7N9 human antibody22, as well as the VH of clone N2_3050 that shares 92% identity with the previously reported influenza A neutralizing antibody FI390, were fused with an identical VL to produce the scFv antibodies. We found that 5 of these 9 constructs could be solubly expressed in E. Coli, while the other 4 scFv only formed inclusion bodies. We next measured their binding to H7N9 HA using a SPR assay. As shown in Fig. 6b, we found that although scFvs e3b3 and m2a1 did not bind H7N9 HA, all the other three scFvs, N2_3050, o1b1 and l1b1, showed evident binding with different affinities (2.7 × 10−7, 2.0 × 10−7 and 5.9 × 10−8, respectively). The relatively low affinity is probably due to the fact that it was impossible to predict the naturally occurring VLs that were paired to VHs in these scFvs. Despite this, these results suggest that functional H7N9-specifc lineage members could be predicted from IGH repertoires.
Table 1. Expression and HA7 binding affinity of sequences clonally related to H7N9 antibodies.
| Sequence ID | V gene | D gene | J gene | CDR3 | Expression | Function |
|---|---|---|---|---|---|---|
| N2_3050 | IGHV1-69*09 | IGHD6-19*01 | IGHJ4*02 | ARASRYSSGWYYFDY | Yes | Yes† |
| o1b1 | IGHV1-69*01 | IGHD1-20*01 | IGHJ3*02 | ARSNYNPLLAAFDI | Yes | Yes |
| l1b1 | IGHV1-69*01 | IGHD5-18*01 | IGHJ5*02 | ARGYSYGLREWFDP | Yes | Yes |
| e3b1 | IGHV1-69*09 | IGHD7-27*01 | IGHJ4*02 | ARDAGSTWGIYFDS | No* | No |
| e3b3 | IGHV1-69*02 | IGHD2-21*02 | IGHJ3*01 | ARDPAGGDRDAFDV | Yes | No |
| c1b1 | IGHV1-69*09 | IGHD4-17*01 | IGHJ5*02 | ARGLNYGDVGWFDP | No | No |
| m2a1 | IGHV4-31*03 | IGHD3-3*01 | IGHJ6*03 | ARDGPYYDFWSGREPDV | Yes | No |
| p2a1 | IGHV4-31*03 | IGHD4-17*01 | IGHJ6*02 | ARANYGDRHTYYYGMDV | No | No |
| p2a3 | IGHV4-31*03 | IGHD4-17*01 | IGHJ6*02 | ARGDYGDRYYYYYGMDV | No | No |
IGH- immunoglobulin heavy chain; TRB – T cell receptor beta chain.
*No Expression means scFv only formed inclusion bodies.
†Antibodies with KD(M) < 10−7 are considered to be functional.
Taken together, these results may suggest that a higher B-cell repertoire diversity in H7N9 infected patients is associated with the efficient production of neutralizing antibodies and, in turn, a better clinical outcomes.
Discussion
New research has highlighted alterations in the T cell and B cell repertoires following vaccination and natural infection with the influenza virus17,23,24. Here we analyzed TRB and Ig repertoires of patients infected with influenza H7N9 virus in 2013, reported their diverse signatures, and examined their relationship with outcome. Furthermore, we report the development of antibodies specific to H7N9 and suggest that their presence may correlate with higher IGH diversity and favorable outcomes.
During acute viral infection, the immune system is activated with clone expansion, resulting in a more oligoclonal library25. In this study we used a novel index for describing and compare the diversity between immune repertoires—D50. In contrast to the previously reported Shannon index and Simpson index, the D50 value is understandable and direct. Meanwhile, relevance between these two indexes is impressive (Supplementary Table S3). Diversity of IGH was positively correlated with outcome. Effective somatic hyper-mutation and affinity maturation of B cells may be a possible mechanism for the high diversity and better outcome in survivors. Moreover, combined with the serologic studies, the IGH diversity showed increasing trend overtime accompanied with the increase of the Nab titer, indicating a more diverse IGH repertoire might be associated with the capacity for producing protective antibodies.
How TCR diversity impacts effective immunity remains unclear. Our results suggest that compared to Ig repertoire, which benefits patients by maintaining the diverse to generate antibodies with higher affinity, distinct correlation of T cell diversity and prognosis attributes to their difference in antiviral mechanisms. Without the somatic hyper-mutation process in B cells, T cell diversity depends on baseline diversity of T cell before infection and level of clonal expansion after antigen simulation. As TCR diversity is thought to be a prerequisite for immune system to recognize various foreign antigens, some earlier studies have suggested that the acquisition of cytotoxicity is independent of TCR usage and its diversity26,27. However, an early and robust H7N9-specific T cell response is critical for quick recovery from infection8. A recent study shows that, impairment of T cell proliferation and survival, not activation or effector function, results in prolonged virus infection in mice because of lacking sufficient antiviral T cells28. The relatively low T cell diversity of H7N9-infected survivors implies a more intense T cell expansion that contributes to virus elimination. This T cell response may be attenuated in the non-survivors. Thus, manipulating the expansion of T cells can be anticipated as a potential therapeutic intervention to benefit these patients29.
Identical TRB sequences between two individuals range from 1–10%30,31. The presence of these public sequences is suggestive to selective pressures of thymic selection and environmental exposure30. We noted that there are more shared CDR3 sequences were highly expressed in survivors, even though they were challenged by the same pathogen as non-survivors. This prognosis associated overlap suggests the effective TRB repertoire response is another possible mechanism for generating public CDR3s.
Importantly, our results provide evidence that functional neutralizing antibodies against the H7N9 virus could be identified using NGS methods. It has been recently recognized that different people may have convergent IGH gene rearrangements in their response to influenza vaccination20. In this study, we found that a number of sequences with stereotypical features of neutralizing antibodies can be identified from their B cell repertoires. We expect that the further improvement of deep sequencing related techniques, as well as a more comprehensive understanding of human antibodies, will enable a novel strategy to identify functional antibodies via immunogenetic analysis without the need of extensive in vitro screening procedures.
Notably, we have successfully identified a panel of sequences that could be clonally related to a previously reported bnAb. To the best of our knowledge, this is the first time that a bnAb against H7N9 virus has been identified in individuals without documented vaccination records. Recently, the discovery of bnAbs has renewed the interest in designing vaccines to elicit similar pan-influenza neutralizing antibodies in vivo21,32,33,34. However, the elicitation of these bnAbs has been found to be extremely challenging, because they only naturally arise in a small fraction of individuals after years of viral infection, and are highly divergent from their putative germline predecessors21. Therefore, a current priority is to understand how these antibodies have been elicited and mature in vivo, and translate this information into vaccine design. Interestingly, our results highlights the capability of deep sequencing techniques to identify and track the nAbs-related B cell clones in vivo, which will provide a foundation for understanding the mechanism of bnAb development and facilitate the development of possible “universal” influenza vaccines.
Our study has several limitations. Blood samples were taken from limited number of patients receiving standard treatment upon first pandemic of H7N9 virus. Some of them were treated with antibiotics, glucocorticoids, or anti-viral agents according to guidelines. However, the effects of these agents on immune repertoires have not been reported. Because H7N9 is a novel avian-origin influenza virus that had not been detected in humans previously, diagnosis in few patients was delayed, and thus immune repertoire changes at early stage of infection remain unclear. Despite of these, our results demonstrated a correlation between T cell and B cell diversity and patient outcome, suggesting a potential role of immune repertoire in influenza infection recovery.
This work represents the first time NGS has been used to analyze the immune repertoires of patients infected with the H7N9. The results reveal specific changes in human immune repertoires in response to H7N9 and provide insights into the immune repertoires that might provide improved antiviral immunity and thus improved recovery from infection. With further research in the field of immune repertoires, these results may have utility in assessing vaccine responses, and also in identifying antigen exposure or clinical diagnosis and evaluation, as well as antibody and vaccine development.
Methods
Patients
From March 2013 to June 2013, 18 individuals were diagnosed with H7N9 infection by viral isolation from throat swabs. Due to patient mortality and difficulty in providing follow-up, a total of 15 patients were enrolled in this study. Clinical characterization of these patients was presented in Supplementary Table S1 in the online data supplement, and management of individuals has been reported in our previous work4. All patients received anti-viral treatment. Among these patients, 5 died and the others survived to hospital discharge. The age, gender, and rate of complications are comparable between survivors and non-survivors. Blood samples were collected at multiple time points during critical illness. This study was approved by institution review board at Shanghai Public Health Clinical Center. All patient managements and blood sample collection were carried out in accordance with the relevant guidelines. Informed consents were obtained from all patients.
Neutralizing antibody assays
We used the previously describedvpseundovirus with non-replicative human immunodeficiency virus backbone carrying influenza A H7 and H9 to measure the neutralizing antibody titers of patient serum35. The highest serum dilution that gave ≥80% inhibitory concentration of the luciferase signal in virus-infected MDCK cells was defined as neutralizing titer.
Isolation of T cells and B cells and extraction of RNA
From anticoagulated whole blood, peripheral blood mononuclear cells (PMBCs) were obtained by Ficoll-gradient according to the manufacturers’ instruction, and the isolated cells were resuspended in RNA protect reagent (Qiagen).
Establishment of immune repertoires
We performed arm-PCR technology to amplify the cDNA semi-quantitatively according to the manufactures’ instructions (iRepertoire, Inc.) as has been described elsewhere36. Briefly, in the first few cycles, we used nested primers (Fo, Fi, Ro, Ri), which were designed specifically for every V and J (or C) genes and shared a common tag sequence. After that, the nested primers were removed from PCR products by exonuclease, and a pair of communal primers, called “superprimer”, was added to pair with the common tag sequence in the products and accomplish the exponent phase. Unique barcoded primers were used to distinguish each sample. After gel purification, the library constructed by previous steps was pooled and underwent high-throughput sequencing based on Illumina MiSeq platform according to the manufactures’ protocol.
Alignment of CDR3 sequences
Complimentary determining regions (CDR3s) were identified as the interval between two conserved amino acid sequences—Y[YFLI]C at the 3′ end of the V gene segment and [FW]GXGT (X stands for 1 of 20 amino acids) within the J segments. Raw data was analyzed by iRepertoire using the previously described IRmap program36. The best matches of germline V and J gene were searched by determining alignments between Illumina platform product and germline sequences in the IMGT/GENE-DB database.
D50 value for diversity comparison
To make the diversity comparison easier and to analyze diversity statistically, we used a new index – D50 value in our work. It is the calculated percentage of dominant unique clones, accumulative reads of which made up for 50% of the total (ranges from 0 to 50 in theory) (Supplementary Table S2, Supplementary Figure S1). To stress clone expansion, avoid the bias from sequencing depth and reduce noise, we used the sequences ranked within 10000 when calculating and excluded others. D50 value of a specific repertoire is positively related to diversity.
Antibody expression
The scFv gene were synthesized by Genscript (Piscataway, NJ) and cloned into pComb3x vector. The plasmids were transferred into HB2151 cells, and freshly single colonies were inoculated into SB medium and induced by 1 mM isopropyl-1-thio-β-D-galactopyranoside for large-scale expression. The antibodies were purified by nickel-nitrilotriacetic acid resin (Qiagen, Valencia, CA) according to the manufacturer’s protocols. Protein purity was estimated as >90% by SDS–polyacrylamide gel electrophoresis, and protein concentration was measured spectrophotometrically (NanoVue, GE Healthcare).
Surface Plasmon Resonance binding experiments
The binding experiments were performed using a ProteOn XRP36 system (Bio-Rad, Hercules, CA) to determine the kinetics of scFvs N2_3050, o1b1 and l1b1 to H7N9 HA antigen (Sino Biological Inc.). H7N9 HA was immobilized on the ProteOn GLM biosensor chip using standard amine coupling chemistry (300 nM in 10 mM sodium acetate buffer, pH 5.0). The surface of sensor chip was activated by 200 mM 1-ethyl-3-dimethyl aminopropylcarbodiimide hydrochloride and 50 mM N-hydroxysulfosuccinimide. O1b1 and l1b1 were prepared in phosphate buffer saline (pH 7.4) containing 0.005% Tween-20 and injected at 50 μl/min for 120 s at different concentrations (10 uM,2 uM, 400 nM, 200 nM, 100 nM and 50 nM for N2_3050 and c1b1; 1 uM, 500 nM, 250 nM, 125 nM, 62.5 nM and 31.5 nM for l1b1, respectively). The dissociation phase was followed for 600 s. The chip surfaces were regenerated by injecting 10 mM glycine HCl, pH 2.0, 100 μl/min for 18 s. The data were analyzed using ProteOn Manager 3.1 software and fitted to a 1:1 interaction model.
Data analysis
When comparing the reads between samples, we used fraction or normalized the reads to a total of 10,000,000 to avoid any bias caused by sample size. The comparison of D50 index was performed using Student’s t test. Expression level of each sequence or V-D-J gene and was compared by Wilcoxon test or Mann-Whitney U test, respectively. Statistical analysis was conducted using SPSS software (version 21.0) and R (3.1.2). Structured Query Language (SQL) was used for immunogenetic analysis. Sequence alignments were made with ClustalW2.
Additional Information
How to cite this article: Hou, D. et al. Immune Repertoire Diversity Correlated with Mortality in Avian Influenza A (H7N9) Virus Infected Patients. Sci. Rep. 6, 33843; doi: 10.1038/srep33843 (2016).
Supplementary Material
Acknowledgments
This research was supported by the National Natural Science Foundation of China (81170056, 81490533, 81100046), and by grant B115 from Shanghai Leading Academic Discipline Project. Dr. YL Song was supported by the State Key Basic Research Program (973) project (2015CB553404), Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Key Medical grant from Shanghai Science and Technology Committee (11411951102, 12JC1402300), and supported by Doctoral Fund of Ministry of Education of China (20130071110044).
Footnotes
Author Contributions D.H. and T.Y. analyzed data and wrote the draft of the manuscript. L.W. and C.C. performed antibody expression and HA binding tests. S.L., X.X. and T.L. collected blood samples. Q.W. performed the PBMC isolation and arm-PCR. Z.Z., J.L. and X.T. collected clinical data. C.W. and M.B.-S. performed Illumina sequencing. C.B., J.Z. and J.X. revised the MS. J.Q. and E.S. revised the MS and provided framework of the MS. J.H. designed the study and provided the primers. Y.S. designed and supervised the study and revised the manuscript.
References
- WHO. Assessment WR. Summary of Surveillance and Investigation findings-Human Cases of Avian Influenza A (H7N9) Virus Infection to Date, http://www.who.int/influenza/human_animal_interface/influenza_h7n9/riskassessment_h7n9_2Oct14.pdf (2014).
- Gao H. N. et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. The New England journal of medicine 368, 2277–2285, 10.1056/NEJMoa1305584 (2013). [DOI] [PubMed] [Google Scholar]
- Yu L. et al. Clinical, virological, and histopathological manifestations of fatal human infections by avian influenza A(H7N9) virus. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 57, 1449–1457, 10.1093/cid/cit541 (2013). [DOI] [PubMed] [Google Scholar]
- Lu S. et al. Prognosis of 18 H7N9 avian influenza patients in Shanghai. PloS one 9, e88728, 10.1371/journal.pone.0088728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Parra S. et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proceedings of the National Academy of Sciences of the United States of America 111, 1049–1054, 10.1073/pnas.1322229111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot A. S. et al. Low immunogenicity predicted for emerging avian-origin H7N9: implication for influenza vaccine design. Human vaccines & immunotherapeutics 9, 950–956, 10.4161/hv.24939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sandt C. E. et al. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. Journal of virology 88, 1684–1693, 10.1128/JVI.02843-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nature communications 6, 6833, 10.1038/ncomms7833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsworth D. J., Castellarin M. & Holt R. A. Sequence analysis of T-cell repertoires in health and disease. Genome medicine 5, 98, 10.1186/gm502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou G. et al. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nature biotechnology 32, 158–168, 10.1038/nbt.2782 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calis J. J. & Rosenberg B. R. Characterizing immune repertoires by high throughput sequencing: strategies and applications. Trends in immunology 35, 581–590, 10.1016/j.it.2014.09.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky B. J. et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nature biotechnology 31, 166–169, 10.1038/nbt.2492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C., Sit R. V., Weinstein J. A., Dekker C. L. & Quake S. R. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proceedings of the National Academy of Sciences of the United States of America 110, 13463–13468, 10.1073/pnas.1312146110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N. et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Science translational medicine 5, 171ra119, 10.1126/scitranslmed.3004794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart P. J. Immunology: the roots of antibody diversity. Nature 419, 29–31, 10.1038/419029a (2002). [DOI] [PubMed] [Google Scholar]
- Zhang A. et al. Kinetics of serological responses in influenza A(H7N9)-infected patients correlate with clinical outcome in China, 2013. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin 18, 20657 (2013). [DOI] [PubMed] [Google Scholar]
- Laserson U. et al. High-resolution antibody dynamics of vaccine-induced immune responses. Proceedings of the National Academy of Sciences of the United States of America 111, 4928–4933, 10.1073/pnas.1323862111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M. M. et al. T cell receptor CDR3 loop length repertoire is determined primarily by features of the V(D)J recombination reaction. European journal of immunology 33, 1568–1575, 10.1002/eji.200323961 (2003). [DOI] [PubMed] [Google Scholar]
- Davis M. M. et al. Ligand recognition by alpha beta T cell receptors. Annual review of immunology 16, 523–544, 10.1146/annurev.immunol.16.1.523 (1998). [DOI] [PubMed] [Google Scholar]
- Jackson K. J. et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell host & microbe 16, 105–114, 10.1016/j.chom.2014.05.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas L. et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 516, 418–422, 10.1038/nature13764 (2014). [DOI] [PubMed] [Google Scholar]
- Chen Z. et al. Human monoclonal antibodies targeting the haemagglutinin glycoprotein can neutralize H7N9 influenza virus. Nature communications 6, 6714, 10.1038/ncomms7714 (2015). [DOI] [PubMed] [Google Scholar]
- Galson J. D., Pollard A. J., Truck J. & Kelly D. F. Studying the antibody repertoire after vaccination: practical applications. Trends in immunology 35, 319–331, 10.1016/j.it.2014.04.005 (2014). [DOI] [PubMed] [Google Scholar]
- Robinson W. H. Sequencing the functional antibody repertoire-diagnostic and therapeutic discovery. Nature reviews. Rheumatology , 10.1038/nrrheum.2014.220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy J. J. & Weyand C. M. T cell development and receptor diversity during aging. Current opinion in immunology 17, 468–475, 10.1016/j.coi.2005.07.020 (2005). [DOI] [PubMed] [Google Scholar]
- Jenkins M. R., Kedzierska K., Doherty P. C. & Turner S. J. Heterogeneity of effector phenotype for acute phase and memory influenza A virus-specific CTL. Journal of immunology 179, 64–70 (2007). [DOI] [PubMed] [Google Scholar]
- La Gruta N. L. et al. Epitope-specific TCRbeta repertoire diversity imparts no functional advantage on the CD8+ T cell response to cognate viral peptides. Proceedings of the National Academy of Sciences of the United States of America 105, 2034–2039, 10.1073/pnas.0711682102 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen K. M. et al. T cell expansion is the limiting factor of virus control in mice with attenuated TCR signaling: implications for human immunodeficiency. Journal of immunology 194, 2725–2734, 10.4049/jimmunol.1400328 (2015). [DOI] [PubMed] [Google Scholar]
- Marchingo J. M. et al. T cell signaling. Antigen affinity, costimulation, and cytokine inputs sum linearly to amplify T cell expansion. Science 346, 1123–1127, 10.1126/science.1260044 (2014). [DOI] [PubMed] [Google Scholar]
- Warren R. L. et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome research 21, 790–797, 10.1101/gr.115428.110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins H. S. et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Science translational medicine 2, 47ra64, 10.1126/scitranslmed.3001442 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856, 10.1126/science.1205669 (2011). [DOI] [PubMed] [Google Scholar]
- Ekiert D. C. et al. Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251, 10.1126/science.1171491 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert D. C. et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850, 10.1126/science.1204839 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C. et al. Safe pseudovirus-based assay for neutralization antibodies against influenza A(H7N9) virus. Emerging infectious diseases 19, 1685–1687, 10.3201/eid1910.130728 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. et al. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proceedings of the National Academy of Sciences of the United States of America 107, 1518–1523, 10.1073/pnas.0913939107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.