

Abstract
Lipopolysaccharide (LPS) is a major component of the outer wall of gram negative bacteria. In high doses LPS contributes to the inflammation in gram negative sepsis, and in low doses contributes to the low grade inflammation characteristic of the metabolic syndrome. We wanted to assess the role of beta2-glycoprotein I (β2GPI) a highly conserved plasma protein and its different biochemical forms in a mouse model of LPS systemic inflammation. Normal and β2GPI deficient mice were administered LPS through their veins and assessed for a range of inflammation markers in their blood and liver. Different biochemical forms of β2GPI were measured in normal mice given either saline or LPS. We show that β2GPI has a significant role in inhibiting LPS induced inflammation. In this study we provide some evidence that β2GPI serves a protective role in a mouse model of LPS inflammation. This resolves the controversy of previous studies which used LPS and β2GPI in test tube based models of LPS induced activation of white cells. We also highlight the potential relevance of a newly discovered biochemical form of β2GPI in LPS mediated inflammation and we speculate that this form has a protective role against LPS induced pathology.
Lipopolysaccharide (LPS) is a major component of the outer membrane of gram negative organisms. In high concentrations in the plasma it has been implicated in the systemic inflammatory response associated with organ dysfunction in the setting of infection1. In low concentrations in the plasma it has been implicated in contributing to initiating obesity and insulin resistance in the metabolic syndrome by inducing a chronic inflammatory state2,3. LPS consists of three parts, lipid A, a core oligosaccharide, and an O side chain4. LPS activates both pro-inflammatory and anti-inflammatory mediators through toll-like receptor 4 (TLR4) signalling5. Dysregulation of both these processes can lead to hyperinflammation and immunosuppression5. LPS in plasma is bound by lipopolysaccharide binding protein which enables the interaction of LPS with its membrane receptor CD146. LPS is subsequently transferred to the MD-2/TLR4 complex6. This leads to oligomerization of MD-2/TLR4 complexes resulting in the recruitment of the intracellular adaptor proteins MyD88 and TRIF6. MyD88 recruitment leads to sequential phosphorylation of IRAK-4 and IRAK-1, activation of TRAF-6, culminating in NF-κB activation and the generation of pro-inflammatory cytokines6. TRIF forms a complex with TRIF related adaptor molecule and TLR4 resulting in the activation of IRF3, and the transcription of IFN-β6.
Beta 2-glycoprotein I (β2GPI/Apolipoprotein H) is an abundant plasma protein that is produced by the liver7. It is composed of five domains (DI-V), and has a molecular weight of approximately 50 kDa7. Domains I to IV each have two disulfide bridges, whereas domain V has three, including a disulfide bridge that incorporates the C-terminal cysteine7. Domain V also contains a positively charged lysine rich region, as well as a hydrophobic flexible loop segment, and both these regions are required for binding of β2GPI to negatively charged macromolecules7.
The domain V disulfide bond is susceptible to cleavage by the oxidoreductases thioredoxin I (TRX-1) and protein disulfide isomerase (PDI) leading to the generation of free thiols at Cysteine (Cys) 288 and Cys 3268,9. A large proportion of plasma β2GPI exists in the free thiol form10. The free thiol form has distinct properties to the oxidized form, the former protecting endothelial cells against hydrogen peroxide mediated cell death11. The implications being that in-vitro studies that look to delineate the biological function of β2GPI have limited in-vivo relevance if they only study the oxidized form and do not take the predominant in-vivo free thiol form into consideration.
A recent study has proposed that β2GPI may be able to attenuate the pro-inflammatory effects of LPS12. Using in-vitro techniques β2GPI bound to LPS, through domain V. The Kd range was between 62 nM and 23 nM depending on the LPS source12. Upon binding LPS, β2GPI undergoes a conformational change from a circular to an open form12. In-vitro β2GPI was able to attenuate LPS induced tissue factor activation and IL-6 release by human monocyte-like THP-1 cells and human umbilical vein endothelial cells12. This effect was negated with the use of receptor-associated protein (RAP), a potent inhibitor of endocytic receptors that belong to the low density lipoprotein (LDL) receptor gene family12. β2GPI has previously been shown to bind several members of the LDL receptor family, the most well studied being LDL receptor-related protein 8 (LRP-8), also known as apolipoprotein E receptor 2 (ApoER2)13. This receptor is expressed during differentiation of monocytes to macrophages14. These findings suggests that β2GPI may function as a scavenger protein for LPS, promoting its endocytosis by monocytes/macrophages through ApoER212. Furthermore the magnitude of fever and plasma inflammatory cytokine rise post LPS injection demonstrated an inverse relationship with serum levels of total β2GPI prior to LPS administration in male healthy human volunteers12.
In a different study β2GPI was shown to specifically bind to LPS15. However monocytes stimulated with LPS in the presence of β2GPI demonstrated comparable levels of pro-inflammatory cytokine production compared to cells stimulated with LPS in the absence of β2GPI15. A third report studying the in-vitro interaction of β2GPI with LPS failed to demonstrate specific binding of β2GPI to LPS16.
In view of these contradictory in-vitro studies we have used C57BL/6 β2GPI−/− (deficient) mice to delineate the in-vivo relevance of β2GPI in LPS pathophysiology. Hence bypassing the limitations of in-vitro studies such as LPS contamination of β2GPI and other components of the in-vitro system. In has been proposed that β2GPI in-vivo has different conformations (open and closed)17, as well as different post-translational redox forms (e.g. free thiol and non-free thiol)10,11, the function of β2GPI can only be accurately delineated in an in-vivo setting.
Results
C57BL/6 β2GPI deficient mice have significantly higher levels of inflammatory cytokines compared to C57BL/6 wild type (WT) mice at 2 and 6 h post LPS challenge
Significantly higher levels of TNFα were noted at both 2 and 6 h post LPS injection in the β2GPI−/− mice compared to the WT mice (Fig. 1A,B). Significantly higher levels of IL-6, IFN-γ, MIP-1α,β and Eotaxin-1 (Fig. 1C–G) were detected in the β2GPI−/− compared to WT mice 6 h post intravenous LPS administration.
Figure 1.

(A–G) β2GPI deficient mice had a significant increase in plasma inflammatory cytokines at 2 h and 6 h time points following LPS challenge. (A) TNFα at 2 h (B) TNFα at 6 h. For IL-6, IFNγ, MIP-1α, MIP-1β and Eotaxin (C–G) at 6 h. (□) = WT = Wild Type mice, (■) = β2GPI−/− = β2-glycoprotein I deficient mice. Mann-Whitney test n = 5 *p < 0.05, **p < 0.01, ***p < 0.001.
The levels of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-9, IL-10, IL-13, IL-17A, GM-CSF, KC, MCP-1, IL-12p40, IL-12p70, G-CSF and RANTES were either the same between WT and β2GPI−/− mice or below the detection limit of the assay (data not shown). Levels of cytokines noted in Fig. 1A–G were the same in WT and β2GPI−/− mice given pyrogen free saline.
Histological changes in the liver after LPS administration
Liver tissue was obtained at 6 h following intravenous LPS administration. β2GPI−/− compared to WT mice demonstrated increased staining with anti-Gr-1 antibody (representative images of Gr-1 staining Fig. 2A–F) consistent with increased neutrophil infiltration into the liver (211.2 vs. 90.2 (positive cells/per 10 high power fields (HPF)), n = 5, p = 0.03) (Fig. 2G). No significant difference was detected in the number of apoptotic cells in the livers of the β2GPI−/− mice 6 h post LPS compared to WT at the identical time point. There was a significant increase in neutrophil infiltration in the β2GPI−/− compared to WT livers when mice were administered pyrogen free saline n = 5, p = 0.03 (Fig. 2G).
Figure 2. Representative images of livers immunostained for Gr-1.

WT (A–C); (A) Normal Saline (B) LPS at 200× magnification (C) LPS at 400× magnification. β2GPI−/− (D–F); (D) Normal Saline (E) LPS at 200× magnification (F) LPS at 400× magnification. Arrows indicate representative positive immunostaining for Gr-1 (brown staining). Quantitation of inflammatory cells staining for Gr-1 (granulocyte marker). (G) The number of anti-Gr-1 positive cells per 10 high power fields (HPF) at 400× magnification. Data represent mean ± SEM, n = 5 ***p < 0.001. Mann-Whitney test.
β2GPI deficient mice have a significant increase in serum alanine aminotransferase (ALT) following LPS administration
Serum ALT, a specific marker of liver damage, was significantly increased in β2GPI−/− mice following LPS injection compared to injection of β2GPI−/− mice with pyrogen free saline n = 8, p = 0.02 (Fig. 3). There was no significant difference seen in WT mice (Fig. 3).
Figure 3. β2GPI deficient mice have an increase in serum ALT following LPS administration.
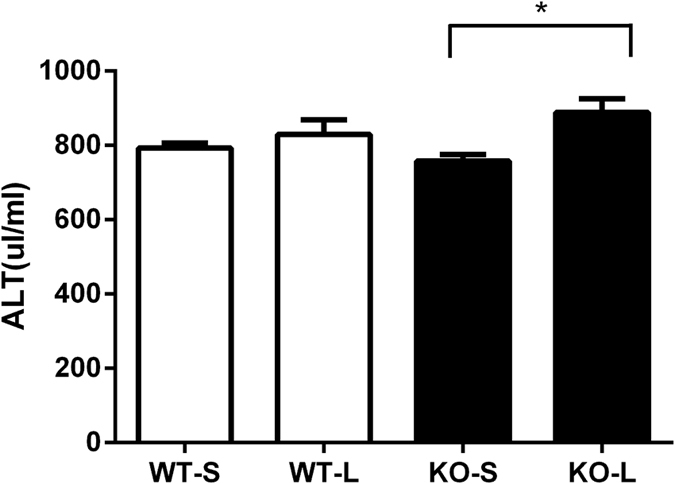
(□) = WT = Wild Type mice, (■) = β2GPI−/− = β2-glycoprotein I deficient mice. n = 8 *p < 0.05, S = Pyrogen Free Saline, L = LPS. Mann-Whitney test.
LPS administration to C57BL/6 WT mice decreased total, but increased free thiol β2GPI serum levels
Total β2GPI levels were significantly decreased in WT mice at 6 h after LPS injection compared to WT mice injected with pyrogen free saline, (86.1 ± 8.5 vs. 101 ± 10.28 mean (μg/ml) ± SD, n = 9, p = 0.004) (Fig. 4A). In contrast. the percentage of serum β2GPI in the free thiol form significantly increased after LPS injection compared to mice injected with pyrogen free saline, 108.4 ± 13.18 vs. 83.5 ± 16.94, mean (% of pooled normal standard) ± SD, n = 9, p = 0.003) (Fig. 4B). As expected there was no detectable β2GPI or free thiol β2GPI in the serum of β2GPI deficient mice (data not shown).
Figure 4. Total and free thiol β2GPI levels in mice following LPS injection.
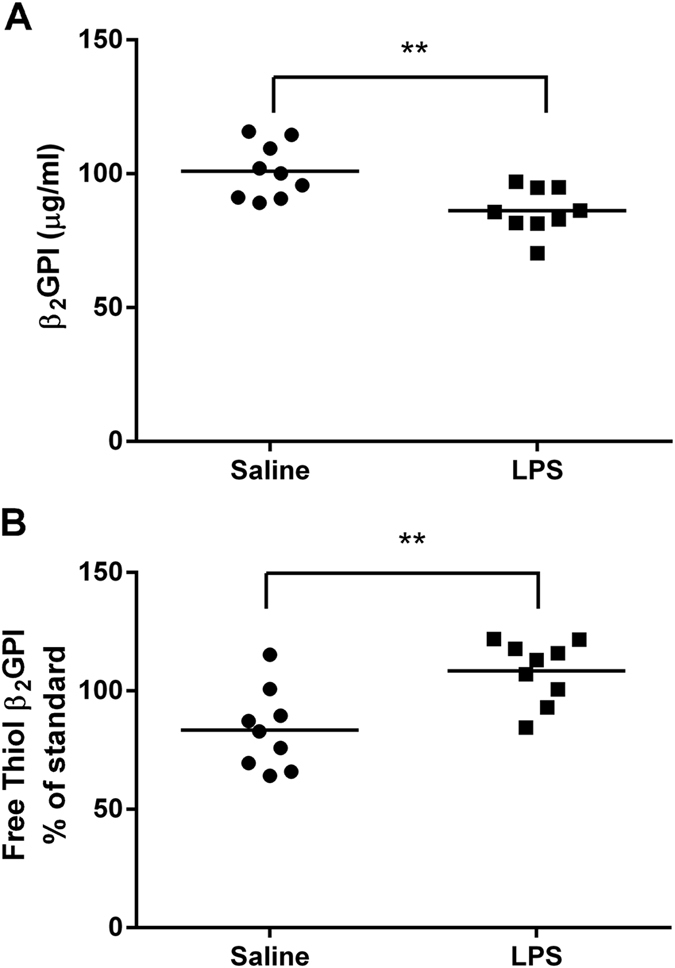
LPS decreased the (A) total but increased (B) free thiol β2GPI levels at 6 hrs after injection of LPS 1 μg/g body weight compared to injection of pyrogen free saline. n = 9 **p < 0.01. Unpaired two tailed Students t-test.
Treatment of oxidized β2GPI with reduced thioredoxin (TRX)-1 in the presence of LPS increases free thiol generation in β2GPI
In view of the increase in the percentage of free thiol β2GPI demonstrated in the serum of LPS treated mice compared to the decrease in total β2GPI levels, we assessed the in-vitro effect of LPS on TRX-1 induced free thiols in β2GPI. LPS but not lipid A at equivalent concentrations significantly increased free thiols in β2GPI (Fig. 5). There was no free thiol β2GPI generation when β2GPI was incubated alone or with LPS in the absence of TRX-1.
Figure 5. LPS but not Lipid A increases free thiol generation in β2GPI in the presence of TRX-1.
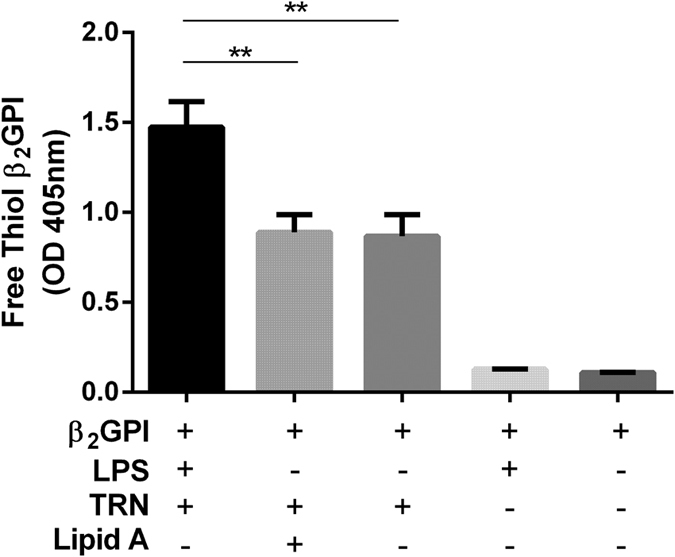
Free thiol β2GPI generation increased only when β2GPI was incubated with LPS, but not Lipid A or PBS in the presence of reduced thioredoxin-1 (TRX-1). There was no free thiol β2GPI generation when β2GPI was incubated with LPS or PBS in the absence of TRX-1. Data are mean ± SEM, n = 3 **p = <0.01. One Way ANOVA with Tukey’s multiple comparison test.
Discussion
The liver plays a major role in the clearance of LPS from blood, removing from the plasma up to 85% of LPS within 15 min of intravenous administration18. LPS is scavenged by hepatic sinusoidal macrophages (Kupffer cells) and hepatocytes19,20. Both cell types express the TLR4/CD14/MD-2 complex and respond to LPS with activation of NF-κB and MAPK, leading to rapid secretion of pro-inflammatory cytokines21. Tumour necrosis factor alpha (TNFα) concentrations have been reported to peak within 2 h of LPS challenge22. In C57BL/6 WT mice given a sublethal dose of LPS pro-inflammatory cytokines and chemokines peaked at 6 h post LPS injection (except TNFα)23. Both TNF-α and IL-1 can act in an autocrine and paracrine manner to regulate their own biosynthesis and that of IL-6 in the liver22. In our current study β2GPI deficient, compared to WT mice, have significantly higher TNFα levels at 2 and 6 h following LPS challenge and, as would be expected, TNFα levels peaked at 2 h. Serum protein levels of the pro-inflammatory cytokines IL-6, IFN-γ and the chemokines MIP-1α and MIP-1β were also significantly increased in β2GPI−/− compared to WT at the 6 h time point after the intravenous administration of LPS. On histological analysis the liver tissue of β2GPI−/− mice administered LPS displayed elevated levels of infiltrating anti-Gr-1 positive inflammatory cells within the liver parenchyma compared to WT mice. The β2GPI−/− mice administered LPS had an increase in serum levels of ALT which was not seen in the WT suggesting that there is a degree of liver injury. Collectively these findings support the concept that β2GPI in plasma binds to LPS and diverts the LPS molecules from activating TLR4 on cell surfaces including hepatocytes and Kupffer cells, thus ameliorating the subsequent activation of downstream transcription factors such as NF-κB that induce cascades of pro-inflammatory cytokine and chemokine production and release. Interestingly we noted that there was a significant increase of neutrophils in the liver of β2GPI deficient compared to WT mice given pyrogen free saline. This raises the possibility of low grade LPS translocation to the liver from the gut microbiome inducing increased neutrophil infiltration in the absence of β2GPI. Metabolic endotoxemia is the phenomenon of increased translocation of LPS from the gut microbiome into the systemic circulation in the setting of a high fat diet3.
It is pertinent to note that β2GPI has been associated with a protective effect against obesity in humans and other animal species24,25. Though the exact mechanism(s) has not been determined we speculate that β2GPI may protect against the inflammatory effects of metabolic endotoxemia by neutralizing LPS.
In this study we also demonstrate that the administration of approximately 1 μg/g of body weight LPS to WT mice, when compared to WT mice administered pyrogen free saline, resulted in a drop of approximately 15% of total serum β2GPI levels in the LPS treated group, from 101 μg/ml to 86 μg/ml. Total human β2GPI responses are more sensitive to LPS administration as it was noted there was a reduction of 25% of baseline total plasma β2GPI levels within 30 min of administration of 4 ng/kg body weight of LPS to healthy adult male volunteers12. In the study involving human volunteers it was proposed that the drop in total β2GPI levels is due to the uptake of the β2GPI-LPS complex by monocytes/macrophages involving the ApoER2 receptor12. It is apparent however that the magnitude of the drop in total β2GPI levels is out of proportion to the relatively small amounts of LPS administered when taking into account a 1:1 stoichiometry of the β2GPI and LPS interaction. This suggests that it is not only β2GPI bound to LPS that is removed from the circulation, but also β2GPI not attached to LPS. We propose that it is the drop in the non free thiol form of β2GPI (oxidized β2GPI) that is likely to account for the fall in total β2GPI levels. Total β2GPI plasma levels are the sum of free thiol plus non-free thiol β2GPI levels. In the study by Agostinis et al. oxidized (perchloric acid treated) human β2GPI when administered to mice intravenously promptly localized in an intracellular location with a granular pattern in the liver26. These findings suggest that the liver may be the major organ responsible for the drop in total plasma β2GPI levels post LPS administration by taking up β2GPI that is oxidized plus also the β2GPI fraction that is bound to LPS. The finding of a higher percentage of free thiol serum β2GPI post LPS injection, despite the concomitant reduction in total β2GPI levels, suggests that free thiol β2GPI may mediate an important role in modulating LPS pathophysiology.
We have previously demonstrated that free thiol β2GPI can be generated when oxidized β2GPI is treated with TRX-18. Furthermore, it was demonstrated that free thiol β2GPI, once generated was able to protect endothelial cells in-vitro from hydrogen peroxide induced cell injury11. In the current study, conversion of free thiol β2GPI by TRX-1 was upregulated by the non-lipid A component of LPS. There are likely to be additional mechanisms that allow oxidized β2GPI to be converted to free thiol β2GPI in the setting of LPS exposure. It has been noted in a previous study that the disulfide reducing enzyme PDI is upregulated and expressed on the surface of splenocytes when mice are exposed to LPS27. We have previously shown that like TRX-1, PDI is able to reduce the Cys288-Cys326 disulfide bond in β2GPI9. Whether other thiol reducing enzymes secreted during LPS stimulation are able to act on β2GPI remains an open question. One possible candidate is gamma interferon-inducible lysosomal thiol reductase (pro-GILT) which is secreted by monocytes treated with LPS28.
The administration of LPS to mice in the study by Metcalfe et al. was noted to lead to the generation of free thiols in a number of diverse proteins on the surface of cells involved in B and T cell regulation and activation, suggesting the existence of a generic, extracellular, cell surface ‘redox-regulator’ mechanism in the setting of immune activation27. It is pertinent to note that both CD14 and MD-2 which form an important part of the LPS-TLR4 recognition/activation complex have been noted to have free thiols, as does LRP-8/ApoER227,29. We speculate that the dampening of the LPS pro-inflammatory response seen in the WT compared to the β2GPI−/− mice may be due to the ability of free thiol β2GPI to potentially interact in vivo with the free thiols present in molecules such as CD-14 or MD-2 or ApoER2, perhaps leading to the formation of covalent disulfide bonds between β2GPI and the respective molecules. For example, if our hypothesis is correct, the interaction between free thiol β2GPI and free thiol ApoER2, may allow for more efficient scavenging of β2GPI-LPS complexes. The interaction between free thiol β2GPI and free thiol MD-2 we speculate may inhibit LPS induced cell activation. In the study by Mancek-Keber et al. it was noted that compounds that form a covalent bond with MD-2 through the free thiol at Cys133 were able to inhibit LPS signaling29. Cys133 is located in the hydrophobic pocket of MD-229, raising the possibility that the hydrophobic flexible loop within domain V of β2GPI may play a role in allowing access of the nearby β2GPI domain V free thiols Cys288 and Cys326. Such speculations warrant further investigation in order to precisely delineate how β2GPI dampens LPS pro-inflammatory responses. This has relevance not only in understanding mechanisms by which hyperinflammation in the setting of severe gram negative sepsis and septic shock can be abated, but also in terms of understanding how low grade endotoxemia encountered in the setting of metabolic syndrome, and which contributes to low grade systemic inflammation2, may be attenuated by understanding the role of redox β2GPI interacting with LPS.
Methods
Materials
E. coli LPS, (serotype 0111:B4), haematoxylin and eosin (H&E), bovine serum albumin (BSA), Tween 20 were purchased from Sigma (Sigma-Aldrich Inc, St Louis, MO). Lipid A was from Hycult biotech (Uden, The Netherlands). Bio-plex ProTM mouse cytokine 23-plex kit was purchased from Bio-Rad (Hercules, CA). Affinity purified murine IgG2 anti-β2GPI monoclonal antibody (moAb) (4B2E7) and affinity purified rabbit polyclonal anti-β2GPI antibody were produced as previously described30,31. Isotype control murine IgG2 and rabbit polyclonal IgG and 70 μm nylon mesh were purchased from BD PharMingen (San Diego, CA). Streptavidin-HRP, anti-rabbit HRP, anti-mouse HRP and anti-goat HRP were purchased from Dako (Carpinteria, CA). N-(3-maleimidylpropionyl) biocytin (MPB) was from Life Technologies (Grand Island, NY). PolyScreen polyvinyldiethylene fluoride (PVDF) transfer membrane, Western blot chemiluminescence reagents were purchased from GE Healthcare (Bucks, UK). An alanine aminotransferase activity assay kit was from Sigma. Streptavidin coated plates were from Thermo Fischer Scientific (Waltham, MA). Recombinant human TRX-1 and sheep polyclonal anti-mouse β2GPI was from R&D Systems (Minneapolis, MN), recombinant thioredoxin reductase (TRX-R) was from Vital Diagnostic (Lincoln, RI), and NADPH was from Merck (Billerica, MA).
Anti-mouse Gr-1 antibody, Anti-Ig HRP detection kit were purchased from BD Bioscience. DeadEndTM Fluorometric TUNEL System was purchased from Promega (Madison, WI).
Mice
Male C57BL/6 (WT) mice, 8–9 weeks old, were purchased from the animal resources centre (ARC) (Perth, WA, Australia). C57BL/6 β2GPI−/− mice were generated in our laboratory and have been previously reported32. All mice were housed in a specific pathogen free environment and health status regularly monitored. Male mice were injected intravenously (IV) with 1 μg/gram body weight (gbw) with E. coli LPS (Sigma-Aldrich) in sterile saline or the same volume of saline. 6 h post injection, mice were euthanized and approximately 1 ml of blood was collected by cardiac puncture. Serum was collected by centrifugation of clotted blood at 1500 g for 10 min. The livers were removed, weighed and fixed in 10% formaldehyde. All animal studies were approved by the Animal Ethics Committee of the University of New South Wales. All methods were performed in accordance with the University of New South Wales Animal Care Ethics Committee guidelines.
Histopathological examination
To evaluate the histological changes in each group, the right lobe of the liver from C57BL/6 and β2GPI−/− mice were fixed in 10% neutral phosphate-buffered formalin solution for 24 h immediately after euthanasia, embedded in paraffin, and cut into 5 μm thick sections. Tissue sections were then stained with either H&E or with anti-Gr-1 antibody to detect and quantitate neutrophils and monocytes or with DeadEnd Fluorometric TUNEL System to assess and quantitate apoptotic cells according to the manufacturers’ instructions. Anti-Gr-1 and apoptotic cells were quantified by counting in ten random high power fields at 400× magnification by an observer blinded to the β2GPI status of the mice and treatments.
ELISA for total and free thiol β2GPI
A sandwich ELISA for quantifying total β2GPI levels in mouse serum samples was performed based on a previously published method10. Briefly, a high binding 96 well plate was coated overnight at 4 °C with sheep polyclonal anti-mouse β2GPI (10 nM/well). Plates were washed 4 times. Unoccupied sites were blocked with 2% BSA in PBS-T. Following washing, 100 μl of rabbit polyclonal anti-mouse β2GPI was added (10 nM/well, diluted in 0.25% BSA/PBS–0.1% Tween 20 (PBST)) and then 100 μl of mouse serum diluted 4,000-fold in PBST was coincubated for 1 h at RT. After washing 4 times, AP-conjugated goat anti-rabbit IgG was added (1:1,500 dilution) and incubated for 1 h at RT, and samples read at OD 405 nm after addition of chromogenic substrate.
The relative amount of β2GPI with free thiols in mouse serum was performed as previously described10,11.
Thioredoxin-1 reduction of oxidized β2GPI
Human recombinant TRX-1 (3.5 μM) was incubated for 1 h at 37 °C with TRX-R (500 nM) plus NADPH (200 μM) diluted in Hanks Balanced Salt Solution. TRX-R activated TRX-1 was diluted with an equal volume of β2GPI (4 μM) with LPS (10–1000 pg/ml) or Lipid A and PBS as control and incubated for another 1 h at 37 °C. β2GPI incubated with LPS or PBS in the absence of TRX-1, TRX-R and NADPH were further controls.
These reactions were performed under argon gas at all times to maintain proteins in their free thiol state. Free thiol β2GPI was labeled by MPB (200 μM) for 30 min at RT in the dark with agitation. After MPB labeling, mixtures were added to β2GPI deficient human plasma at a final concentration of 200 μg/ml. Unbound MPB was then removed by acetone precipitation. The protein pellet was resuspended in PBS-0.05% Tween 20 (final dilution 100 fold). The samples were then diluted a further 40 fold, added in duplicate to a streptavidin coated 96 well plate (100 μl/well), and incubated for 90 min at RT. Prior to addition of MPB labeled serum samples, streptavidin coated plates were washed 3 times with PBST and blocked with 2% BSA/PBST. After washing 3 times with PBST, a rabbit polyclonal anti-β2GPI was added (25 nM) and incubated for 1 h at RT. After 3 further washings with PBST, AP conjugated goat anti-rabbit IgG (1:1,500) was added for 1 h at RT and samples read at 405 nm after addition of chromogenic substrate.
Cytokine Assays
Cytokine assays were performed at 2 and 6 h time points following LPS or pyrogen free saline injection. At 2 h the levels of proinflammatory cytokines IL-6, MCP-1, IFNγ, TNFα, IL-12p70 and anti-inflammatory cytokine Il-10, were quantified in the plasma sample using BD Cytometric Bead Array (CBA) Mouse Inflammation Kit and BD FACSCantoll according to manufacturer’s instructions.
At 6 h the levels of various cytokines and chemokines (IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-17A, Eotaxin, G-CSF, GM-CSF, IFN-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES, TNF-α) were quantified by Bio-plex ProTM mouse cytokine 23-plex Group I kit according to the manufacturer’s instructions. Briefly, 50 μl of diluted samples were incubated with 50 μl of the magnetic beads covalently coated with Abs specifically reacting with each of the analytes. Discrimination of individual analytes within a multiplex suspension was achieved by fluorescence with a distinct spectral address. The suspension was incubated for 30 min at RT in the dark on a shaker at 850 rpm. After washing 3 times, 25 μl of biotinylated detection Abs were added to create a sandwich complex and incubated at RT for 30 min in the dark on a shaker at 850 rpm. After washing a further 3 times, 50 μl of streptavidin phycoerythrin (SA-PE) conjugate was added and incubated for 30 min in the dark with agitation followed by 3 washes and 125 μl washing buffer was added into each well to resuspend the sandwich complex. Data were acquired and analyzed on a Bio-Plex 200 System (Bio-Rad Laboratories). The minimum detection level for each analyte was 1 pg/ml.
Statistics
Graph Pad Prism 6 evaluation software (Graph Pad, San Diego, CA) was used for data processing and analysis. Values for all measurements are expressed as mean ± standard error of the mean (SEM). One-way ANOVA with the Tukey’s multiple comparison test was used when there were more than 2 groups to compare. The unpaired two tailed Student’s t test was used to evaluate significant differences when two groups were being compared when the data was normally distributed. The Mann-Whitney test was used to evaluate for significant differences between 2 groups when the data was non-parametric. p values of <0.05 were considered statistically significant.
Additional Information
How to cite this article: Zhou, S. et al. Gram Negative Bacterial Inflammation Ameliorated by the Plasma Protein Beta 2-Glycoprotein I. Sci. Rep. 6, 33656; doi: 10.1038/srep33656 (2016).
Acknowledgments
The study was partly funded by a grant to B.G. and F.E.-A. from the St. George and Sutherland Medical Research Foundation and the National Health and Medical Research Council of Australia to S.A.K. and B.G.
Footnotes
Author Contributions S.A.K. and B.G. conceived the project, designed experiments and analyzed data. S.Z., M.Q., F.E.-A., Y.W., S.D., G.C., J.C.W. and J.B. designed, performed experiments and analyzed data. S.Z., M.Q., F.E.-A., Y.W., S.D., G.C., L.C., D.Y., J.C.W., J.B., S.A.K. and B.G. had input in writing the paper.
References
- Angus D. C. & van der Poll T. Severe sepsis and septic shock. The New England journal of medicine 369, 2063 (2013). [DOI] [PubMed] [Google Scholar]
- Cani P. D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007). [DOI] [PubMed] [Google Scholar]
- Cani P. D. et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008). [DOI] [PubMed] [Google Scholar]
- Raetz C. R. & Whitfield C. Lipopolysaccharide endotoxins. Annual review of biochemistry 71, 635–700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paracha R. Z. et al. Formal Modelling of Toll like Receptor 4 and JAK/STAT Signalling Pathways: Insight into the Roles of SOCS-1, Interferon-beta and Proinflammatory Cytokines in Sepsis. Plos One 9, e108466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. C., Yeh W. C. & Ohashi P. S. LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151 (2008). [DOI] [PubMed] [Google Scholar]
- Miyakis S., Giannakopoulos B. & Krilis S. A. Beta 2 glycoprotein I--function in health and disease. Thrombosis research 114, 335–346 (2004). [DOI] [PubMed] [Google Scholar]
- Passam F. H. et al. Beta 2 glycoprotein I is a substrate of thiol oxidoreductases. Blood 116, 1995–1997 (2010). [DOI] [PubMed] [Google Scholar]
- Passam F. H. et al. Redox control of beta2-glycoprotein I-von Willebrand factor interaction by thioredoxin-1. Journal of thrombosis and haemostasis 8, 1754–1762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou Y. et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen beta2-glycoprotein I. Arthritis and rheumatism 63, 2774–2782 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou Y. et al. Naturally occurring free thiols within beta 2-glycoprotein I in vivo: nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood 116, 1961–1970 (2010). [DOI] [PubMed] [Google Scholar]
- Agar C. et al. beta(2)-glycoprotein I: a novel component of innate immunity. Blood 117, 6939–6947 (2011). [DOI] [PubMed] [Google Scholar]
- Pennings M. T. et al. Interaction of beta2-glycoprotein I with members of the low density lipoprotein receptor family. Journal of thrombosis and haemostasis 4, 1680–1690 (2006). [DOI] [PubMed] [Google Scholar]
- Watanabe Y. et al. Induction of LDL receptor-related protein during the differentiation of monocyte-macrophages. Possible involvement in the atherosclerotic process. Arteriosclerosis and thrombosis 14, 1000–1006 (1994). [DOI] [PubMed] [Google Scholar]
- Laplante P. et al. Interaction of beta2-glycoprotein I with lipopolysaccharide leads to Toll-like receptor 4 (TLR4)-dependent activation of macrophages. The Journal of biological chemistry 286, 42494–42503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gries A. et al. Biophysical analysis of the interaction of the serum protein human beta2GPI with bacterial lipopolysaccharide. FEBS open bio 4, 432–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agar C. et al. Beta2-glycoprotein I can exist in 2 conformations: implications for our understanding of the antiphospholipid syndrome. Blood 116, 1336–1343 (2010). [DOI] [PubMed] [Google Scholar]
- Zlydaszyk J. C. & Moon R. J. Fate of 51Cr-labeled lipopolysaccharide in tissue culture cells and livers of normal mice. Infection and immunity 14, 100–105 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathison J. C. & Ulevitch R. J. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. Journal of immunology 123, 2133–2143 (1979). [PubMed] [Google Scholar]
- Mimura Y., Sakisaka S., Harada M., Sata M. & Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology 109, 1969–1976 (1995). [DOI] [PubMed] [Google Scholar]
- Scott M. J., Liu S., Shapiro R. A., Vodovotz Y. & Billiar T. R. Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology 49, 1695–1708 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster M. I., Germolec D. R., Yoshida T., Kayama F. & Thompson M. Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology 19, 480–488 (1994). [PubMed] [Google Scholar]
- Srinivasan S., Leeman S. E. & Amar S. Beneficial Dysregulation of the Time Course of Inflammatory Mediators in Lipopolysaccharide-Induced Tumor Necrosis Factor Alpha Factor-Deficient Mice. Clinical and Vaccine Immunology 17, 699–704 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasstedt S. J., Coon H., Xin Y., Adams T. D. & Hunt S. C. APOH interacts with FTO to predispose to healthy thinness. Human genetics 135, 201–207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T. et al. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC physiology 10, 21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostinis C. et al. In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood 118, 4231–4238 (2011). [DOI] [PubMed] [Google Scholar]
- Metcalfe C., Cresswell P., Ciaccia L., Thomas B. & Barclay A. N. Labile disulfide bonds are common at the leucocyte cell surface. Open biology 1, 110010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackman R. L. & Cresswell P. Exposure of the promonocytic cell line THP-1 to Escherichia coli induces IFN-gamma-inducible lysosomal thiol reductase expression by inflammatory cytokines. Journal of immunology 177, 4833–4840 (2006). [DOI] [PubMed] [Google Scholar]
- Mancek-Keber M., Gradisar H., Inigo Pestana M., Martinez de Tejada G. & Jerala R. Free thiol group of MD-2 as the target for inhibition of the lipopolysaccharide-induced cell activation. The Journal of biological chemistry 284, 19493–19500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y. et al. Detection of ‘antiphospholipid’ antibodies: a single chromogenic assay of thrombin generation sensitively detects lupus anticoagulants, anticardiolipin antibodies, plus antibodies binding beta(2)-glycoprotein I and prothrombin. Clinical and experimental immunology 124, 502–508 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddel S. W., Wang Y. X., Sheng Y. H. & Krilis S. A. Epitope studies with anti-beta 2-glycoprotein I antibodies from autoantibody and immunized sources. Journal of autoimmunity 15, 91–96 (2000). [DOI] [PubMed] [Google Scholar]
- Sheng Y. et al. Impaired thrombin generation in beta 2-glycoprotein I null mice. The Journal of biological chemistry 276, 13817–13821 (2001). [DOI] [PubMed] [Google Scholar]