

Abstract
A series of novel hexadentate enterobactin analogues, which contain three catechol chelating moieties attached to different molecular scaffolds with flexible alkyl chain lengths, were prepared. The solution thermodynamic stabilities of the complexes with uranyl, ferric(III), and zinc(II) ions were then investigated. The hexadentate ligands demonstrate effective binding ability to uranyl ion, and the average uranyl affinities are two orders of magnitude higher than 2,3-dihydroxy-N1,N4-bis[(1,2-hydroxypyridinone-6-carboxamide)ethyl]terephthalamide [TMA(2Li-1,2-HOPO)2] ligand with similar denticity. The high affinity of hexadentate ligands could be due to the presence of the flexible scaffold, which favors the geometric agreement between the ligand and the uranyl coordination preference. The hexadentate ligands also exhibit higher antiradical efficiency than butylated hydroxyanisole (BHA). These results provide a basis for further studies on the potential applications of hexadentate ligands as therapeutic chelating agents.
The development of civilian energy generation and atomic weapon requires further research on various environment and health issues of uranium1. Uranium is introduced into the environment via atomic weapon tests and accidents in nuclear facilities and is then absorbed by humans through ingestion, inhalation, or wounds. The hexavalent uranyl ion [UO22+, U(VI)] is the most stable form of this element in vivo2 and form complexes with chelating agents, such as proteins or carbonates, in the body. Tissues, especially kidney and bones, accumulate uranium for months to years. The radiological accumulation of uranium in tissues causes long-term damage and may induce cancer in the deposition site3,4,5. Uranium with high specific alpha activity can produce harmful free-radicals that activate apoptosis6,7. Thus, uranium should be excreted from the body by administration of nontoxic chelating agents, which can form stable complexes with the uranyl ion. This treatment results in rapid excretion of the deposited uranium from the blood and target organs, thereby reducing uranium concentration and radiation. In addition, production of harmful free radicals will be inhibited.
The uranyl ion, a hard Lewis acid, exhibits high affinity for hard donor groups. Equatorial pentacoordination or hexacoordination generally occurs between 5- and 6-membered chelated rings with bidentate ligands8,9,10,11. In animals, uranium levels can be reduced by injection of bidentate tiron12,13, moreover, the stability of the U(VI)-catechol complex (log KML = 15.9)14 indicates that multidentate ligands containing the catechol moiety as binding units are effective for chelation of the uranyl ion. Therefore, a rational approach for design of multidentate sequestering agents for uranyl ion was inspired by enterobactin15,16, a naturally occurring microbial iron(III)-sequestering agent. A common feature of the design of actinide-sequestering agents is the use of anionic oxygen donors in functional groups, such as catechol from enterobactin. The molecular scaffold should be attached to the catechol moiety in an ortho position relative to phenolate oxygen via amide linkages. The ligands should adopt the correct geometry for metal binding, and the amides contribute to the stability of the iron complex through hydrogen bonding17,18,19. The structure-activity relationship emphasizes that different linker lengths affect the conformation of the complexes20. The most potent enterobactin (pFe3+ values for Fe3+ complex of enterobactin up to 35.5)21 is hexadentate, which contains three catechol moieties attached to the molecular scaffold, this feature allows the formation of a coordination cavity suitable for Fe3+ 22. However, the ionic radius of uranyl ion (0.95 Å)23 is larger than that of Fe3+ (0.65 Å)24. Therefore, a rational design of hexadentate enterobactin analogue ligands with different molecular scaffolds of flexible alkyl chain lengths must be developed, this design is essential to achieve the coordinative saturation and conformational flexibility of uranyl, thereby allowing the formation of a large coordination cavity suitable for the uranyl ion. To the best of our knowledge, the new hexadentate enterobactin analogues have not been synthesized.
Studies have been performed to obtain a ligand with good chelating ability and antioxidant capacity. The first part of this study involved synthesis of a series of hexadentate enterobactin analogues. The second part involved studying the solution thermodynamic behaviors of these ligands and their complexes with uranyl, iron(III) and zinc(II) ions in aqueous solution. The third part involved evaluation of the antioxidant capacity of the derivatives by 2,2-diphenyl-1-picrylhydrazyl (DPPH·) antioxidant assay25,26,27.
Results and Discussion
Synthesis of hexadentate ligands
The preparation of hexadentate enterobactin analogues 7a–c (L1–3H6) is shown in Fig. 1. 2,3-bis(dibenzyloxy)benzonic acid 2 (80%) was generated from commercially available 2,3-bis(hydroxyl)benzonic acid 128. Aminoalcohol 3a–c and 2 were condensed using HOBt/DCC to obtain the desired benzamides (4a–c) with up to 90% yield29. 1,3,5-Benzenetricarbonyl trichloride was then added to benzamides 4a–c in the presence of Et3N in anhydrous CH2Cl2. The reaction generated benzyl-protected derivatives 6a–c with up to 71% yield. Deprotection of the hydroxyl groups under typical catalytic hydrogenation conditions with removal of the benzyl group (room temperature, 130 mL/min H2, atmospheric pressure, and Pd/C in THF) produced 5a–c (L7–9H2) and 7a–c (L1–3H6) with up to 99% yield.
Figure 1. Synthesis of hexadentate enterobactin analogues 7a–c (L1–3H6) and 5a–c (L7–9H2).

Solution thermodynamics
In the presence of dissolved metal ions (Ma+) and protonated ligands (LHi, where L is a ligand with i removable protons), the pH-dependent metal-ligand complex of general formula MmLlHh forms according to the equilibrium shown in Eq. 1. The relative amount of each species in solution is determined by Eq. 2, whose rearrangement provides the standard formation constant notation of log βmlh (Eq. 3). The log βmlh value describes a cumulative formation constant, and a stepwise formation constant (log K) can be calculated from log βmlh values by Eq. 4. When addressing protonation constants, the stepwise formation constants are commonly reported as log KiH (i = 1, 2, 3 …).
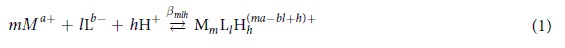 |
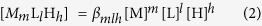 |
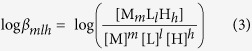 |
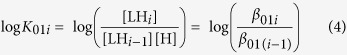 |
Proton titration/affinity
The protonation constants log KiH (i = 1, 2, 3 …) of ligands L1–3H6 were determined from spectrophotometric titration measurements in aqueous KCl solution an ionic strength of 0.10 M at 298.2 K. The protonation constants for the intermediate L7–9H2 were also measured and compared with that for ligands L1–3H6. The determined log KiH values (i = 4–6) and estimated values of the related ligands are listed in Table 1 30,31,32 (Fig. 2 for the corresponding structures). The values of L7–9H2 are listed in Table S1.
Table 1. Protonation constants log KiH of L1–3H6 and other related compounds.
| Ligand |
||||||
|---|---|---|---|---|---|---|
| L1H6a | L2H6a | L3H6a | L4H6b | L5H6c | L6H6d | |
| log K1H | 12.9e | 12.9e | 12.9e | 12.9e | 12.9e | 12.9e |
| log K2H | 12.1e | 12.1e | 12.1e | 12.1e | 12.1e | 12.1e |
| log K3H | 11.3e | 11.3e | 11.3e | 11.26 | 11.3e | 11.3e |
| log K4H | 8.98 (5) | 8.91 (6) | 8.86 (8) | 8.75 | 8.55 | 9.26 |
| log K5H | 7.56 (8) | 7.52 (4) | 7.43 (2) | 8.61 | 7.5 | 8.65 |
| log K6H | 6.16 (7) | 6.13 (5) | 6.0 (5) | 6.71 | 6.0 | 7.86 |
| log K7H | — | — | — | 5.88 | — | — |
| Averagef | 7.57 | 7.52 | 7.43 | 7.49 | 7.36 | 8.59 |
aDetermined by spectrophotometric titration: [L1–3H6] = 2 × 10−5 M; μ = 0.10 M KCl; T = 298.2 K; pH range = 6.5–10.0; 5.0 vol % methanol aqueous solution.
bRef. 30, μ = 0.10 M KNO3.
cRef. 31, 5.0 vol % methanol aqueous solution.
dRef. 32, μ = 0.10 M KNO3.
eEstimated values.
fAverage KiH of the three more acidic catecholamide protonation constants: ∑(log K4H + log K5H + log K6H)/3.
Figure 2. Molecular structure of related compounds.

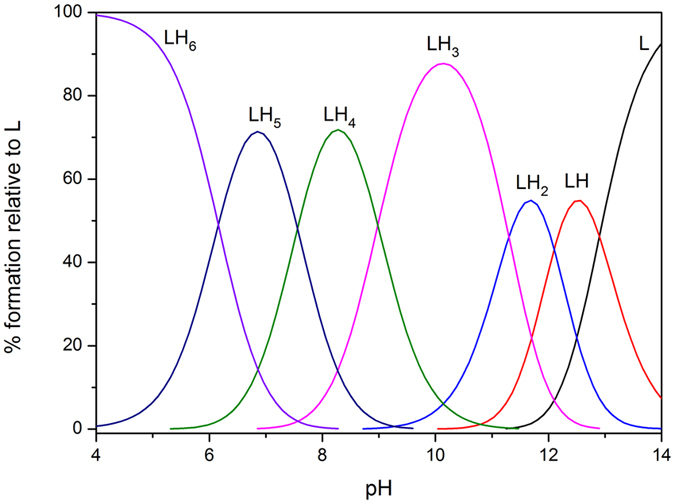
The species distribution diagram of L1H6 was selected for illustration because it is similar to that of L1–3H6, as shown in Fig. 3. The species distribution diagram of L2–3H6 is shown in Figures S1 and S2. These species distribution diagrams were obtained using HySS program33. The compounds contain six basic sites form the phenolate oxygen atoms of the catechol moiety. However, only three protonation constants could be accurately determined under our experimental conditions. Indeed, the three values of the first protonation constants of each catechol moiety is very high and cannot be determined by potentiometry.
Figure 3. Species distribution curves calculated for the ligand L1H6, the charge number are omitted for clarity; conditions: [L1H6] = 2 × 10−5 M.
The catechol derivatives L7–12H2 differ between the intrinsic acidity of the two dissociable protons of the phenolic oxygen atoms (Table S1). This finding is explained by electronic effects and intramolecular hydrogen bond formation among neighboring amides17,18,19.
The first three protonation constants of L1–3H6, corresponding to the first protonation of each catechol moiety, cannot be directly determined because of their very high values and the possible oxidation of the ligand at pH = 12.0, as observed in other catecholamide derivatives34. The values of the constants are approximately 13.0 for tris- and bis-catechol compounds31,32,35,36. Meanwhile, considering the ineluctable statistical factor and the actual practice30,35, we used the estimated values of log K1H = 12.9, log K2H = 12.1 and log K3H = 11.3 (Table 1).
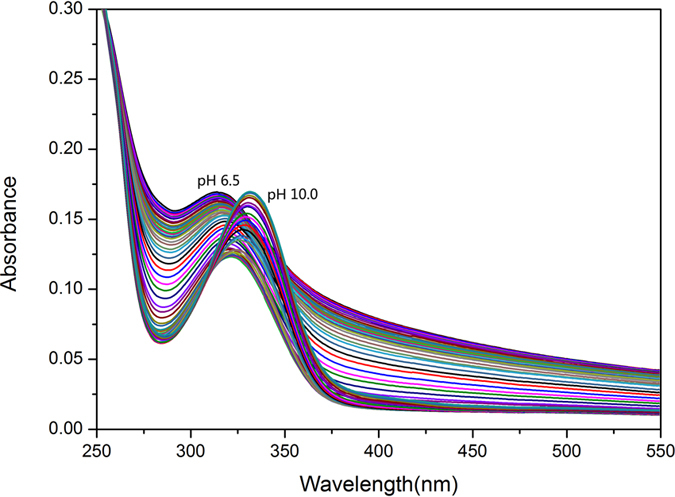
The spectrophotometric titration curves of L1–3H6 from pH 6.5 to 10.0 are shown in Fig. 4 and Figures S3 and S4. The initial absorbance at high energy shifts to low energy with increasing pH. The half-peak width gradually narrows, and the intensity of the peak at 330 nm increases upon deprotonation. At pH 10.0, about 90.0% of L1–3H6 are in the anionic form (L1–3H3)3− (Fig. 3 and Figures S1 and S2), corresponding each catechol moiety lost one more acidic proton.
Figure 4. Spectrophotometric titration curves of L1H6, conditions: [L1H6] = 2 × 10−5 M; μ = 0.10 M KCl; T = 298.2 K; pH range = 6.5–10.0; 5.0 vol % methanol aqueous solution.
The values of log K4H - log K6H are ascribed to the three consecutive protonations of the less basic oxygen atoms of the catecholamide dianions with functions similar to amide carbonyl. The average values of the three constants are comparable with the corresponding values for trencam30, enterobacin31 and 3,3,4-cycam36 (Table 1). This finding is in good agreement with the value of log K2H = 7.31–7.63 for L7–10H2 (Table S1). The average value of the three more acidic protonation constants of 3,3,4-cycam is 8.5932, which is similar to that of L11H2 (8.42; Table 1 and Table S1). In fact, theoretical calculations, analysis of crystal structures, and experimental potentiometric data for series of catecholamide derivatives indicated that the presence of amide on the catecholamide molecules increased the second protonation constant of the nearby catechol by about one log unit37.
Uranyl titrations/affinity
The uranyl affinities of ligands L1–3H6 were determined by performing spectrophotometric titrations using a 1:1 metal to ligand ratio to avoid the decomposition of free ligand at high pH values. Maintaining this ratio may also ensure the formation of mononuclear complexes. The poor solubility of the uranyl complexes requires 2 × 10−5 M analyte and 5 vol % starting methanol for solvating the neutral uranyl complexes during titration. The uranyl titration spectra with L1–3H6 ligands generally exhibit similar absorption spectra within 250–400 nm, with a maximum absorption within 280–350 nm range and subtle shoulder at long wavelengths (Fig. 5 and Figures S5 and S6). These features resemble those of the free ligands and are attributed to π → π* transitions. The uranyl complexes routinely form [UO2(L1–3H4)] have been generated at pH 4.5, uranyl titration with all ligands displayed increased intensity from pH 4.5 to pH 7.5 for L1H6, pH 8.1 for L2H6, and pH 8.4 for L3H6. These finding indicated the deprotonation of more acidic two protons of the ligands and complexation of the uranyl ion [UO2(L1–3H2)]2−. Subsequently, the intensity rapidly decreased until around pH 9.0 and slowly increased until around pH 10.0 with red shift of the absorption peaks. This result revealed the complete deprotonation of the ligands and binding to the uranyl ion [UO2(L1–3)]4−. The acid titrations (pH 4.6 to 2.0) were also carried out for each ligand. The intensity decreased until around pH 3.0 and slowly increased until around pH 2.0 with blue shift of the absorption peaks, which indicated that the protonation of the ligands and binding to the uranyl ion [UO2(L1–3H5)]+.
Figure 5. Spectrophotometric titration curves for uranyl with L1H6, conditions: [UO22+] = [L1H6] = 2 × 10−5 M; μ = 0.10 M KCl; T = 298.2 K; pH range = 2.1–10.2; 5.0 vol % methanol aqueous solution.

The uranyl titration spectra significantly differ between L1–3H6 and other reported tetradentate complexes at high pH. The coordinative saturated [UO2(L)]4− complexes can form at relatively low pH, and no partial hydrolysis of the uranyl ion occurs with increasing hydroxide concentration. By contrast, the coordination modes of uranyl with tetradentate ligands do not saturate the uranyl coordination plane, hence, the partial hydrolysis of the uranyl ion is predicted to occur at high pH values38,39,40.
The uranyl formation constants log βmlh for ligands L1–3H6 are reported in Table 2. A species independent metric is needed to compare uranyl affinities of the bis- and tris-bidentate ligands because log βmlh values are species dependent. In this regard, pM is the metric employed, where pM = −log[Mfree]. “Mfree” refers to solvated metal ions free of complexation by ligands or hydroxides, high pM corresponds to low concentrations of uncomplexed metal ions in the solution. As a reference index, pUO22+ under oceanic conditions is 16.8, which could be due to the high affinity of carbonate for the uranyl ion41,42. In this study, pUO22+ values are calculated using standard conditions of [UO22+] = 10−6 M and [L] = 10−5 M. Typically, these values are reported at physiological pH and can be calculated at any pH upon determination of log KiH and log βmlh values. The pUO22+ values at pH 3.0, 7.4, and 9.0 are listed for ligands L1–3H6 and related compounds in Table 2.
Table 2. Formation constants log βmlh and pUO22+ values of L1–3H6 and other related compounds.
| ligand | logβ11-1 | logβ110 | logβ111 | logβ112 | logβ113 | logβ114 | logβ115 | pUO22+ a |
||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 3.0 | pH 7.4 | pH 9.0 | ||||||||
| L1H6 | — | 31.21 (4) | 40.98 (3) | 48.50 (3) | 55.40 (4) | 61.12 (3) | 63.08 (5) | 9.10 (3) | 18.88 (1) | 23.44 (2) |
| L2H6 | — | 32.72 (5) | 42.00 (2) | 49.43 (3) | 56.71 (2) | 62.31 (3) | 64.02 (3) | 10.41 (2) | 20.03 (1) | 24.64 (3) |
| L3H6 | — | 33.10 (3) | 42.41 (1) | 49.68 (4) | 57.11 (2) | 62.42 (1) | 64.21 (3) | 10.79 (3) | 20.99 (4) | 25.00 (1) |
| TMA(2Li-1,2-HOPO)2b | — | 21.95 | 26.86 | 30.79 | — | — | 6.9 | 18.2 | 21.0 | |
| PEG-4li-bis-Me- 3,2-HOPOc | 6.97 | 13.90 | — | — | — | — | 8.98 | 15.39 | 16.93 | |
The species distribution diagram of L1H6 was selected for illustration (Fig. 6) because the species distribution diagrams of the uranyl complexes with ligands L1–3H6 are similar, moreover, the diagrams of ligands L2–3H6 are shown in Figures S7 and S8.
Figure 6. Species distribution curves calculated for uranyl complexes with ligand L1H6, the charge number are omitted for clarity; conditions: [UO22+] = [L1H6] = 2 × 10−5 M.
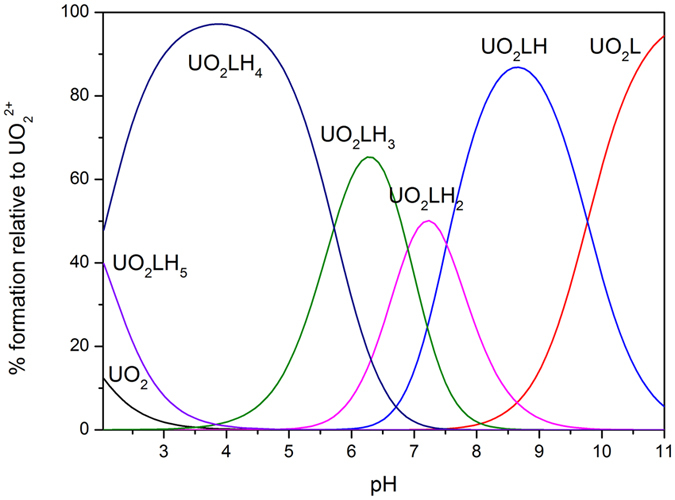
The pUO22+ values of hexadentate L1–3H6 ligands are significantly higher than those of the tetradentate bis-Me-3,2-HOPO40 at all pH values. This finding commonly occurs in L1–3H6 ligands because of their high denticity. However, the pUO22+ values are slightly higher than those of the hexadentate TMA(2Li-1,2-HOPO)243, indicating that denticity is not the sole reason. Meanwhile, changes in minor log KiH values are an insignificant factor in determining uranyl affinity in L1–3H6 ligands, the higher affinity is presumably due to favorable geometric agreement between the ligand and the uranyl coordination preference. The fact that the pUO22+ value of L3H6 is higher than L1–2H6 indicated that scaffold flexibility favors higher uranyl affinity, as predicted.
Ferric(III) and zinc(II) ion titrations/affinity
Metal affinity studies have focused on ferric(III) ion, however, the presence of zinc(II) ion in biological systems leads us to evaluate ligands with zinc(II) ion. The ferric(III) and zinc(II) ion affinities of ligands L1–3H6 were determined through spectrophotometric titrations under the same conditions above. The ferric(III) and zinc(II) ion titration spectra of L1–3H6 are shown in Figures S9–S14. The species distribution diagrams of ferric(III) and zinc(II) complexes with L1–3H6 are shown in Figures S15–S20. The ferric(III) and zinc(II) formation constants log βmlh and pM values at pH 3.0, 7.4, and 9.0 are listed for L1–3H6 and related compounds in Tables 3 and 4, respectively.
Table 3. Formation constants log β mlh and pFe3+ values of L1–3H6 and other related compounds.
| ligand | logβ110 | logβ111 | logβ112 | logβ113 | logβ114 | logβ115 | pFe3+ a |
||
|---|---|---|---|---|---|---|---|---|---|
| pH 3.0 | pH 7.4 | pH 9.0 | |||||||
| L1H6 | 41.66 (4) | 49.60 (1) | 57.26 (3) | 63.56 (3) | 66.12 (5) | 67.18 (6) | 14.39 (6) | 27.58 (2) | 33.06(1) |
| L2H6 | 40.81 (6) | 48.74 (4) | 56.34 (2) | 62.48 (3) | 65.01 (7) | 66.05 (8) | 13.62 (7) | 26.78 (2) | 32.24 (3) |
| L3H6 | 40.13 (8) | 48.25 (5) | 56.02 (7) | 61.62 (2) | 64.08 (7) | 65.06 (7) | 13.02 (6) | 26.47 (4) | 31.61 (1) |
| MECAMb | 43.0 | 50.2 | 56.23 | 60.73 | 64.53 | — | 13.20 | 29.40 | 34.56 |
| Enterobactinb | 49.0 | 53.95 | 57.47 | 59.97 | — | — | 12.28 | 35.50 | 40.52 |
| DTPAc | — | — | — | — | — | — | — | 24.60 | — |
Table 4. Formation constants log β mlh and pZn2+ values of L1–3H6 and other related compounds.
| ligand | logβ110 | logβ111 | logβ112 | logβ113 | pZn2+ a | ||
|---|---|---|---|---|---|---|---|
| pH 3.0 | pH 7.4 | pH 9.0 | |||||
| L1H6 | 14.28 (3) | 23.01 (4) | 31.30 (5) | 34.17 (7) | 6.0 | 6.0 | 6.24 (2) |
| L2H6 | 15.30 (6) | 24.21 (4) | 32.1 (2) | 35.4 (3) | 6.0 | 6.0 (1) | 7.01 (2) |
| L3H6 | 14.81 (8) | 23.46 (5) | 31.56 (7) | 34.52 (2) | 6.0 | 6.0 (1) | 6.55 (1) |
| DOTAb | — | — | — | — | — | 17.9 | — |
| DTPAb | — | — | — | — | — | 14.8 | — |
apZn2+ = −log[Zn2+free], [Zn2+] = 10−6 M and [L] = 10−5 M.
bRef. 44.
The pFe3+ values of hexadentate L1–3H6 ligands are higher than those of the efficient chelator diethylenetriaminepentaacetic acid (DTPA)44 at pH 7.4 but lower than those of enterobactin21,45 and N,N’,N”-tris(2,3-dihydroxybenzoyl)-1,3,5-tris(aminomethyl)benzene (MECAM)45, which is an efficient siderophore with high ferric(III) affinity. Enterobactin employs three catechol moieties to tightly encapsulate ferric(III) ion in the hexadentate coordination sphere46,47. However, the enterobactin analogues L1-3H6 with longer alkyl chain cannot adequately encapsulate it. This phenomenon could explain the low pFe3+ values of L1–3H6 ligands.
As shown in Table 4, the pZn2+ values of hexadentate L1–3H6 ligands are significantly lower than those of the efficient chelators 1,4,7,10-tetraazacyclododecane-N,N′,N″,N″′-tetraacetic acid (DOTA)44 and DTPA44 at pH 7.4. The low pZn+ values are similar to those of hexadentate catechol ligands48,49,50, indicated the formation of catechol derivatives with weak zinc(II) affinity, as predicted.
Antioxidant activity studies
DPPH· is a radical-generating substance widely used to monitor the free radical scavenging abilities of various antioxidants25,26,27. The assays were carried out in methanol, and the results are expressed as EC50, which represents the antioxidant concentration required to decrease the initial DPPH· concentration by 50%. Low EC50 values indicate high radical scavenging capacity. This parameter is widely used to measure antioxidant capacity but does not consider the reaction time. The time needed to reach the steady state to the concentration corresponding at EC50 (TEC50) was calculated, and antiradical efficiency (AE) was introduced as a parameter to characterize the antioxidant compounds27. AE is determined by Eq. 5.
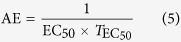 |
The kinetic curves of phenolic L1–3H6 with different concentrations are shown in Figures S21–S23. The time of reaction reached a steady state with great difference in the condition of different antioxidant concentrations. The TEC50 of phenolic L1–3H6 were obtained by plotting the times at the steady state against the concentration.
The EC50 values of phenolic L1–3H6 were determined from the curves of the percentage of DPPH at the steady state against the molar ratio of antioxidant to DPPH (Figures S24–S26). The EC50 and AE values calculated for L1–3H6 and other compounds are listed in Table 5.
Table 5. Effective concentration (EC50) and antiradical efficiency (AE) obtained with DPPH· assay.
The structures of phenolic derivatives have great influence on the activity51,52. The phenolic compounds L1–3H6 exhibit similar antioxidant capacity due to the similar molecular structures. Meanwhile, it is known that the polyphenols are more efficient than monophenols52. The DPPH assay results indicated that the hexaphenols L1–3H6 exihibited lower EC50 and shorter TEC50 values than diphenols catechol53 and monophenol BHA27, which confers them higher AE values. This result is as expected.
Conclusions
Coordinative saturation of the uranyl ion is achieved by development of hexadentate enterobactin analogues with different molecular scaffolds containing flexible alkyl chain lengths. The dominance of the hexadentate ligands in uranyl binding is supported by solution phase thermodynamic measurements. The flexible alkyl chain molecular scaffold exhibits conformational flexibility and forms a large coordination cavity suitable for the uranyl ion. The antioxidant capacity is determined by DPPH· assay, the hexadentate ligands are more active than catechol and BHA. Finally, considering the high uranyl affinity of the hexadentate enterobactin analogues, we conclude that these ligands may be more applicable for actinyl ions with larger radius, such as UO2+ and NpO2+, in these ions, ligand distortions may be lessened and could be encapsulated.
Experimental Section
General
The organic reagents used were pure commercial products from Aladdin. The solvents were purchased from Chengdu Kelong Chemical Reagents Co. Anhydrous CH2Cl2 was distilled prior to use. The 300–400 mesh silica gels was purchased from Qingdao Hailang Chemical Reagents Co. 1H NMR and 13C NMR spectra were recorded on Bruker Avance 300, Avance 400, or Avance 600 spectrometer. The FTIR spectra were obtained from Nicolet 380 FTIR spectrophotometer (Thermo Fisher Nicolet, USA) with a resolution of 4 cm−1 from 400 cm−1 to 4000 cm−1. UV-vis spectrophotometer (Thermo Scientific Evolution 201, USA) used had a double-beam light source from 190 nm to 1100 nm. Mass spectral analysis was conducted using Varian 1200 LC/MS.
2,3-bis(benzyloxy)benzoic Acid (2)
A solution of 2,3-dihydroxybenzoic acid (10.20 g, 65.9 mmol), benzyl bromide (22.2 g, 130.0 mmol), and K2CO3 (18.0 g, 130.0 mmol) in acetone (220 mL) was refluxed and stirred for 24 h. After filtration, the solution was concentrated in vacuo to obtain the crude product as clear oil. The crude product was dissolved in methanol (200 mL), and LiOH·H2O (360.0 mmol, 15.10 g) was slowly added. The mixture was refluxed and stirred for 3 h. Then, the solution was acidified with 3.0 M HCl to pH 2.0 and filtered to obtain the product 2 as white solid (yield of 80%). 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.50–7.10 (m, 12H, Ar-H), 7.03 (t, J = 8.0 Hz, 1H, Ar-H), 5.12 (s, 2H, O-CH2-Ar), 5.09 (s, 2H, O-CH2-Ar). 13C NMR (150 MHz, CDCl3): δ (ppm) = 165.38 (C=O), 151.54 (ArC), 147.32 (ArC), 136.07 (ArCH), 134.87 (ArCH), 129.51 (ArCH), 129.06 (ArCH), 129.03 (ArCH), 128.77 (ArCH), 128.00 (ArCH), 125.25 (ArCH), 124.67 (ArCH), 123.27 (ArCH), 119.21 (ArCH), 71.77 (CH2). FTIR (KBr, cm−1): 3100, 2700, 1683, 1035. APCI-MS (m/z): 333.4 [M-H]−.
2,3-bis(benzyloxy)-N-(hydroxyethyl)benzamide (4a)
A solution of 2,3-bis(benzyloxy) benzoic acid 2 (1.67 g, 5.0 mmol), HOBt (0.12 g, 0.9 mmol) and DCC (1.24 g, 6.0 mmol) in CH2Cl2 (50 mL) was stirred for 30 min at room temperature. Ethanolamine (0.34 g, 5.5 mmol) was added dropwise over 3 min and the mixture stirred 10 h. The solution was filtered to remove the dicyclohexyl urea (DCU). The filtrate was concentrated in vacuo and the residue purified by flash column chromatography to give the product 4a as clear oil (80%). Rf = 0.4 (volume ratio 2:3 acetone/hexane). 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.30 (br s, 1H, CO-NH), 7.71 (m, 1H, Ar-H), 7.50–7.10 (m, 12H, Ar-H), 5.15 (s, 2H, O-CH2-Ar), 5.10 (s, 2H, O-CH2-Ar), 3.62 (t, J = 5.4 Hz, 2H, CH2), 3.40 (m, 2H, CH2), 2.87 (br s, 1H, OH). 13C NMR (150 MHz, CDCl3): δ (ppm) = 165.78 (C=O), 150.95 (ArC), 146.15 (ArC), 135.60 (ArC), 127.99 (ArCH), 127.94 (ArCH), 127.52 (ArCH), 126.92 (ArCH), 123.68 (ArCH), 122.49 (ArCH), 116.50 (ArCH), 75.72 (CH2), 70.56 (CH2), 61.88 (CH2), 42.10 (CH2). FTIR (KBr, cm−1): 3347, 1625, 1577, 1555, 1498, 1028. APCI-MS (m/z): 378.0 [M+H]+.
2,3-bis(benzyloxy)-N-(3-hydroxypropyl)benzamide (4b)
A solution of 2,3-bis(benzyloxy) benzoic acid 2 (1.67 g, 5.0 mmol), HOBt (0.12 g, 0.9 mmol), and DCC (1.24 g, 6.0 mmol) in CH2Cl2 (50 mL) was stirred for 30 min at room temperature. 3-Amino-1-propanol (0.41 g, 5.5 mmol) was added dropwise over 3 min and the mixture stirred 10 h. The solution was filtered to remove the dicyclohexyl urea (DCU). The filtrate was concentrated in vacuo and the residue purified by flash column chromatography to give the product 4b as clear oil (85%). Rf = 0.5 (volume ratio 2:3 acetone/hexane). 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.12 (br s, 1H, CO-NH), 7.72 (m, 1H, Ar-H), 7.50–7.12 (m, 12H, Ar-H), 5.16 (s, 2H, O-CH2-Ar), 5.09 (s, 2H, O-CH2-Ar), 3.50 (t, J = 5.4 Hz, 2H, CH2), 3.39 (m, 2H, CH2), 1.52 (m, 2H, CH2). 13C NMR (150 MHz, CDCl3): δ (ppm) = 165.69 (C=O), 150.94 (ArC), 146.11 (ArC), 135.58 (ArC), 128.08 (ArCH), 127.98 (ArCH), 127.54 (ArCH), 126.89 (ArCH), 123.71 (ArCH), 122.54 (ArCH), 116.43 (ArCH), 75.76 (CH2), 70.55 (CH2), 57.89 (CH2), 34.89 (CH2), 31.69 (CH2). FTIR (KBr, cm−1): 3327, 1635, 1577, 1540, 1452, 1028. APCI-MS (m/z): 392.0 [M+H]+.
2,3-bis(benzyloxy)-N-(4-hydroxybutyl)benzamide (4c)
A solution of 2,3-bis(benzyloxy) benzoic acid 2 (1.67 g, 5.0 mmol), HOBt (0.12 g, 0.9 mmol) and DCC (1.24 g, 6.0 mmol) in CH2Cl2 (50 mL) was stirred for 30 min at room temperature. 4-Amino-1-butanol (0.49 g, 5.5 mmol) was added dropwise over 3 min and the mixture stirred 10 h. The solution was filtered to remove the dicyclohexyl urea (DCU). The filtrate was concentrated in vacuo and the residue purified by flash column chromatography to give the product 4c as clear oil (90%). Rf = 0.5 (volume ratio 2:3 acetone/hexane). 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.01 (br s, 1H, CO-NH), 7.74 (m, 1H, Ar-H), 7.50–7.10 (m, 12H, Ar-H), 5.16 (s, 2H, O-CH2-Ar), 5.09 (s, 2H, O-CH2-Ar), 3.58 (m, 2H, CH2), 3.32 (m, 2H, CH2), 1.46 (m, 4H, CH2-CH2). 13C NMR (150 MHz, CDCl3): δ (ppm) = 164.47 (C=O), 150.93 (ArC), 145.99 (ArC), 135.64 (ArC), 127.99 (ArCH), 127.50 (ArCH), 126.90 (ArCH), 123.68 (ArCH), 122.52 (ArCH), 116.17 (ArCH), 75.59 (CH2), 70.52 (CH2), 61.56 (CH2), 38.57 (CH2), 29.06 (CH2), 25.03 (CH2). FTIR (KBr, cm−1): 3327, 1635, 1577, 1540, 1452, 1033. APCI-MS (m/z): 406.2 [M+H]+.
2,3-bis(hydroxy)-N-(hydroxyethyl)benzamide (5a)
A mixture of 4a (0.75 g, 2.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over celite, evaporated to dryness and dried under vacuum to give 5a as grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.15 (br s, 1H, CO-NH), 7.29 (d, J = 8.1 Hz, 1H, Ar-H), 6.98 (dd, J = 7.8, 1.4 Hz, 1H, Ar-H), 6.73 (t, J = 8.0 Hz, 1H, Ar-H), 3.74 (t, J = 5.7 Hz, 2H, CH2), 3.55 (q, J = 5.6 Hz, 2H, CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.50 (C=O), 149.70 (ArC), 146.28 (ArC), 118.32 (ArCH), 118.17 (ArCH), 116.84 (ArCH), 114.63 (ArC), 60.20 (CH2), 42.09 (CH2). FTIR (KBr, cm−1): 3370, 2930, 1627, 1593, 1540, 1396, 1338, 1252, 1055. APCI-MS (m/z): 198.2 [M+H]+.
2,3-bis(hydroxy)-N-(3-hydroxypropyl)benzamide (5b)
A mixture of 4b (0.79 g, 2.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over celite, evaporated to dryness and dried under vacuum to give 5b as grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.26 (br s, 1H, CO-NH), 7.19 (d, J = 8.1 Hz, 1H, Ar-H), 6.92 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 6.67 (t, J = 8.0 Hz, 1H, Ar-H), 3.64 (t, J = 6.0 Hz, 2H, CH2), 3.50 (m, 2H, CH2), 1.79 (m, 2H, CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.37 (C=O), 149.79 (ArC), 146.37 (ArC), 118.38 (ArCH), 118.22 (ArCH), 116.73 (ArCH), 114.67 (ArC), 59.43 (CH2), 36.83 (CH2), 31.91 (CH2). FTIR (KBr, cm−1): 3336, 2932, 1639, 1589, 1547, 1488, 1460, 1334, 1262, 1179, 1070. APCI-MS (m/z): 212.3 [M+H]+.
2,3-bis(hydroxy)-N-(4-hydroxybutyl)benzamide (5c)
A mixture of 4c (0.81 g, 2.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over celite, evaporated to dryness and dried under vacuum to give 5c as a grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.31 (br s, 1H, CO-NH), 7.28 (d, J = 8.1 Hz, 1H, Ar-H), 6.97 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 6.71 (t, J = 8.0 Hz, 1H, Ar-H), 3.62 (m, 2H, CH2), 3.45 (m, 2H, CH2), 1.72 (m, 2H, CH2), 1.63 (m, 2H, CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.25 (C=O), 149.82 (ArC), 146.33 (ArC), 118.34 (ArCH), 118.17 (ArCH), 116.80 (ArCH), 114.78 (ArC), 61.27 (CH2), 39.17 (CH2), 30.01 (CH2), 25.51 (CH2). FTIR (KBr, cm−1): 3409, 3238, 2954, 1644, 1583, 1542, 1474, 1385, 1276, 1236, 1070. APCI-MS (m/z): 225.4 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(benzyloxy)-N-(hydroxyethyl)benzamide] ester (6a)
A solution of 1,3,5-benzenetricarbonyl trichloride (0.265 g, 1.0 mmol) in CH2Cl2 (10 mL) was droped in the solution of 2,3-bis(benzyloxy)-N-(hydroxyethyl) benzamide 4a (0.75 g, 2.0 mmol), Et3N (2 mL) in CH2Cl2 (20 mL) under ice bath and vigorous stirring conditions. The mixture stirred at room temperature for 16 h. After evaporation of solvent and the residue purified by flash column chromatography to give the product 6a as clear oil (yield of 70%). Rf = 0.7 (volume ratio 1:30 methanol/CHCl3). 1H NMR (600 MHz, CDCl3): δ (ppm) = 8.62 (s, 3H, Ar-H), 8.19 (t, J = 5.8 Hz, 3H, CO-NH), 7.63 (m, 3H, Ar-H), 7.47 (m, 6H, Ar-H), 7.38 (m, 9H, Ar-H), 7.21 (m, 6H, Ar-H), 7.11 (m, 15H, Ar-H), 5.12 (s, 6H, CH2), 5.04 (s, 6H, CH2), 4.29 (t, J = 5.5 Hz, 6H, CH2), 3.63 (q, J = 5.6 Hz, 6H, CH2). 13C NMR (150 MHz, CDCl3): δ (ppm) = 164.39 (C=O), 163.35 (C=O), 150.57 (ArC), 145.60 (ArC), 135.33 (ArC), 135.15 (ArC), 133.54, (ArCH), 129.82 (ArC), 127.64 (ArC), 127.61 (ArCH), 127.56 (ArCH), 127.45 (ArCH), 127.28 (ArCH), 126.77 (ArCH), 126.11 (ArCH), 123.37 (ArCH), 122.06 (ArCH), 75.48 (CH2), 70.13 (CH2), 63.31 (CH2), 37.43 (CH2). FTIR (KBr, cm−1): 3399, 2973, 2928, 1730, 1647, 1576, 1453, 1263, 1244, 1049, 741, 698. APCI-MS (m/z): 1289.5 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(benzyloxy)-N-(3-hydroxypropyl)benzamide] ester (6b)
A solution of 1,3,5-benzenetricarbonyl trichloride (0.265 g, 1.0 mmol) in CH2Cl2 (10 mL) was droped in the solution of 2,3-bis(benzyloxy)-N-(3-hydroxypropyl) benzamide 4b (0.79 g, 2.0 mmol), Et3N (2 mL) in CH2Cl2 (20 mL) under ice bath and vigorous stirring conditions. The mixture stirred at room temperature for 16 h. After evaporation of solvent and the residue purified by flash column chromatography to give the product 6a as clear oil (yield of 68%). Rf = 0.7 (volume ratio 1:30 methanol/CHCl3). 1H NMR (600 MHz, CDCl3): δ (ppm) = 8.67 (s, 3H, Ar-H), 7.98 (t, J = 7.1 Hz, 3H, CO-NH), 7.62 (m, 3H, Ar-H), 7.38 (m, 6H, Ar-H), 7.30 (m, 9H, Ar-H), 7.23 (m, 6H, Ar-H), 7.04 (m, 15H, Ar-H), 5.05 (s, 6H, CH2), 5.01 (s, 6H, CH2), 4.19 (t, J = 6.4 Hz, 6H, CH2), 3.32 (q, J = 6.7 Hz, 6H, CH2), 1.74 (m, 6H, CH2). 13C NMR (150 MHz, CDCl3): δ (ppm) = 165.28 (C=O), 164.78 (C=O), 151.65 (ArC), 146.86 (ArC), 136.43 (ArC), 134.56 (ArC), 131.15, (ArCH), 128.72 (ArC), 128.71 (ArC), 128.69 (ArCH), 128.26 (ArCH), 127.65 (ArCH), 127.21 (ArCH), 124.35 (ArCH), 123.27 (ArCH), 116.95 (ArCH), 76.48 (CH2), 71.26 (CH2), 63.46 (CH2), 36.59 (CH2), 28.53(CH2). FTIR (KBr, cm−1): 3390, 2929, 1726, 1654, 1575, 1533, 1453, 1262, 1241, 1026, 740, 697. APCI-MS (m/z): 1330.4 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(benzyloxy)-N-(4-hydroxybutyl)benzamide] ester (6c)
A solution of 1,3,5-benzenetricarbonyl trichloride (0.265 g, 1.0 mmol) in CH2Cl2 (10 mL) was droped in the solution of 2,3-bis(benzyloxy)-N-(3-hydroxypropyl) benzamide 4c (0.81 g, 2.0 mmol), Et3N (2 mL) in CH2Cl2 (20 mL) under ice bath and vigorous stirring conditions. The mixture stirred at room temperature for 16 h. After evaporation of solvent and the residue purified by flash column chromatography to give the product 6a as clear oil (yield of 71%). Rf = 0.7 (volume ratio 1:30 methanol/CHCl3). 1H NMR (600 MHz, CDCl3): δ (ppm) = 8.72 (s, 3H, Ar-H), 7.90 (t, J = 5.6 Hz, 3H, CO-NH), 7.66 (m, 3H, Ar-H), 7.40 (m, 6H, Ar-H), 7.33 (m, 9H, Ar-H), 7.25 (m, 6H, Ar-H), 7.07 (m, 15H, Ar-H), 5.08 (s, 6H, CH2), 5.02 (s, 6H, CH2), 4.19 (t, J = 6.7 Hz, 6H, CH2), 3.34 (q, J = 6.9 Hz, 6H, CH2), 1.61 (m, 6H, CH2), 1.36 (m, 6H, CH2). 13C NMR (150 MHz, CDCl3): δ (ppm) = 165.12 (C=O), 164.93 (C=O), 151.69 (ArC), 146.85 (ArC), 136.44 (ArC), 134.48 (ArC), 131.39, (ArCH), 128.72 (ArC), 128.71 (ArC), 128.28 (ArCH), 127.67 (ArCH), 127.25 (ArCH), 124.43 (ArCH), 123.36 (ArCH), 116.99 (ArCH), 76.46 (CH2), 71.31 (CH2), 65.28 (CH2), 39.13 (CH2), 26.20 (CH2), 25.85 (CH2). FTIR (KBr, cm−1): 3390, 2929, 1726, 1654, 1575, 1533, 1453, 1262, 1241, 1026, 740, 697. APCI-MS (m/z): 1372.8 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(hydroxy)-N-(hydroxyethyl)benzamide] ester (7a)
A mixture of 6a (1.29 g, 1.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over Celite, evaporated to dryness and dried under vacuum to give 7a as grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.82 (s, 3H, Ar-H), 7.26 (dd, J = 8.1, 1.4 Hz, 3H, Ar-H), 6.98 (dd, J = 7.8, 1.4 Hz, 3H, Ar-H), 6.73 (t, J = 8.0 Hz, 3H, Ar-H), 4.59 (t, J = 5.7 Hz, 6H, CH2), 3.87 (m, 6H, CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.70 (C=O), 164.48 (C=O), 149.60 (ArC), 146.28 (ArC), 134.20 (ArCH), 131.46 (ArC), 118.50, (ArCH), 118.32 (ArCH), 116.86 (ArCH), 114.56 (ArC), 64.07 (CH2), 38.31 (CH2). FTIR (KBr, cm−1): 3429, 2925, 1723, 1638, 1547, 1460, 1384, 1261, 1042.FTIR (KBr, cm−1): 3430, 2955, 1730, 1640, 1544, 1454, 1246, 1157, 1029. APCI-MS (m/z): 748.6 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(hydroxy)-N-(3-hydroxypropyl)benzamide] ester (7b)
A mixture of 6b (1.33 g, 1.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over Celite, evaporated to dryness and dried under vacuum to give 7b as grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.75 (s, 3H, Ar-H), 7.20 (dd, J = 8.1, 1.3 Hz, 3H, Ar-H), 6.92 (dd, J = 7.8, 1.3 Hz, 3H, Ar-H), 6.65 (t, J = 8.0 Hz, 3H, Ar-H), 4.51 (t, J = 6.2 Hz, 6H, CH2), 3.67 (m, 6H, CH2), 2.18 (m, 6H, CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.43 (C=O), 164.41 (C=O), 149.65 (ArC), 146.25 (ArC), 133.78 (ArCH), 131.45 (ArC), 125.17, (ArCH), 118.29 (ArCH), 118.16 (ArCH), 116.66 (ArCH), 114.52 (ArC), 63.46 (CH2), 36.31 (CH2), 29.82 (CH2). FTIR (KBr, cm−1): 3429, 2925, 1723, 1638, 1547, 1460, 1384, 1261, 1042. APCI-MS (m/z): 790.8 [M+H]+.
1,3,5-benzenetricarboxylic acid tris[2,3-bis(hydroxy)-N-(4-hydroxybutyl)benzamide] ester (7c)
A mixture of 6c (1.37 g, 1.0 mmol) and Pd/C (5%) (200 mg) in THF (50 mL) was stirred under H2 (130 mL/min) atmosphere for 6 h. The resulting mixture was filtered over Celite, evaporated to dryness and dried under vacuum to give 7c as grey power (yield of 99%). 1H NMR (600 MHz, (CD3)2CO): δ (ppm) = 8.81 (s, 3H, Ar-H), 7.24 (d, J = 8.1 Hz, 3H, Ar-H), 6.94 (dd, J = 7.8, 1.3 Hz, 3H, Ar-H), 6.70 (t, J = 8.0 Hz, 3H, Ar-H), 4.46 (t, J = 6.2 Hz, 6H, CH2), 3.52 (m, 6H, CH2), 1.91 (m, 12H, CH2CH2). 13C NMR (150 MHz, (CD3)2CO): δ (ppm) = 170.68 (C=O), 164.48 (C=O), 149.62 (ArC), 146.30 (ArC), 134.03 (ArCH), 131.63 (ArC), 118.26, (ArCH), 118.14 (ArCH), 116.66 (ArCH), 114.61 (ArC), 65.14 (CH2), 38.69 (CH2), 26.00 (CH2), 25.76 (CH2). FTIR (KBr, cm−1): 3410, 2954, 1724, 1639, 1598, 1545, 1459, 1330, 1247, 1167, 1044. APCI-MS (m/z): 832.4 [M+H]+.
Titration solutions and methods
INESA ZDJ-4B automatic potential titrator was used to measure the pH of the experimental solutions. Meanwhile, it was used for incremental additions of base standard solution to the titration cup under N2 atmosphere. Titrations were performed in 0.10 M KCl supporting electrolyte. The temperature of the experimental solution was maintained at 298.2 K by an externally thermostat water bath. UV-visible spectra for incremental titrations and batch titrations were recorded on a Thermo Scientific Evolution 201 UV-vis spectrophotometer. Solid reagents were weighed on a Sartorius BT25S analytical balance accurate to 0.01 mg. All titration solutions were prepared using distilled water from Ulupure ULUP-IV ultra water system and degassed by ultrasonic device. Standard solution of 0.10 M KOH and HNO3 were purchased from Aladdin. Ligand stock solutions were made by dissolving a weighed amount of ligand accurate to 0.01 mg in 5.0 vol % methanol aqueous solution in volumetric flask. A stock solution of 0.01 M metal ion [uranyl, ferric(III), and zinc(II) ion] were made by dissolving a weighed amount of corresponding metal salt in 5.0 vol % HNO3 standard solution. All metal ion titrations were conducted with a 1:1 ligand:metal ratio. Metal-to-ligand ratios were controlled by carefully addition of a ligand solution of known concentration and a metal ion stock solution to the titration cup. All titrations were repeated a minimum of three times.
Titration date treatment
Spectrophotometric titration data were analyzed using the HypSpec 2014 program54, utilizing nonlinear leastsquares regression to determine formation constants. Wavelengths between 250–550 nm were typically used for data refinement except ferric(III) titration. The number of absorbing species to be refined upon was determined by factor analysis within the HypSpec 2014 program54. Speciation diagrams were generated by using HySS program33 titration simulation software and the protonation and metal complex formation constants determined by potentiometric and spectrophotometric titration experiments.
Antioxidant assay methods
The antioxidant assay was carried out in dim room. DPPH· methanol solution was made by dissolving a weighed amount of DPPH· in volumetric flask which was wrapped by tinfoil. An aliquot of methanol (0.1 mL), different aliquot stock methanol solution of 5 × 10−5 M antioxidant were added to 2.5 mL methanol solution of 6 × 10−5 M DPPH·, and the volume adjusted to a final value of 3.0 mL with methanol. Absorbances at 515 nm were measured immediately at 10 s intervals on a Thermo Scientific Evolution 201 UV-vis spectrophotometer until the reaction reached steady state. Five different concentrations were measured for each assay. Then the EC50 values were plotted to obtain from graph of the percentage of DPPH· remaining at the steady state against the molar ratio antioxidant to DPPH·. Moreover, the time needed to reach the steady state to EC50 concentration (TEC50) and the AE values were also calculated.
Additional Information
How to cite this article: Zhang, Q. et al. Novel enterobactin analogues as potential therapeutic chelating agents: Synthesis, thermodynamic and antioxidant studies. Sci. Rep. 6, 34024; doi: 10.1038/srep34024 (2016).
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (project no. 51572230), Open Project of State Key Laboratory Cultivation Base for Nonmetal Composites and Functional (project no. 14zdfk05), Major Project of the Education Department of Sichuan Province (project no. 13ZA0172), Southwest University of Science and Technology Outstanding Youth Fundation (project no. 13zx9107).
Footnotes
Author Contributions B.J. and R.P. directed the project. Q.Z. and B.J. proposed and designed the project, Q.Z., Z.S., X.W., Q.L. and S.L. carried out the experiment and analyzed the data through discussions with B.J. and R.P. All the authors discussed the results and contributed to final version of the manuscript.
References
- Allard B., Olofsson U. & Torstenfelt B. Environmental actinide chemistry. Inorg. Chim. Acta 94, 205–221 (1984). [Google Scholar]
- Hamilton J. G. The metabolic properties of the fission products and actinide elements. Rev. Mod. Phys. 20, 718–728 (1948). [Google Scholar]
- Galle P. Toxiques Nucléaires 185–205 (Masson, 1997). [Google Scholar]
- Brugge D., de Lemos J. L. & Oldmixon B. Exposure pathways and health effects associated with chemical and radiological toxicity of natural uranium: a review. Rev. Environ. Health 20, 177–193 (2005). [DOI] [PubMed] [Google Scholar]
- Durbin P. W. In The Chemistry of the Actinide and Transactinide Elements 3rd edn, vol. 5 (eds Morss L. R. et al.) 3329 (Springer Science & Business Media, 2006). [Google Scholar]
- Pellmar T. C., Keyser D. O., Emery C. & Hogan J. B. Electrophysiological changes in hippocampal slices isolated from rats embedded with depleted uranium fragments. Neurotoxicology 20, 785–792 (1999). [PubMed] [Google Scholar]
- Periyakaruppan A., Kumar F., Sarkar S., Sharma C. S. & Ramesh G. T. Uranium induces oxidative stress in lung epithelial cells. Arch. Toxicol 81, 389–395 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. T. et al. UO22+-amino hybrid materials: structural variation and photocatalysis properties. CrystEngComm 17, 642–652 (2015). [Google Scholar]
- Sather A. C., Berryman O. B., Moore C. E. & Jr J. R. Uranyl ion coordination with rigid aromatic carboxylates and structural characterization of their complexes. Chem. Commun. 49, 6379–6381 (2013). [DOI] [PubMed] [Google Scholar]
- Frasson E., Bombieri G. & Panattoni C. Stereochemistry of uranyl acetylacetonate monohydrate. Coord. Chem. Rev. 1, 145–150 (1966). [Google Scholar]
- Harrowfield J. M., Kepert D. L., Patrick J. M., White A. H. & Lincoln S. F. Crystal structure of pentakis (dimethyl sulphoxide-O) dioxouranium (VI) bis(perchlorate). J. Chem. Soc., Dalton Trans. 2, 393–396 (1983). [Google Scholar]
- Domingo J. L., Ortega A., Llobet J. M., Paternain J. L. & Corbella J. The effects of repeated parenteral administration of chelating agents on the distribution and excretion of uranium. Res. Commun. Chem. Pathol. Pharmacol. 64, 161–164 (1989). [PubMed] [Google Scholar]
- Stradling G. N., Gray S. A., Moody J. C. & Ellender M. Efficacy of tiron for enhancing the excretion of uranium from the rat. Hum. Exp. Toxicol. 10, 195–198 (1991). [DOI] [PubMed] [Google Scholar]
- Sylwester E. R., Allen P. G., Dharmawardana U. R. & Sutton M. Structural studies of uranium and thorium complexes with 4,5-dihydroxy-3,5-benzenesdisulfonate (Tiron) at low and neutral pH by X-ray absorption spectroscopy. Inorg. Chim. 40, 2835–2841 (2001). [DOI] [PubMed] [Google Scholar]
- Harris W. R. et al. Coordination chemistry of microbial iron transport compounds. 19. Stability constants and electrochemical behavior of ferric enterobactin and model complexes. J. Am. Chem. Soc. 101, 6097–6104 (1979). [Google Scholar]
- Harris W. R., Carrano C. J. & Raymond K. N. Spectrophotometric determination of the proton-dependent stability constant of ferric enterobactin. J. Am. Chem. Soc. 101, 2213–2214 (1979). [Google Scholar]
- Huang S. P., Franz K. J., Olmstead M. M. & Fish R. H. Synthetic and structural studies of a linear bis-catechol amide, N, N’-bis(2,3-dihydroxybenzoyl)-1,7-diazaheptane (5-LICAM), and its complexes with Ni2+ and Co2+: utilization of a polymer-supported, sulfonated analog, 5-LICAMS, as a biomimetic ligand for divalent metal ion removal from aqueous solution. Inorg. Chim. 34, 2820–2825 (1995). [Google Scholar]
- Xu J., Kullgren B., Durbin P. W. & Raymond K. N. Specific sequestering agents for the actinides. 28. Synthesis and initial evaluation of multidentate 4-carbamoyl-3-hydroxy-1-methyl-2(1H)-pyridinone ligands for in vivo plutonium (IV) chelation. J. Med. Chem. 38, 2606–2614 (1995). [DOI] [PubMed] [Google Scholar]
- Xu J., O’Sulliva B. & Raymond K. N. Hexadentate Hydroxypyridonate Iron Chelators Based on TREN-Me-3, 2-HOPO: Variation of Cap Size. Inorg. Chim. 41, 6731–6742 (2002). [DOI] [PubMed] [Google Scholar]
- Xu J. & Raymond K. N. Uranyl sequestering agents: correlation of properties and efficacy with structure for UO22+ complexes of linear tetradentate 1-methyl-3-hydroxy-2(1H)-pyridinone ligands. Inorg. Chem. 38, 308–315 (1999). [Google Scholar]
- Harris W. R., Raymond K. N. & Weitl F. L. Ferric ion sequestering agents. 6. The spectrophotometric and potentiometric evaluation of sulfonated tricatecholate ligands. J. Am. Chem. Soc. 103, 2667–2675 (1981). [Google Scholar]
- Raymond K. N. & Smith W. L. In Structure and Bonding vol. 43 (ed. Goodenough J. B.) (Springer-Verlag, 1981). [Google Scholar]
- Dean N. E., Hancock R. D., Cahill C. L. & Frisch M. Affinity of the highly preorganized ligand PDA (1,10-phenanthroline-2,9-dicarboxylic acid) for large metal ions of higher charge. A crystallographic and thermodynamic study of PDA complexes of thorium (IV) and the uranyl (VI) ion. Inorg. Chim. 47, 2000–2010 (2008). [DOI] [PubMed] [Google Scholar]
- Raymond K. N., Freeman G. E. & Kappel M. J. Actinide-specific complexing agents: their structural and solution chemistry. Inorg. Chim. Acta 94, 193–204 (1984). [Google Scholar]
- Romano C. S., Abadi K., Repetto V., Vojnov A. A. & Moreno S. Synergistic antioxidant and antibacterial activity of rosemary plus butylated derivatives. Food Chem. 115, 456–461 (2009). [Google Scholar]
- Sharma O. P. & Bhat T. K. DPPH antioxidant assay revisited. Food Chem. 113, 1202–1205 (2009). [Google Scholar]
- Concepción S. M., Larrauri J. A. & Calixto F. S. A procedure to measure the antiradical efficiency of polyphenols. J. Sci. Food Agric. 76, 270–276 (1998). [Google Scholar]
- Laursen B., Denieul M. P. & Skrydstrup T. Formal total synthesis of the PKC inhibitor, balanol: preparation of the fully protected benzophenone fragment. Tetrahedron 58, 2231–2238 (2002). [Google Scholar]
- Gardner R. A., Kinkade R., Wang C. & Phanstiel IV O. Total Synthesis of petrobactin and its homologues as potential growth stimuli for marinobacter hydrocarbonoclasticus, an Oil-Degrading Bacteria. J. Org. Chem. 69, 3530–3537 (2004). [DOI] [PubMed] [Google Scholar]
- Rodgers S. J., Lee C. W., Ng C. Y. & Raymond K. N. Ferric ion sequestering agents. 15. Synthesis, solution chemistry, and electrochemistry of a new cationic analog of enterobactin. Inorg. Chim. 26, 1622–1625 (1987). [Google Scholar]
- Loomis L. D. & Raymond K. N. Solution equilibria of enterobactin and metal-enterobactin complexes. Inorg. Chim. 30, 906–911 (1991). [Google Scholar]
- Harris W. H. & Raymond K. N. Ferric ion sequestering agents. 3. The spectrophotometric and potentiometric evaluation of two new enterobactin analogs: 1,5,9-N,N’,N”-tris(2,3-dihydroxybenzoyl) cyclotriazatridecane and 1,3,5-N,N’,N”-tris(2,3-dihydroxybenzoyl) triaminomethylbenzene. J. Am. Chem. Soc. 101, 6534–6541 (1979). [Google Scholar]
- Alderighi L. et al. Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 184, 311–318 (1999). [Google Scholar]
- Imbert D. et al. Synthesis and iron (III) complexing ability of CacCAM, a new analog of enterobactin possessing a free carboxylic anchor arm. Comparative studies with TRENCAM. New J. Chem. 24, 281–288 (2000). [Google Scholar]
- Harris W. R., Raymond K. N. & Weitl F. L. Ferric ion sequestering agents. 6. The spectrophotometric and potentiometric evaluation of sulfonated tricatecholate ligands. J. Am. Chem. Soc. 103, 2667–2675 (1981). [Google Scholar]
- Hou Z., Stack T. D. P., Sunderland C. J. & Raymond K. N. Enhanced iron (III) chelation through ligand predisposition: syntheses, structures and stability of tris-catecholate enterobactin analogs. Inorg. Chim. Acta 263, 341–355 (1997). [Google Scholar]
- Hay B. P., Dixon D. A., Vargas R., Garza J. & Raymond K. N. Structural criteria for the rational design of selective ligands. 3. Quantitative structure-stability relationship for iron (III) complexation by tris-catecholamide siderophores. Inorg. Chim. 40, 3922–3935 (2001). [DOI] [PubMed] [Google Scholar]
- Szigethy G. & Raymond K. N. Influence of linker geometry on uranyl complexation by rigidly linked bis(3-hydroxy-N-methyl-pyridin-2-one). Inorg. Chim. 49, 6755–6765 (2010). [DOI] [PubMed] [Google Scholar]
- Xu J. & Raymond K. N. Uranyl sequestering agents: correlation of properties and efficacy with structure for UO22+ complexes of linear tetradentate 1-methyl-3-hydroxy-2(1H)-pyridinone ligands. Inorg. Chem. 38, 308–315 (1999). [Google Scholar]
- Szigethy G. & Raymond K. N. The influence of linker geometry in Bis(3-hydroxy-N-methyl-pyridin-2-one) ligands on solution phase uranyl affinity. Chem.-Eur. J. 17, 1818–1827 (2011). [DOI] [PubMed] [Google Scholar]
- Schwochau K. Topics in Current Chemistry 91–133 (Spinger, 1984). [Google Scholar]
- Martell A. E. & Smith R. M. In Critical Stability Constants, vol. 5 (Plenum, 1977). [Google Scholar]
- Szigethy G. & Raymond K. N. Hexadentate terephthalamide (bis-hydroxypyridinone) ligands for uranyl chelation: structural and thermodynamic consequences of ligand variation. J. Am. Chem. Soc. 133, 7942–7956 (2011). [DOI] [PubMed] [Google Scholar]
- Santos M. A., Gama S., Gano L., Cantinho G. & Farkas E. A new bis(3-hydroxy-4-pyridinone)-IDA derivative as a potential therapeutic chelating agent. Synthesis, metal-complexation and biological assays. Dalton Trans. 3772–3781 (2004). [DOI] [PubMed] [Google Scholar]
- Hou Z., Stack T. D. P., Sunderland C. J. & Raymond K. N. Enhanced iron(III) chelation through ligand predisposition: syntheses, structures and stability of tris-catecholate enterobactin analogs. Inorg. Chim. Acta 263, 341–355 (1997). [Google Scholar]
- Isied S. S., Kuo G. & Raymond K. N. Coordination isomers of biological iron transport compounds. V. The preparation and chirality of the chromium (III) enterobactin complex and model tris(catechol) chromium (III) analogues. J. Am. Chem. Soc. 98, 1763–1767 (1976). [DOI] [PubMed] [Google Scholar]
- Scarrow R. C., Ecker D. J., Ng C., Liu S. & Raymond K. N. Iron (III) coordination chemistry of linear dihydroxyserine compounds derived from enterobactin. Inorg. Chim. 30, 900–906 (1991). [Google Scholar]
- Guerra K. P. & Delgado R. Homo-and heterodinuclear complexes of the tris(catecholamide) derivative of a tetraazamacrocycle with Fe3+, Cu2+ and Zn2+ metal ions. Dalton Trans. 4, 539–550 (2008). [DOI] [PubMed] [Google Scholar]
- Kappel M. J. & Raymond K. N. Ferric ion sequestering agents. 10. Selectivity of sulfonated poly(catechoylamides) for ferric ion. Inorg. Chim. 21, 3437–3442 (1982). [Google Scholar]
- Biaso F., Baret P., Pierre J. L. & Serratrice G. Comparative studies on the iron chelators O-TRENSOX and TRENCAMS: selectivity of the complexation towards other biologically relevant metal ions and Al3+. J. Inorg. Biochem. 89, 123–130 (2002). [DOI] [PubMed] [Google Scholar]
- Cuvelier M. E., Richard H. & Berset C. Comparison of the antioxidative activity of some acid-phenols: structure-activity relationship. Biosci. Biotechnol. Biochem. 56, 324–325 (1992). [Google Scholar]
- Shahidi F., Janitha P. K. & Wanasundara P. D. Phenolic antioxidants. Crit. Rev. Food Sci. Nutr. 32, 67–103 (1992). [DOI] [PubMed] [Google Scholar]
- Bortolomeazzi R., Sebastianutto N., Toniolo R. & Pizzariello A. Comparative evaluation of the antioxidant capacity of smoke flavouring phenols by crocin bleaching inhibition, DPPH radical scavenging and oxidation potential. Food Chem. 100, 1481–1489 (2007). [Google Scholar]
- Gans P., Sabatini A. & Vacca A. Determination of equilibrium constants from spectrophometric data obtained from solutions of known pH: the program pHab. Ann. Chim. 89, 45–49 (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.