

Abstract
IL-7 and thymic stromal lymphopoietin (TSLP) are two major cytokines controlling murine B cell development. IL-7 has been studied extensively, but only recently has it become possible to unravel the role of TSLP in detail. We studied the biological activities of TSLP in B cell development at distinct ages in the mouse. On the one hand, TSLP is able to give rise to a measurable B1 cell compartment derived from fetal liver pro-B cells, although, as is the case for B2 cells, it does not play a prevalent role in the development of this subset. On the other hand, TSLP drives the proliferation of pro-B cells from the fetal and neonatal liver, but in the bone marrow environment, B cell precursors require pre-B cell receptor expression for TSLP responsiveness.
The process of B cell development ensures the continuous production of B cells expressing Ig antigen receptors (1). During this process, precursor populations at the pro-B cell stage rearrange the gene segments of the Ig heavy chain (IgH) (2, 3). Productively rearranged IgHs pair with the surrogate light chain, consisting of λ5 and Vpre-B, thereby forming the pre-B cell receptor (pBCR). Cells expressing the pBCR (pre-B cells) go on to rearrange their Ig light chain loci (2, 4, 5). Mice lacking λ5 are devoid of pre-B cells because of the inability of the B cell precursors to form the pBCR (3, 6). However, these mice still contain low numbers of mature B cells that accumulate with age (6). The pre-B cell compartment is further separated in large pre-B cells, containing dividing cells, and small, resting pre-B cells (2, 7). Both large and small pre-B cells are absent from recombination-activating gene 2-deficient (Rag2°) mice (8, 9) because of the inability to rearrange the IgH locus, and no B lymphocytes are produced in these mice.
In the bone marrow (BM), but not in the fetal liver (FL), pre-B cells are represented in much higher numbers than pro-B cells, indicating that a mechanism must operate in the BM to selectively enrich for cells expressing the pBCR. Expression of the pBCR was suggested to be sufficient to drive pre-B cell proliferative expansion (10), but the ability of pre-B cells to respond to lower concentrations of IL-7 has also been implicated as the cause for this selective enrichment (11, 12). However, these mechanisms do not exclude that pre-B cells would, in addition, acquire reactivity to other cytokines in the BM milieu, thus leading to a selective expansion of pBCR-expressing precursors.
Once produced in primary lymphoid organs, B lymphocytes accumulate in the periphery, where they can be divided into two major subpopulations, designated B1 and B2 (13). B1 cells differ in several respects from B2 (or conventional B) cells, including their anatomical location, cell surface phenotype, antibody repertoire, and developmental origin (reviewed in refs. 14–16).
Thymic stromal lymphopoietin (TSLP) is a cytokine that can drive B lymphopoiesis from FL or BM precursors (17–19). The activity of TSLP on FL and BM B cell precursors can, however, be distinguished on the basis of the target populations. TSLP is active on fetal pro-B cells and drives an IL-7-independent pathway of B cell production (20), whereas it does not induce proliferation of BM-derived pro-B cells (19).
B1 cells are most efficiently generated during fetal life (14), and it is known that the IL-7-independent pathway is only active in fetal/perinatal life (19, 21). In this work, we therefore assessed to what extent TSLP can drive the generation of the B1 compartment from early progenitors. We find that TSLP can drive the generation of B1 cells in the absence of IL-7 but that, under physiologic conditions, IL-7 is responsible for the development of the majority of cells in this subset.
In adult B lymphopoiesis the activity of TSLP is restricted to cells that have passed the pro-B cell stage (19), but the exact developmental stage in which TSLP is active is not known. We show in this work that in the liver environment, B cell precursors proliferate in response to TSLP even in the absence of pBCR expression, whereas BM-derived pro-B cells rapidly lose their response to this cytokine. We also find that BM precursors lacking λ5 are completely unresponsive to the activity of TSLP, demonstrating that pBCR expression is necessary for the response of adult BM progenitors to TSLP. To directly study the activity of TSLP on cells that contain a rearranged IgH gene and thus are able to express the pBCR, we analyzed mice carrying an IgH transgene (IgH-tg) on the Rag2° background. This expression of an IgH-tg allows B cell differentiation beyond the pro-B cell stage, although B cell development is arrested at the pre-B cell stage before expression of the complete Ig molecule because of the lack of Ig light chain rearrangements. Using this system, we show directly that TSLP is active on adult BM cells only at the large pre-B stage of differentiation.
Materials and Methods
Mice. H3 IgH-tg, λ5°, common γ chain (γc)°, IL-7°, IL-7 receptor α (IL-7Rα)°, and T cell antigen receptor β° (TCRβ°) mice are described in refs. 6 and 22–26. H3 IgH-tg mice were crossed onto the Rag2° background. λ5° mice were on the BALB/c background. BALB/c and BALB/c.Rag2° mice were purchased from CDTA (Orleans, France). We intercrossed γc°, IL7Rα°, and TCRβ° mice to generate γc°TCRβ°, IL-7Rα°TCRβ°, and γc°IL-7Rα°TCRβ° multiple mutants. Rag2°γc° mice are described in ref. 27 and were used as recipients in adoptive transfer experiments after irradiation (600 rads). All mice were kept under specific pathogen-free conditions (Institut Pasteur, Paris). All animal experiments were done in accordance with the guidelines of the Institut Pasteur, which are approved by the French Ministry of Agriculture.
Flow Cytometric Analysis. Cells isolated from FL, BM, and spleen were prepared as described in ref. 21. Before staining, cells were treated with Fc-Block (anti-CD16/CD32) (Pharmingen).
The following mAb were used as conjugates to fluorescein, phycoerythrin, allophycocyanin, peridinin chlorophyll-a protein-cyanin 5.5 (PerCP-Cy5.5), or biotin: anti-TER119, anti-CD19 (1D3), anti-B220 (RA3–6B2), anti-CD43 (S7), anti-CD24 (30F1), anti-BP-1 (BP-1), and CD5 (53–7.3) (all from Pharmingen). Anti-IgM antiserum was purchased from Southern Biotechnologies Associates. Anti-CD25 (PC61.5) was purchased from eBioscience (San Diego). Biotinylated antibodies were revealed with either streptavidin-allophycocyanin or streptavidin-PerCP-Cy5.5 (Pharmingen). Stained cells were analyzed by using a FACSCalibur flow cytometer and cellquest 3.3software (Becton Dickinson). The TSLP receptor (TSLP-R) antibody was purchased from R & D Systems.
Adoptive Transfer Experiments. FL cells were depleted of TER119+ cells by using a magnet-activated cell sorter according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA) and then stained for CD19 and B220. The CD19– and B220– population was sorted on a Moflo sorter (DakoCytomation). Purity was higher than 95% for all samples. FL cells (105) were injected into irradiated recipients, and the mice were analyzed 5 weeks later.
Analysis of B Cell Precursor Frequencies. BM precursors were purified by cell sorting on a Moflo sorter. The cells were plated under limiting dilution conditions in 96-well plates (48 wells per dilution) as described in ref. 19, with small modifications: irradiated (2,000 rads) NIH 3T3 cells (15,000 cells per well) were used. Culture medium [Opti-MEM (GIBCO/BRL) with 10% FCS/penicillin/streptomycin/2-mercaptoethanol] was supplemented with saturating concentrations of IL-7 (a supernatant from J558 cells transfected with an IL-7 cDNA, kindly given by F. Melchers, University of Basel, Basel) as determined by the proliferation of a factor-dependent cell line (2E8, a kind gift of P. W. Kincade, Oklahoma Medical Research Foundation, Oklahoma City, OK) or with recombinant TSLP [20 ng/ml, a concentration above the maximal activity of this cytokine (18)]. Recombinant TSLP was a kind gift of the DNAX Research Institute (Palo Alto, CA). Growth of lymphocyte colonies was scored at day 7 of culture.
Analysis of Cell Phenotypes After in Vitro Culture. BM precursors were purified as described above but cultured in bulk in the presence of IL-7 or with recombinant TSLP (20 ng/ml). At day 7 the cells were harvested and stained for CD19 and IgM.
Statistical Analysis. The Mann–Whitney t test was used to perform statistical analysis of significance. P < 0.05 was considered significant.
The frequency of precursors responding to soluble factors and the 95% confidence limits of the response were calculated with l-calc for Windows (StemSoft Software, Vancouver).
Results
TSLP Can Drive the Generation of B1 Cells from FL Precursors. The IL-7 receptor consists of IL-7Rα and the γc (28), whereas TSLP binds to a receptor composed of IL-7Rα and a chain homologous to γc designated TSLP-R (18, 29, 30). Thus, precursors from γc-deficient mice do not respond to IL-7 but respond to TSLP, whereas precursors isolated from IL-7Rα-deficient mice respond to neither IL-7 nor TSLP (31).
B1 cells are mainly generated during fetal life (14). Because TSLP is selectively active on fetal pro-B cells (19), we reasoned that it might be involved in the generation of this B cell subpopulation. We therefore determined the number of peritoneal B1 cells in control mice or in mice lacking γc, IL7Rα, or both. All strains of mice were in the TCRβ° background, to avoid the age-related, T cell-dependent B cell loss that occurs in the absence of γc (32).
Adult mice lacking γc have a number of peritoneal CD5+ B1 cells similar to control mice, whereas IL-7Rα° mice have ≈10-fold less peritoneal B1 cells when compared to γc° mice (P < 0.001). The number of peritoneal B1 cells is not reduced further in mice lacking both γc and IL-7Rα (Fig. 1A). This difference in the number of B1 cells present in γc° and IL-7Rα° is indicative of a role for TSLP in the generation of B1 cells. However, because this population has the ability to accumulate and self-replenish in the periphery (14), the cell numbers recovered from adult mice may reflect not only the capacity of the precursors to generate B1 cells, but also the secondary expansion/accumulation of these cells.
Fig. 1.

TSLP supports the generation of B1 cells. (A) Number of B1 cells in the peritoneal cavity of control, γc°, IL-7Rα°, and γc°IL-7Rα° mice. The number of CD5+IgM+ cells found in the peritoneal cavity of the indicated mouse strains at 12 weeks of age is shown. n, number of mice analyzed; *, P < 0.01. (B) Fetal liver precursors from γc° and IL7Rα° mice have a 10- and 100-fold reduced capacity, respectively, to generate B1 cells upon transfer into alymphoid recipient mice. The number of B220+IgM+ cells found in the peritoneal cavity of Rag2°γc° mice 5 weeks after reconstitution with 105 sorted, day-15 FL precursors from donor mice of the indicated genotypes is shown. *, below detection limit.
To directly study the potential of the FL precursors to develop into B1 cells in the absence of IL-7 signals, we isolated CD19–B220– cells from γc°, IL7Rα°, γc°IL7Rα°, or control fetuses and transferred equal numbers of them into alymphoid (Rag2°γc°) recipient mice. We analyzed the recipients 5 weeks after transfer to minimize the effect of peripheral expansion on the number of B1 lymphocytes recovered from the peritoneal cavity of the recipients. We found that FL precursors from γc° embryos gave rise to 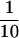 the number of B1 cells of controls. The number of B1 cells generated upon transfer of FL precursors from IL-7Rα° embryos was further reduced 10-fold as compared to those derived from γc° precursors. These differences in the recovery of B1 cells are statistically significant (Fig. 1B). These results demonstrate that, in the absence of IL-7, TSLP is able to drive generation of B1 cells from FL precursors but that IL-7 is the major factor in the development of this subset. In γc° mice the peripheral B1 compartment reaches control numbers because of the ability of B1 cells to self-replenish in the periphery.
the number of B1 cells of controls. The number of B1 cells generated upon transfer of FL precursors from IL-7Rα° embryos was further reduced 10-fold as compared to those derived from γc° precursors. These differences in the recovery of B1 cells are statistically significant (Fig. 1B). These results demonstrate that, in the absence of IL-7, TSLP is able to drive generation of B1 cells from FL precursors but that IL-7 is the major factor in the development of this subset. In γc° mice the peripheral B1 compartment reaches control numbers because of the ability of B1 cells to self-replenish in the periphery.
TSLP Supports Proliferation of PreBCR-Negative Precursors in the Liver Environment but Does Not in the BM Environment. TSLP supports the IL-7-independent proliferation of fetal, but not adult, pro-B cells (19). We studied the kinetics of this age-related difference in the pro-B cell response to TSLP and also investigated whether the liver or the BM environments determine the responsiveness to TSLP. Therefore, we studied the TSLP responsiveness of liver- and BM-derived B cell precursors in neonatal mice. To restrict our analysis to pro-B cells, we used Rag2° mice, which lack the more mature stages of B cell development because of their inability to rearrange their Ig gene segments (8). We isolated pro-B cells from the liver of mice at 1 week old, when a small number of pro-B cells can still be found in this organ, and from the BM of 1- and 3-week-old mice. We found that liver-derived pro-B cells show a frequency of TSLP responsiveness of 1/9, not significantly different from what we had found previously for day-15 FL-derived pro-B cells (1/7) (19). The frequency of TSLP responsiveness of BM-derived pro-B cells from the same 1-week-old animals was 1/59. This response was further diminished to 1/292 by 3 weeks of age, until it was undetectable in adult BM precursors, as shown in ref. 19 (Table 1).
Table 1. Frequency of B cell precursors responding to growth factors.
| Age | Organ | Cells | IL-7 | TSLP |
|---|---|---|---|---|
| 1 week | Liver | CD19+ | 1/7 [5:9] | 1/9 [7:12] |
| BM | CD19+ | 1/7 [5:9] | 1/59 [43:82] | |
| 3 weeks | BM | CD19+ | 1/18 [16:21] | 1/292 [216:454] |
| Adult | BM | B220+CD19+ | 1/60 [47:78] | <1/1,000 |
Cell populations were purified by cell sorting from the BM or the liver of C57BL/6.Rag2° mice at the indicated ages and plated into wells containing irradiated NIH 3T3 feeder cells and medium supplemented with either IL-7 or TSLP. The growth of colonies was scored 7 days later, and the frequency of cells proliferating in response to each factor is indicated (in square brackets we show the 95% confidence limits of the response). One representative result of at least two independent experiments is shown.
Because this loss of the TSLP response parallels the progressive disappearance of B cell precursors in IL-7° animals (21), we sought to determine whether TSLP could support the generation of IgM+ cells in young IL-7° mice. For that purpose we cultured CD19+CD43+ pro-B cells from 1- to 2-week-old IL-7° mice in the presence of either TSLP or IL-7. After 7 days of culture, pro-B cells from young mice gave rise to IgM+ cells independently of the culture conditions. (Fig. 2).
Fig. 2.

TSLP and IL-7 drive the generation of IgM+ cells. Phenotypic analysis of BM-derived C57BL/6(Upper) and C57BL/6.IL-7° (Lower) pro-B cells from 1- to 2-week-old mice cultured for 7 days in the presence of NIH 3T3 feeder cells supplemented with IL-7 (Left) or TSLP (Right). Cultures with IL-7 typically yield 100 times, and TSLP cultures 30 times, more cells than at the start of the culture (not shown). Numbers indicate the percentage of IgM+ cells among CD19+ cells.
TSLP Does Not Support the Growth of B Cell Precursors from λ5-Deficient Mice. In the adult BM, TSLP responsiveness is restricted to B cell precursors beyond the pro-B cells stage (Table 1) (19). To determine at which stage TSLP acts, we analyzed genetically modified mice in which B cell development is arrested at different stages.
λ5° mice do not express the pBCR and show a developmental arrest at the transition from pro-B cell to pre-B cell (3, 6). The BM of λ5° mice, similar to that of Rag2° mice, lacks B220+CD43+BP-1+CD24+++ cells [Hardy's fraction C′, (2)]. In addition, the B220+IgM–CD25+ population [large and small pre-B cells, according to the Osmond and Melchers scheme (33)] is severely depleted in these mice (data not shown).
We determined the responsiveness to TSLP of B220+CD43+ cells from λ5° mice. Because we had available only λ5° mice in the BALB/c background, we used as controls BALB/c.Rag2° and BALB/c wild-type mice. We found that the response of cells from BALB/c.Rag2° to TSLP is below detection (<1/2000), confirming what was observed for B cell precursors from Rag2° mice in a C57BL/6 background (19) (Table 2). B cell precursors isolated from λ5° mice are similarly unresponsive to TSLP (<1/2000), demonstrating that pBCR expression is required for cells to be able to respond to TSLP (Fig. 3). Precursors from BALB/c, BALB/c.Rag2°, and BALB/c.λ5° mice respond to IL-7 with similar frequencies (1/8, 1/10, and 1/18, respectively).
Table 2. Frequency of BM B cell precursors responding to soluble factors.
| Factors
|
|||
|---|---|---|---|
| Mice | BM population | IL-7 | TSLP |
| C57BL/6 | CD19+CD43+ | ND | 1/12; 1/10 |
| H3 × Rag2° | CD19+CD43+ | 1/30; 1/33 | 1/60; 1/25; 1/31 |
| Rag2° | CD19+CD43+ | 1/60 | <1/5,000; <1/473 |
The indicated cell populations were purified by cell sorting from the BM of adult C57BL/6, H3 × Rag2°, or Rag2° mice. Cells were plated into wells containing irradiated NIH-3T3 feeder cells with medium supplemented with either IL-7 or TSLP. The growth of colonies was scored 7 days later. The multiple results shown per genotype were obtained in different experiments. ND, not determined.
Fig. 3.

TSLP does not support the growth of BM B cell precursors from adult λ5° mice. Shown is a limiting dilution analysis of pro-B cells, isolated from adult BM of λ5° (▴), BALB/c.Rag2° (○), and BALB/c mice (•) in the presence of TSLP. The best-fit line crossing the origin is plotted. The dashed line intercepts the vertical axis at 37%. Data are representative of two experiments. No growth was observed in cultures not supplemented with either IL-7 or TSLP (not shown).
TSLP Supports the Proliferation of Adult Large Pre-B, but Not Small Pre-B, Cells. Both large and small pre-B cells are absent from Rag2° mice (8, 34). Small pre-B cells express IL-7Rα, yet they are unresponsive to IL-7 (35). The presence of IL-7Rα on small pre-B cells could allow their response to TSLP, because these cells also express the TSLP-R, as determined by RT-PCR on sorted small pre-B cells (data not shown). To determine whether the TSLP-R is expressed on the surface we stained BM precursors with antibodies to CD19, CD43, CD25, and TSLP-R. As shown in Fig. 4, the majority of both pro-B (CD19+CD43+CD25–) and pre-B (CD19+CD25+) cells express TSLP-R on their surface (Fig. 4). In contrast, peripheral T lymphocytes did not express detectable levels of TSLP-R (data not shown). This result confirms at the protein level our previous RT-PCR analysis of BM pro-B cells from C57BL/6.Rag2° mice (19). However, TSLP was not able to support the growth of small pre-B cells (IgM–CD19+CD43–) from C57BL/6 mice in vitro (<1/500 cells gave rise to B cell colonies in response to TSLP; data not shown). Therefore, to determine whether large pre-B cells are able to proliferate in response to TSLP, we introduced an IgH-tg on the Rag2° background (H3 × Rag2° mice) (22). The ectopic expression of an already rearranged IgH-tg enables Rag° B cell precursors to differentiate beyond the pro-B cell stage, leading to an enrichment of fraction C′/large pre-B (B220+CD43+BP-1+CD24+++) cells and a reduction of the pro-B cell compartment in the BM of these mice (Fig. 5). The paucity of pro-B cells in mice expressing a rearranged heavy-chain gene early in their development has been previously described and attributed to rapid transit through the early precursor stages (36). This accelerated development of IgH-tg cells might also explain the lower levels of BP-1 expression on their surface (shown in Fig. 5). Further differentiation of precursor B cells past the pre-B cell stage is prevented by the absence of functional Ig light-chain rearrangements in H3 × Rag2° mice.
Fig. 4.

Expression of TSLP-R on BM precursor B cells from C57BL/6 and C57BL/6.Rag2° mice. BM cells from 8-week-old mice were stained for CD19, CD43, CD25, and TSLP-R. Shown are living (propidium iodide-negative) CD19+ BM cells from C57BL/6(Left) and C57BL/6.Rag2° (Right) mice. The expression of TSLP-R on pro-B cells (CD43+CD25–)(Upper) and pre-B cells (CD25+)(Lower) are shown as histograms. Gray shading indicates isotype control antibody.
Fig. 5.

The BM pro-B cell compartment in C57BL/6, C57BL/6.Rag2°, and H3 × Rag2° mice. BM cells from the indicated mice were stained for B220, CD43, HSA (30F1), and BP-1. Shown are the profiles of HSA versus BP-1 expression on gated B220+CD43+ BM cells. Cells of Hardy's fraction C (pro-B cells) and C′ (large pre-B cells) are boxed. Note the reduced level of expression of BP-1 on cells from fraction C′ in the BM of H3 × Rag2° mice.
Therefore, CD19+CD43+ cells from C57BL/6 mice contain a mixture of pro-B cells and large pre-B cells. As shown in Table 2, these cells respond to TSLP with a frequency of 1/10–12. The same population isolated from C57BL/6.Rag2° mice only contains pro-B cells and does not respond to TSLP (<1/473–5,000). In H3 × Rag2° mice CD19+CD43+ cells are essentially all large pre-B cells, and the expression of the IgH-tg allows the response to TSLP to be restored (1/25–60) (Table 2). Taken together, these results demonstrate that TSLP is selectively active on large pre-B cells (fraction C′) from adult BM in vitro.
Discussion
TSLP was initially described as a cytokine able to drive B lymphocyte differentiation (17). In the past 3 years a renewed interest in the biological activities of this cytokine occurred, after the publication of its sequence (37), the characterization of its receptor complex (30, 31), and the description of its activity on human dendritic cells (38, 39). We reported recently that TSLP is able to support B lymphocyte production from FL, but not adult BM, pro-B cells (19) and showed that TSLP is responsible for the vast majority of the B lymphocytes that are generated in the absence of IL-7 signaling (19, 21, 32). Here, we confirm and extend these findings, showing that BM pro-B cells can also respond to TSLP but that the window of responsiveness is restricted to the first 3 weeks after birth (Table 1). Furthermore, we show that pre-BCR-negative precursors respond to TSLP in the liver environment but not in the BM at the same age. It is tempting to speculate that the liver provides signals necessary for pro-B cells to proliferate in response to TSLP but that such signals require pre-BCR expression in the BM.
An IL-7-independent pathway, only active in fetal/perinatal life, is able to give rise to an almost full compartment of B1 cells in IL-7° mice (21). The kinetics of loss of TSLP responsiveness by pro-B cells (Table 1) is similar to that of the progressive reduction of B lymphopoiesis in both IL-7° and γc° mice (21, 32). Because B1 cells are predominantly produced in fetal life (14), it was reasonable to expect a major role for TSLP in the generation of this B cell subset. Our results confirm that TSLP can drive B1 cell production from FL precursors but also show that the absence of IL-7 signaling leads to a 90% reduction in the size of the B1 compartment after adoptive transfer of FL precursors (Fig. 2). This result demonstrates that, as is the case for conventional B cells (B2), IL-7 is the predominant factor controlling B1 cell generation. The fact that the number of peritoneal cavity B1 cells isolated from γc° or from IL-7° mice (21, 32) is only marginally reduced reflects the ability of this population to self-replenish and accumulate (14, 40). It should be noted that, besides IL-7 and TSLP, other factors can drive B1 generation, because a sizeable B1 compartment exists even in IL-7Rα° mice (Fig. 2). As recently shown, Flt3/Flk2 ligand is one such factor (41).
The mechanism behind the unresponsiveness of adult pro-B cells to TSLP remains enigmatic because these cells express IL7Rα and also TSLP-R, as determined both by RT-PCR analysis (19) and surface staining with a TSLP-R-specific antibody (Fig. 4). In line with our results, a BM-derived B cell line is unable to proliferate in response to TSLP, although it expresses a functional TSLP-R complex (42). The molecular basis for the differential response to TSLP of FL versus BM pro-B cells remains a key question. This issue can only be resolved when detailed knowledge of the signaling pathway(s) downstream of the TSLP-R in FL and BM pro-B cells is available.
The only adult precursors that proliferate in response to TSLP in control mice are large pre-B cells, characterized by a high frequency of cycling cells (7) and by the expression of the pBCR complex, composed of a rearranged heavy chain, Vpre-B, and λ5 (43). BM B cell precursors from mice unable to express this complex because of either the absence of Ig rearrangements, as in Rag° mice, or the lack of pBCR components, as in λ5° mice, do not proliferate in response to this cytokine (Fig. 3). On the other hand, when an IgH-tg is introduced in the Rag2° background, thus allowing development of pBCR-expressing precursors, the response to TSLP is restored (Table 2). Collectively, our results demonstrate that, besides a functional TSLP-R complex, B cell precursors in the BM require expression of the pBCR to expand in response to TSLP.
A small number of precursors can express on the surface a μH chain even in the absence of λ5, and this μH chain-only receptor can prolong the survival of the cells (44, 45). Because λ5° precursors do not respond to TSLP (<1/2000 CD19+CD43+ cells formed colonies in vitro; Fig. 3), it is clear that this μH chain-only receptor is unable to confer TSLP responsiveness.
The various subsets of B cell precursors are differentially represented in the FL and in the BM. In the BM, the small pre-B subset, direct progeny of large pre-B cells, is predominant and constitutes up to 50% of all B lineage cells in this organ. This predominance is best explained by postulating that cells that have successfully rearranged one heavy-chain locus, and thus are able to express the pBCR, have a proliferative advantage. We now show that BM pBCR-expressing cells have such an advantage, conferred by their response to TSLP. Fetal liver pro-B cells, however, respond to TSLP (19), which explains why pre-B cells are not in the majority in the FL (46).
Expression of the pBCR was suggested to be sufficient to induce the proliferative expansion of pre-B cells, in the absence of exogenous growth factors (10). It should be pointed out, however, that in our cultures no colonies were formed in the absence of exogenous factors. Furthermore, as discussed in ref. 10, pre-B cells isolated ex vivo may have entered cell cycle before the start of in vitro culture. Thus, they may already have received all of the signals necessary for driving their proliferative expansion in the absence of added factors. In addition, it is clear that the presence of IL-7 and stromal cells results in a significant increase in the number of pre-B cells recovered after culture (10). This last observation raises the question of the biological significance of this “factor-independent pathway” of pre-B cell expansion.
Another mechanism suggested for the predominance of pre-B cells in the BM is the lower threshold for IL-7 reactivity found in pBCR-expressing cells (11). This mechanism would only operate if the cells, as they differentiated, moved into putative BM niches with lower concentrations of IL-7 (47). TSLP is expressed in the adult BM (37); therefore, BM pre-B cells always have an advantage over BM pro-B cells, regardless of whether regional differences in IL-7 concentration exist.
The ability of TSLP to drive the expansion of pre-BCR-negative precursors in the liver readily explains why mice transgenic for a heavy-chain gene that does not associate with λ5 (48) are only able to efficiently generate transgenic B cells during the neonatal stage (49). Such cells are inefficiently produced later in life because, during adult lymphopoiesis, TSLP expands endogenous heavy-chain-expressing, pre-BCR-positive cells.
Acknowledgments
We thank Drs. M. Goodhardt and A. Freitas for H3 × Rag2° mice and Dr F. Huetz for the λ5 knockout mice. We also thank Drs. S. Dias and M. Garcia-Ojeda for helpful discussions and critical reading of the manuscript. This work was supported by the Institut Pasteur, the Association pour la Recherché Contre le Cancer, the Institut Nationale de la Santè et Recherche Medicale (INSERM), and the Ligue Nationale Contre Le Cancer as an Associated Laboratory. C.A.J.V. was supported by fellowships from the Fondation pour la Recherche Médicale and INSERM.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BCR, B cell receptor; BM, bone marrow; FL, fetal liver; γc, common γ chain; IgH, Ig heavy chain; IgH-tg, IgH transgene; IL-7Rα, IL-7 receptor α; PerCP-Cy5.5, peridinin chlorophyll-a protein-cyanin 5.5; Rag, recombination-activating gene; TCRβ°, T cell antigen receptor β°; TSLP, thymic stromal lymphopoietin; TSLP-R, TSLP receptor.
References
- 1.Osmond, D. G. & Nossal, G. J. (1974) Cell Immunol. 13, 132–145. [DOI] [PubMed] [Google Scholar]
- 2.Hardy, R. R., Carmack, C. E., Shinton, S. A., Kemp, J. D. & Hayakawa, K. (1991) J. Exp. Med. 173, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehlich, A., Schaal, S., Gu, H., Kitamura, D., Muller, W. & Rajewsky, K. (1993) Cell 72, 695–704. [DOI] [PubMed] [Google Scholar]
- 4.Coffman, R. L. & Weissman, I. L. (1983) J. Mol. Cell. Immunol. 1, 31–41. [PubMed] [Google Scholar]
- 5.Reth, M., Petrac, E., Wiese, P., Lobel, L. & Alt, F. W. (1987) EMBO J. 6, 3299–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitamura, D., Kudo, A., Schaal, S., Muller, W., Melchers, F. & Rajewsky, K. (1992) Cell 69, 823–831. [DOI] [PubMed] [Google Scholar]
- 7.Owen, J. J., Wright, D. E., Habu, S., Raff, M. C. & Cooper, M. D. (1977) J. Immunol. 118, 2067–2072. [PubMed] [Google Scholar]
- 8.Shinkai, Y., Rathbun, G., Lam, K. P., Oltz, E. M., Stewart, V., Mendelsohn, M., Charron, J., Datta, M., Young, F., Stall, A. M., et al. (1992) Cell 68, 855–867. [DOI] [PubMed] [Google Scholar]
- 9.Rolink, A., Karasuyama, H., Haasner, D., Grawunder, U., Martensson, I. L., Kudo, A. & Melchers, F. (1994) Immunol. Rev. 137, 185–201. [DOI] [PubMed] [Google Scholar]
- 10.Rolink, A. G., Winkler, T., Melchers, F. & Andersson, J. (2000) J. Exp. Med. 191, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall, A. J., Fleming, H. E., Wu, G. E. & Paige, C. J. (1998) J. Immunol. 161, 6038–6045. [PubMed] [Google Scholar]
- 12.Fleming, H. E. & Paige, C. J. (2001) Immunity 15, 521–531. [DOI] [PubMed] [Google Scholar]
- 13.Kantor, A. B. & Herzenberg, L. A. (1993) Annu. Rev. Immunol. 11, 501–538. [DOI] [PubMed] [Google Scholar]
- 14.Herzenberg, L. A. (2000) Immunol. Rev. 175, 9–22. [PubMed] [Google Scholar]
- 15.Hayakawa, K. & Hardy, R. R. (2000) Curr. Opin. Immunol. 12, 346–353. [DOI] [PubMed] [Google Scholar]
- 16.Hardy, R. R., Li, Y. S., Allman, D., Asano, M., Gui, M. & Hayakawa, K. (2000) Immunol. Rev. 175, 23–32. [PubMed] [Google Scholar]
- 17.Ray, R. J., Furlonger, C., Williams, D. E. & Paige, C. J. (1996) Eur. J. Immunol. 26, 10–16. [DOI] [PubMed] [Google Scholar]
- 18.Levin, S. D., Koelling, R. M., Friend, S. L., Isaksen, D. E., Ziegler, S. F., Perlmutter, R. M. & Farr, A. G. (1999) J. Immunol. 162, 677–683. [PubMed] [Google Scholar]
- 19.Vosshenrich, C. A. J., Cumano, A., Muller, W., Di Santo, J. P. & Vieira, P. (2003) Nat. Immunol. 4, 773–779. [DOI] [PubMed] [Google Scholar]
- 20.Carpino, N., Thierfelder, W. E., Chang, M. S., Saris, C., Turner, S. J., Ziegler, S. F. & Ihle, J. N. (2004) Mol. Cell. Biol. 24, 2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho, T. L., Mota-Santos, T., Cumano, A., Demengeot, J. & Vieira, P. (2001) J. Exp. Med. 194, 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto, M., Murakami, M., Shimizu, A., Ozaki, S., Tsubata, T., Kumagai, S. & Honjo, T. (1992) J. Exp. Med. 175, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Santo, J. P., Muller, W., Guy-Grand, D., Fischer, A. & Rajewsky, K. (1995) Proc. Natl. Acad. Sci. USA 92, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Freeden-Jeffry, U., Vieira, P., Lucian, L. A., McNeil, T., Burdach, S. E. & Murray, R. (1995) J. Exp. Med. 181, 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peschon, J. J., Morrissey, P. J., Grabstein, K. H., Ramsdell, F. J., Maraskovsky, E., Gliniak, B. C., Park, L. S., Ziegler, S. F., Williams, D. E., Ware, C. B., et al. (1994) J. Exp. Med. 180, 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mombaerts, P., Clarke, A. R., Rudnicki, M. A., Iacomini, J., Itohara, S., Lafaille, J. J., Wang, L., Ichikawa, Y., Jaenisch, R., Hooper, M. L., et al. (1992) Nature 360, 225–231. [DOI] [PubMed] [Google Scholar]
- 27.Colucci, F., Soudais, C., Rosmaraki, E., Vanes, L., Tybulewicz, V. L. & Di Santo, J. P. (1999) J. Immunol. 162, 2761–2765. [PubMed] [Google Scholar]
- 28.Noguchi, M., Nakamura, Y., Russell, S. M., Ziegler, S. F., Tsang, M., Cao, X. & Leonard, W. J. (1993) Science 262, 1877–1880. [DOI] [PubMed] [Google Scholar]
- 29.Isaksen, D. E., Baumann, H., Trobridge, P. A., Farr, A. G., Levin, S. D. & Ziegler, S. F. (1999) J. Immunol. 163, 5971–5977. [PubMed] [Google Scholar]
- 30.Pandey, A., Ozaki, K., Baumann, H., Levin, S. D., Puel, A., Farr, A. G., Ziegler, S. F., Leonard, W. J. & Lodish, H. F. (2000) Nat. Immunol. 1, 59–64. [DOI] [PubMed] [Google Scholar]
- 31.Park, L. S., Martin, U., Garka, K., Gliniak, B., Di Santo, J. P., Muller, W., Largaespada, D. A., Copeland, N. G., Jenkins, N. A., Farr, A. G., et al. (2000) J. Exp. Med. 192, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vosshenrich, C. A. J., Sharara, L. I., Guy-Grand, D., Rajewsky, K., Muller, W. & Di Santo, J. P. (2000) Eur. J. Immunol. 30, 1614–1622. [DOI] [PubMed] [Google Scholar]
- 33.Osmond, D. G., Rolink, A. & Melchers, F. (1998) Immunol. Today 19, 65–68. [DOI] [PubMed] [Google Scholar]
- 34.Rolink, A., Grawunder, U., Winkler, T. H., Karasuyama, H. & Melchers, F. (1994) Int. Immunol. 6, 1257–1264. [DOI] [PubMed] [Google Scholar]
- 35.Loffert, D., Schaal, S., Ehlich, A., Hardy, R. R., Zou, Y. R., Muller, W. & Rajewsky, K. (1994) Immunol. Rev. 137, 135–153. [DOI] [PubMed] [Google Scholar]
- 36.Sonoda, E., Pewzner-Jung, Y., Schwers, S., Taki, S., Jung, S., Eilat, D. & Rajewsky, K. (1997) Immunity 6, 225–233. [DOI] [PubMed] [Google Scholar]
- 37.Sims, J. E., Williams, D. E., Morrissey, P. J., Garka, K., Foxworthe, D., Price, V., Friend, S. L., Farr, A., Bedell, M. A., Jenkins, N. A., et al. (2000) J. Exp. Med. 192, 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soumelis, V., Reche, P. A., Kanzler, H., Yuan, W., Edward, G., Homey, B., Gilliet, M., Ho, S., Antonenko, S., Lauerma, A., et al. (2002) Nat. Immunol. 3, 673–680. [DOI] [PubMed] [Google Scholar]
- 39.Gilliet, M., Soumelis, V., Watanabe, N., Hanabuchi, S., Antonenko, S., de Waal-Malefyt, R. & Liu, Y. J. (2003) J. Exp. Med. 197, 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao, Z. & Rajewsky, K. (2001) J. Exp. Med. 194, 1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sitnicka, E., Brakebusch, C., Martensson, I. L., Svensson, M., Agace, W. W., Sigvardsson, M., Buza-Vidas, N., Bryder, D., Cilio, C. M., Ahlenius, H., et al. (2003) J. Exp. Med. 198, 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isaksen, D. E., Baumann, H., Zhou, B., Nivollet, S., Farr, A. G., Levin, S. D. & Ziegler, S. F. (2002) J. Immunol. 168, 3288–3294. [DOI] [PubMed] [Google Scholar]
- 43.Hardy, R. R. & Hayakawa, K. (2001) Annu. Rev. Immunol. 19, 595–621. [DOI] [PubMed] [Google Scholar]
- 44.Su, Y. W., Flemming, A., Wossning, T., Hobeika, E., Reth, M. & Jumaa, H. (2003) J. Exp. Med. 198, 1699–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuh, W., Meister, S., Roth, E. & Jack, H. M. (2003) J. Immunol. 171, 3343–3347. [DOI] [PubMed] [Google Scholar]
- 46.Delassus, S., Darche, S., Kourilsky, P. & Cumano, A. (1998) J. Immunol. 160, 3274–3280. [PubMed] [Google Scholar]
- 47.Fleming, H. E. & Paige, C. J. (2002) Semin. Immunol. 14, 423–430. [DOI] [PubMed] [Google Scholar]
- 48.Wasserman, R., Li, Y. S., Shinton, S. A., Carmack, C. E., Manser, T., Wiest, D. L., Hayakawa, K. & Hardy, R. R. (1998) J. Exp. Med. 187, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardy, R. R., Wei, C. J. & Hayakawa, K. (2004) Immunol. Rev. 197, 60–74. [DOI] [PubMed] [Google Scholar]