

Abstract
H2 metabolism is proposed to be the most ancient and diverse mechanism of energy-conservation. The metalloenzymes mediating this metabolism, hydrogenases, are encoded by over 60 microbial phyla and are present in all major ecosystems. We developed a classification system and web tool, HydDB, for the structural and functional analysis of these enzymes. We show that hydrogenase function can be predicted by primary sequence alone using an expanded classification scheme (comprising 29 [NiFe], 8 [FeFe], and 1 [Fe] hydrogenase classes) that defines 11 new classes with distinct biological functions. Using this scheme, we built a web tool that rapidly and reliably classifies hydrogenase primary sequences using a combination of k-nearest neighbors’ algorithms and CDD referencing. Demonstrating its capacity, the tool reliably predicted hydrogenase content and function in 12 newly-sequenced bacteria, archaea, and eukaryotes. HydDB provides the capacity to browse the amino acid sequences of 3248 annotated hydrogenase catalytic subunits and also contains a detailed repository of physiological, biochemical, and structural information about the 38 hydrogenase classes defined here. The database and classifier are freely and publicly available at http://services.birc.au.dk/hyddb/
Microorganisms conserve energy by metabolizing H2. Oxidation of this high-energy fuel yields electrons that can be used for respiration and carbon-fixation. This diffusible gas is also produced in diverse fermentation and anaerobic respiratory processes1. H2 metabolism contributes to the growth and survival of microorganisms across the three domains of life, including chemotrophs and phototrophs, lithotrophs and heterotrophs, aerobes and anaerobes, mesophiles and extremophiles alike1,2. On the ecosystem scale, H2 supports microbial communities in most terrestrial, aquatic, and host-associated ecosystems1,3. It is also proposed that H2 was the primordial electron donor4,5. In biological systems, metalloenzymes known as hydrogenases are responsible for oxidizing and evolving H21,6. Our recent survey showed there is a far greater number and diversity of hydrogenases than previously thought2. It is predicted that over 55 microbial phyla and over a third of all microorganisms harbor hydrogenases2,7. Better understanding H2 metabolism and the enzymes that mediate it also has wider implications, particularly in relation to human health and disease3,8, biogeochemical cycling9, and renewable energy10,11.
There are three types of hydrogenase, the [NiFe], [FeFe], and [Fe] hydrogenases, that are distinguished by their metal composition. Whereas the [Fe]-hydrogenases are a small methanogenic-specific family12, the [NiFe] and [FeFe] classes are widely distributed and functionally diverse. They can be classified through a hierarchical system into different groups and subgroups/subtypes with distinct biochemical features (e.g. directionality, affinity, redox partners, and localization) and physiological roles (i.e. respiration, fermentation, bifurcation, sensing)1,6. It is necessary to define the subgroup or subtype of the hydrogenase to predict hydrogenase function. For example, while Group 2a and 2b [NiFe]-hydrogenases share >35% sequence identity, they have distinct roles as respiratory uptake hydrogenases and H2 sensors respectively13,14. Likewise, discrimination between Group A1 and Group A3 [FeFe]-hydrogenases is necessary to distinguish fermentative and bifurcating enzymes2,15. Building on previous work16,17, we recently created a comprehensive hydrogenase classification scheme predictive of biological function2. This scheme was primarily based on the topology of phylogenetic trees built from the amino acid sequences of hydrogenase catalytic subunits/domains. It also factored in genetic organization, metal-binding motifs, and functional information. This analysis identified 22 subgroups (within four groups) of [NiFe]-hydrogenases and six subtypes (within three groups) of [FeFe]-hydrogenases, each proposed to have unique physiological roles and contexts2.
In this work, we build on these findings to develop the first web database for the classification and analysis of hydrogenases. We developed an expanded classification scheme that captures the full sequence diversity of hydrogenase enzymes and predicts their biological function. Using this information, we developed a classification tool based on the k-nearest neighbors’ (k-NN) method. HydDB is a user-friendly, high-throughput, and functionally-predictive tool for hydrogenase classification that operates with precision exceeding 99.8%.
Results and Discussion
A sequence-based classification scheme for hydrogenases
We initially developed a classification scheme to enable prediction of hydrogenase function by primary sequence alone. To do this, we visualized the relationships between all hydrogenases in sequence similarity networks (SSN)18, in which nodes represent individual proteins and the distances between them reflect BLAST E-values. As reflected by our analysis of other protein superfamilies19,20, SSNs allow robust inference of sequence-structure-function relationships for large datasets without the problems associated with phylogenetic trees (e.g. long-branch attraction). Consistent with previous phylogenetic analyses2,16,17, this analysis showed the hydrogenase sequences clustered into eight major groups (Groups 1 to 4 [NiFe]-hydrogenases, Groups A to C [FeFe]-hydrogenases, [Fe]-hydrogenases), six of which separate into multiple functionally-distinct subgroups or subtypes at narrower logE filters (Fig. 1; Figure S1). The SSNs demonstrated that all [NiFe]-hydrogenase subgroups defined through phylogenetic trees in our previous work2 separated into distinct clusters, which is consistent with our evolutionary model that such hydrogenases diverged from a common ancestor to adopt multiple distinct functions2. The only exception were the Group A [FeFe]-hydrogenases, which, as previously-reported2,17, cannot be classified by sequence alone as they have principally diversified through changes in domain architecture and quaternary structure. It remains necessary to analyze the organization of the genes encoding these enzymes to determine their specific function, e.g. whether they serve fermentative or electron-bifurcating roles.
Figure 1. Sequence similarity network of hydrogenase sequences.

Nodes represent individual proteins and the edges show the BLAST E-values between them at the logE filter defined at the bottom-left of each panel. The sequences are colored by class as defined in the legends. Figure S1 shows the further delineation of the encircled [NiFe] hydrogenase classes.
The SSN analysis revealed that several branches that clustered together on the phylogenetic tree analysis2 in fact separate into several well-resolved subclades (Fig. 1). We determined whether this was significant by analyzing the taxonomic distribution, genetic organization, metal-binding sites, and reported biochemical or functional characteristics of the differentiated subclades. On this basis, we concluded that 11 of the new subclades identified are likely to have unique physiological roles. We therefore refine and expand the hydrogenase classification to reflect the hydrogenases are more diverse in both primary sequence and predicted function than accounted for by even the latest classification scheme2. The new scheme comprises 38 hydrogenase classes, namely 29 [NiFe]-hydrogenase subclasses, 8 [FeFe]-hydrogenase subtypes, and the monophyletic [Fe]-hydrogenases (Table 1).
Table 1. Expanded classification scheme for hydrogenase enzymes.
| [NiFe] Group 1: Respiratory H2-uptake [NiFe]-hydrogenases | |||
| 1a | Periplasmic | Electron input for sulfate, metal, and organohalide respiration. [NiFeSe] variants. | 2 |
| 1b | Prototypical | Electron input for sulfate, fumarate, metal, and nitrate respiration. | 2 |
| 1c | Hyb-type | Electron input for fumarate, nitrate, and sulfate respiration. Physiologically reversible. | 2 |
| 1d | Oxygen-tolerant | Electron input for aerobic respiration and oxygen-tolerant anaerobic respiration. | 2 |
| 1e | Isp-type | Electron input primarily for sulfur respiration. Physiologically reversible. | 2 |
| 1f | Oxygen-protecting | Unresolved role. May liberate electrons to reduce reactive oxygen species. | 2 |
| 1g | Crenarchaeota-type | Electron input primarily for sulfur respiration. | 2 |
| 1h | Actinobacteria-type | Electron input for aerobic respiration. Scavenges electrons from atmospheric H2. | 2,46 |
| 1i | Coriobacteria-type (putative) | Undetermined role. May liberate electrons for anaerobic respiration. | This work |
| 1j | Archaeoglobi-type | Electron input for sulfate respirationπ. | This work |
| 1k | Methanophenazine-reducing | Electron input for methanogenic heterodisulfide respiration22. | This work |
| [NiFe] Group 2: Alternative and sensory uptake [NiFe]-hydrogenases | |||
| 2a | Cyanobacteria-type | Electron input for aerobic respiration. Recycles H2 produced by other cellular processes. | 16 |
| 2b | Histidine kinase-linked | H2 sensing. Activates two-component system controlling hydrogenase expression. | 16 |
| 2c | Diguanylate cyclase-linked (putative) | Undetermined role. May sense H2 and regulate processes through cyclic di-GMP production. | 2 |
| 2d | Aquificae-type | Unresolved role. May generate reductant for carbon fixation or have a regulatory role. | 2 |
| 2e | Metallosphaera-type (putative) | Undetermined role. May liberate electrons primarily for aerobic respiration26. | This work |
| [NiFe] Group 3: Cofactor-coupled bidirectional [NiFe]-hydrogenases | |||
| 3a | F420-coupled | Couples oxidation of H2 to reduction of F420 during methanogenesis. Physiologically reversible. [NiFeSe] variants. | 16 |
| 3b | NADP-coupled | Couples oxidation of NADPH to evolution of H2. Physiologically reversible. May have sulfhydrogenase activity. | 16 |
| 3c | Heterodisulfide reductase-linked | Bifurcates electrons from H2 to heterodisulfide and Fdox in methanogens. [NiFeSe] variants. | 16 |
| 3d | NAD-coupled | Interconverts electrons between H2 and NAD depending on cellular redox state. | 16 |
| [NiFe] Group 4: Respiratory H2-evolving [NiFe]-hydrogenases | |||
| 4a | Formate hydrogenlyase | Couples formate oxidation to fermentative H2 evolution. May be H+-translocating. | 2 |
| 4b | Formate-respiring | Respires formate or carbon monoxide using H+ as electron acceptor. Na+-translocating via Mrp23. | This work |
| 4c | Carbon monoxide-respiring | Respires carbon monoxide using H+ as electron acceptor. H+-translocating. | 2 |
| 4d | Ferredoxin-coupled, Mrp-linked | Couples Fdred oxidation to H+ reduction. Na+-translocating via Mrp complex24. | This work |
| 4e | Ferredoxin-coupled, Ech-type | Couples Fdred oxidation to H+ reduction. Physiologically reversible via H+/Na+ translocation. | 2 |
| 4f | Formate-coupled (putative) | Undetermined role. May couple formate oxidation to H2 evolution and H+ translocation. | 2 |
| 4g | Ferredoxin-coupled (putative) | Undetermined role. May couple Fdred oxidation to proton reduction and H+/Na+ translocation. | This work |
| 4h | Ferredoxin-coupled, Eha-type | Couples Fdred oxidation to H+ reduction in anaplerotic processes. H+/Na+-translocating25. | This work |
| 4i | Ferredoxin-coupled, Ehb-type | Couples Fdred oxidation to H+ reduction in anabolic processes. H+/Na+-translocating25. | This work |
| [FeFe] Hydrogenases | |||
| A1 | Prototypical | Couples ferredoxin oxidation to fermentative or photobiological H2 evolution. | 2,17 |
| A2 | Glutamate synthase-linked (putative) | Undetermined role. May couple H2 oxidation to NAD reduction, generating reductant for glutamate synthase. | 2,17 |
| A3 | Bifurcating | Reversibly bifurcates electrons from H2 to NAD and Fdox in anaerobic bacteria. | 2,17 |
| A4 | Formate dehydrogenase-linked | Couples formate oxidation to H2 evolution. Some bifurcate electrons from H2 to ferredoxin and NADP. | 2,17 |
| B | Colonic-type (putative) | Undetermined role. May couple Fdred oxidation to fermentative H2 evolution. | 17 |
| C1 | Histidine kinase-linked (putative) | Undetermined role. May sense H2 and regulate processes via histidine kinases2. | This work |
| C2 | Chemotactic (putative) | Undetermined role. May sense H2 and regulate processes via methyl-accepting chemotaxis proteins2. | This work |
| C3 | Phosphatase-linked (putative) | Undetermined role. May sense H2 and regulate processes via serine/threonine phosphatases2. | This work |
| [Fe] Hydrogenases | |||
| All | Methenyl-H4MPT dehydrogenase | Reversibly couples H2 oxidation to 5,10-methenyltetrahydromethanopterin reduction. | 16 |
The majority of the classes were defined in previous work2,16,17,46. The [NiFe] Group 1i, 1j, 2e, 4d, 4g, 4h, and 4i enzymes and [FeFe] Groups C1, C2, and C3 enzymes were defined in this work based on their separation into distinct clusters in the SSN analysis (Fig. 1). HydDB contains detailed information on each of these classes, including their taxonomic distribution, genetic organization, biochemistry, and structures, as well a list of primary references.
Three lineages originally classified as Group 1a [NiFe]-hydrogenases were reclassified as new subgroups, namely those affiliated with Coriobacteria (Group 1i), Archaeoglobi (Group 1j), and Methanosarcinales (Group 1i). Cellular and molecular studies show these enzymes all support anaerobic respiration of H2, but differ in the membrane carriers (methanophenazine, menaquinone) and terminal electron acceptors (heterodisulfide, sulfate, nitrate) that they couple to21,22. The previously-proposed 4b and 4d subgroups2 were dissolved, as the SSN analysis confirmed they were polyphyletic. These sequences are reclassified here into five new subgroups: the formate- and carbon monoxide-respiring Mrp-linked complexes (Group 4b)23, the ferredoxin-coupled Mrp-linked complexes (Group 4d)24, the well-described methanogenic Eha (Group 4h) and Ehb (Group 4i) supercomplexes25, and a more loosely clustered class of unknown function (Group 4g). Enzymes within these subgroups, with the exception of the uncharacterized 4g enzymes, sustain well-described specialist functions in the energetics of various archaea23,24,25. Three crenarchaeotal hydrogenases were also classified as their own family (Group 2e); these enzymes enable certain crenarchaeotes to grow aerobically on O226,27 and hence may represent a unique lineage of aerobic uptake hydrogenases currently underrepresented in genome databases. The Group C [FeFe]-hydrogenases were also separated into three main subtypes given they separate into distinct clusters even at relatively broad logE values (Fig. 1); these subtypes are each transcribed with different regulatory elements and are likely to have distinct regulatory roles2,17,28 (Table 1).
HydDB reliably predicts hydrogenase class using the k-NN method and CDD referencing
Using this information, we built a web tool to classify hydrogenases. Hydrogenase classification is determined through a three-step process following input of the catalytic subunit sequence. Two checks are initially performed to confirm if the inputted sequence is likely to encode a hydrogenase catalytic subunit/domain. The Conserved Domain Database (CDD)29 is referenced to confirm that the inputted sequence has a hydrogenase catalytic domain, i.e. “Complex1_49kDa superfamily” (cl21493) (for NiFe-hydrogenases), “Fe_hyd_lg_C superfamily” (cl14953) (for FeFe-hydrogenases), and “HMD” (pfam03201) (for Fe-hydrogenases). A homology check is also performed that computes the BLAST E-value between the inputted sequence and its closest homolog in HydDB. HydDB classifies any inputted sequence that lacks hydrogenase conserved domains or has low homology scores (E-value > 10−5) as a non-hydrogenase (Table S1).
In the final step, the sequence is classified through the k-NN method that determines the most similar sequences listed in the HydDB reference database. To determine the optimal k for the dataset, we performed a 5-fold cross-validation for k = 1…10 and computed the precision for each k. The results are shown in Fig. 2. The classifier predicted the classes of the 3248 hydrogenase sequences with 99.8% precision and high robustness when performing a 5-fold cross-validation (as described in the Methods section) for k = 4. The six sequences where there were discrepancies between the SSN and k-NN predictions are shown in Table S2. The classifier has also been trained to detect and exclude protein families that are homologous to hydrogenases but do not metabolize H2 (Nuo, Ehr, NARF, HmdII1,2) using reference sequences of these proteins (Table S1).
Figure 2. Evaluating the k-NN classifier for k = 1…10.
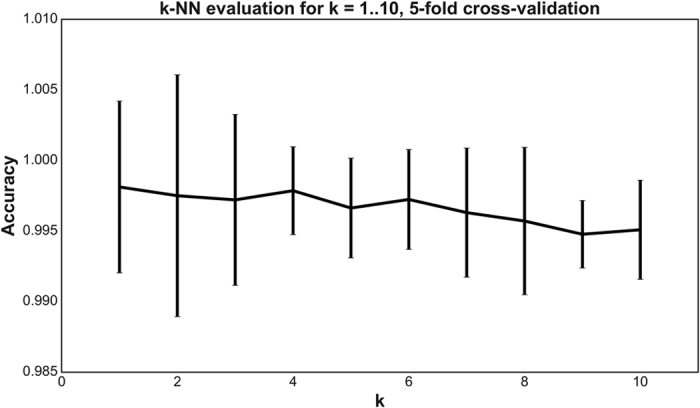
For each k, a 5-fold cross-validation was performed. The mean precision ± two standard deviations of the folds is shown in the figure (note the y-axis). k = 1 provides the most accurate classifier. However, k = 4 provides almost the same precision and is more robust to errors in the training set (reflected by the lower standard deviation). In general, the standard deviation is very small, indicating that the predictions are robust to changes in the training data.
Sequences of the [FeFe] Group A can be classified into functionally-distinct subtypes (A1, A2, A3, A4) based on genetic organization2. The classifier can classify such hydrogenases if the protein sequence immediately downstream from the catalytic subunit sequence is provided. The classifier references the CDD to search for conserved domains in the downstream protein sequence. A sequence is classified as [FeFe] Group A2 if one of the domains “GltA”, “GltD”, “glutamate synthase small subunit” or “putative oxidoreductase”, but not “NuoF”, is found in the sequence. Sequences are classified as [FeFe] Group A3 if the domain “NuoF” is found and [FeFe] Group A4 if the domain “HycB” is present. If none of the domains are found, the sequence is classified as A1. These classification rules were determined by collecting 69 downstream protein sequences. The sequences were then submitted to the CDD and the domains which most often occurred in each subtype were extracted.
In addition to its precision, the classifier is superior to other approaches due to its usability. It is accessible as a free web service at http://services.birc.au.dk/hyddb/ HydDB allows the users to paste or upload sequences of hydrogenase catalytic subunit sequences in FASTA format and run the classification (Figure S2). When analysis has completed, results are presented in a table that can be downloaded as a CSV file (Figure S3). This provides an efficient and user-friendly way to classify hydrogenases, in contrast to the previous standard which requires visualization of phylogenetic trees derived from multiple sequence alignments30.
HydDB infers the physiological roles of H2 metabolism
As summarized in Table 1, hydrogenase class is strongly correlated with physiological role. As a result, the classifier is capable of predicting both the class and function of a sequenced hydrogenase. To demonstrate this capacity, we used HydDB to analyze the hydrogenases present in 12 newly-sequenced bacteria, archaea, and eukaryotes of major ecological significance. The classifier correctly classified all 24 hydrogenases identified in the sequenced genomes, as validated with SSNs (Table 2). On the basis of these classifications, the physiological roles of H2 metabolism were predicted (Table 2). For five of the organisms, these predictions are confirmed or supported by previously published data27,31,32,33,34. Other predictions are in line with metabolic models derived from metagenome surveying35,36,37. In some cases, the capacity for organisms to metabolize H2 was not tested or inferred in previous studies despite the presence of hydrogenases in the sequenced genomes32,38,39,40.
Table 2. Predictive capacity of the HydDB.
| Organism | Phylum | Hydrogenase accession no. | HydDB classification | SSN classification | Predicted H2 metabolism | Confirmed H2 metabolism |
|---|---|---|---|---|---|---|
| Pyrinomonas methylaliphatogenes | Acidobacteria | WP_041979300.1 | [NiFe] Group 1h | [NiFe] Group 1h | Persistence by aerobic respiration of atmospheric H2 | Confirmed experimentally31 |
| Phaeodactylibacter xiamenensis | Bacteroidetes | WP_044227713.1 WP_044216927.1 WP_044227053.1 | [NiFe] Group 1d [NiFe] Group 2a [NiFe] Group 3d | [NiFe] Group 1d [NiFe] Group 2a [NiFe] Group 3d | Chemolithoautotrophic growth by aerobic H2 oxidation | Bacterium grows aerobically, but H2 oxidation untested32 |
| Bathyarchaeota archaeon BA1 | Bathyarchaeota | KPV62434.1 KPV62673.1 KPV62298.1 | [NiFe] Group 3c [NiFe] Group 3c [NiFe] Group 4g | [NiFe] Group 3c [NiFe] Group 3c [NiFe] Group 4g | Couples Fdred oxidation to H2 evolution in energy-conserving and bifurcating processes | Unconfirmed but consistent with metagenome-based models36 |
| Lenisia limosa | Obazoa (Breviatea class) | LenisMan28 | [FeFe] Group A1 | [FeFe] Group A | Fermentative evolution of H2 | Confirmed experimentally47 |
| Acidianus copahuensis | Crenarchaeota | WP_048100721.1 WP_048100713.1 WP_048100378.1 WP_048100359.1 | [NiFe] Group 1g [NiFe] Group 1g [NiFe] Group 1h [NiFe] Group 2e | [NiFe] Group 1g [NiFe] Group 1g [NiFe] Group 1h [NiFe] Group 2e | Chemolithoautotrophic growth by H2 oxidation using O2 or S0 as electron acceptors | Partially confirmed experimentally27 |
| Arcobacter sp. E1/2/3 | Proteobacteria (Epsilon class) | Arc.peg.2312 | [NiFe] Group 1b | [NiFe] Group 1b | Chemolithoautotrophic growth by anaerobic H2 oxidation | Confirmed experimentally47 |
| Methanoperedens nitroreducens | Euryarchaeota (ANME) | WP_048088262.1 WP_048090768.1 | [NiFe] Group 3b [NiFe] Group 3b | [NiFe] Group 3b [NiFe] Group 3b | Secondary role for H2 metabolism limited to fermentative evolution of H2 | Unconfirmed but consistent with metagenome-based models35 |
| Kryptonium thompsoni | Kryptonia | CUU03002.1 CUU06124.1 | [NiFe] Group 1d [NiFe] Group 3b | [NiFe] Group 1d [NiFe] Group 3b | Chemolithoautotrophic growth by aerobic H2 oxidation, fermentative evolution of H2. | Untested, candidate phylum identified by metagenomics39 |
| Lokiarchaeum sp. GC14_75 | Lokiarchaeota | KKK40681.1 | [NiFe] Group 3c | [NiFe] Group 3c | Bifurcates electrons between H2, heterodisulfide, and ferredoxin | Unconfirmed but consistent with metagenome-based models48 |
| Nitrospira moscoviensis | Nitrospirae | WP_053379275.1 | [NiFe] Group 2a | [NiFe] Group 2a | Chemolithoautotrophic growth by aerobic H2 oxidation | Confirmed experimentally33 |
| Bacterium GW2011_GWE1_35_17 | Moranbacteria | KKQ46070.1 KKQ45273.1 | [NiFe] Group 1a [NiFe] Group 3b | [NiFe] Group 1a [NiFe] Group 3b | Chemolithoautotrophic growth by anaerobic H2 oxidation, fermentative evolution of H2 | Unconfirmed but consistent with metagenome-based models37 |
| Bacterium GW2011_GWA2_33_10 | Peregrinibacteria | KKP36897.1 | [FeFe] Group A3 | [FeFe] Group A | Bifurcates electrons between H2, NADH, and ferredoxin | Unconfirmed but consistent with metagenome-based models37 |
| Entotheonella sp. TSY1 | Tectomicrobia | ETW97737.1 ETW94065.1 | [NiFe] Group 1h [NiFe] Group 3b | [NiFe] Group 1h [NiFe] Group 3b | Persistence by aerobic respiration of atmospheric H2, fermentative evolution of H2 | Untested, candidate phylum identified by metagenomics40 |
HydDB accurately determined hydrogenase content and predicted the physiological roles of H2 metabolism in 12 newly-sequenced archaeal and bacterial species.
While HydDB serves as a reliable initial predictor of hydrogenase class and function, further analysis is recommended to verify predictions. Hydrogenase sequences only provide organisms with the genetic capacity to metabolise H2; their function is ultimately modulated by their expression and integration within the cell1,41. In addition, some classifications are likely to be overgeneralized due to lack of functional and biochemical characterization of certain lineages and sublineages. For example, it is not clear if two distant members of the Group 1h [NiFe]-hydrogenases (Robiginitalea biformata, Sulfolobus islandicus) perform the same H2-scavenging functions as the core group9. Likewise, it seems probable that the Group 3a [NiFe]-hydrogenases of Thermococci and Aquificae use a distinct electron donor to the main class42. Prominent cautions are included in the enzyme pages in cases such as these. HydDB will be updated when literature is published that influences functional assignments.
HydDB contains interfaces for hydrogenase browsing and analyzing
In addition to its classification function, HydDB is designed to be a definitive repository for hydrogenase retrieval and analysis. The database presently contains entries for 3248 hydrogenases, including their NCBI accession numbers, amino acid sequences, hydrogenase classes, taxonomic affiliations, and predicted behavior (Figure S4). To enable easy exploration of the data set, the database also provides access to an interface for searching, filtering, and sorting the data, as well as the capacity to download the results in CSV or FASTA format. There are individual pages for the 38 hydrogenase classes defined here (Table 1), including descriptions of their physiological role, genetic organization, taxonomic distribution, and biochemical features. This is supplemented with a compendium of structural information about the hydrogenases, which is integrated with the Protein Databank (PDB), as well as a library of over 500 literature references (Figure S5).
Conclusions
To summarize, HydDB is a definitive resource for hydrogenase classification and analysis. The classifier described here provides a reliable, efficient, and convenient tool for hydrogenase classification and functional prediction. HydDB also provides browsing tools for the rapid analysis and retrieval of hydrogenase sequences. Finally, the manually-curated repository of class descriptions, hydrogenase structures, and literature references provides a deep but accessible resource for understanding hydrogenases.
Methods
Sequence datasets
The database was constructed using the amino acid sequences of all curated non-redundant 3248 hydrogenase catalytic subunits represented in the NCBI RefSeq database in August 20142 (Dataset S1). In order to test the classification tool, additional sequences from newly-sequenced archaeal and bacterial phyla were retrieved from the Joint Genome Institute’s Integrated Microbial Genomes database43.
Sequence similarity networks
Sequence similarity networks (SSNs)18 constructed using Cytoscape 4.144 were used to visualize the distribution and diversity of the retrieved hydrogenase sequences. In this analysis, each node represents one of the 3248 hydrogenase sequences in the reference database (Dataset S1). Each edge represents the sequence similarity between them as determined by E-values from all-vs-all BLAST analysis, with all self and duplicate edges removed. Three networks were constructed, namely for the [NiFe]-hydrogenase large subunit sequences (Dataset S2), [FeFe]-hydrogenase catalytic domain sequences (Dataset S3), and [Fe]-hydrogenase sequences (Dataset S4). To control the degree of separation between nodes, logE cutoffs that were incrementally decreased from −5 to −200 until no major changes in clustering was observed. The logE cutoffs used for the final classifications are shown in Fig. 1 and Figure S1.
Classification method
The 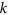 -NN method is a well-known machine learning method for classification45. Given a set of data points x1, x2, … xN (e.g. sequences) with known labels y1, y2, …, yN (e.g. type annotations), the label of a point,
-NN method is a well-known machine learning method for classification45. Given a set of data points x1, x2, … xN (e.g. sequences) with known labels y1, y2, …, yN (e.g. type annotations), the label of a point, 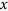 , is predicted by computing the distance from
, is predicted by computing the distance from 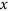 to x1, x2, … xN and extracting the
to x1, x2, … xN and extracting the 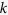 labeled points closest to
labeled points closest to 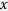 , i.e. the neighbors. The predicted label is then determined by majority vote of the labels of the neighbors. The distance measure applied here is that of a BLAST search. Thus, the classifier corresponds to a homology search where the types of the top
, i.e. the neighbors. The predicted label is then determined by majority vote of the labels of the neighbors. The distance measure applied here is that of a BLAST search. Thus, the classifier corresponds to a homology search where the types of the top 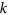 results are considered. However, formulating the classification method as a machine learning problem allows the use of common evaluation methods to estimate the precision of the method and perform model selection. The classifier was evaluated using
results are considered. However, formulating the classification method as a machine learning problem allows the use of common evaluation methods to estimate the precision of the method and perform model selection. The classifier was evaluated using 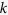 -fold cross-validation. The dataset is first split in to
-fold cross-validation. The dataset is first split in to 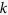 parts of equal size.
parts of equal size.  parts (the training set) are then used for training the classifier and the labels of the data points in the remaining part (the test set) are then predicted. This process, called a fold, is repeated
parts (the training set) are then used for training the classifier and the labels of the data points in the remaining part (the test set) are then predicted. This process, called a fold, is repeated 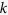 times. The predicted labels of each fold are then compared to the known labels and a precision can be computed.
times. The predicted labels of each fold are then compared to the known labels and a precision can be computed.
Additional Information
How to cite this article: Søndergaard, D. et al. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 6, 34212; doi: 10.1038/srep34212 (2016).
Supplementary Material
Acknowledgments
We thank A/Prof Colin J. Jackson, Dr. Hafna Ahmed, Dr. Andrew Warden, Dr. Stephen Pearce, and the two anonymous reviewers for their helpful advice and comments regarding this manuscript. This work was supported by a PUMPkin Centre of Excellence PhD Scholarship awarded to DS and a CSIRO Office of the Chief Executive Postdoctoral Fellowship awarded to CG.
Footnotes
Author Contributions C.G. and D.S. designed experiments. D.S. and C.G. performed experiments. C.G., D.S. and C.N.S.P. analyzed data. C.N.S.P. supervised students. C.G. and D.S. wrote the paper.
References
- Schwartz E., Fritsch J. & Friedrich B. H2-metabolizing prokaryotes (Springer Berlin Heidelberg, 2013). [Google Scholar]
- Greening C. et al. Genome and metagenome surveys of hydrogenase diversity indicate H2 is a widely-utilised energy source for microbial growth and survival. Isme J. 10, 761–777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook G. M., Greening C., Hards K. & Berney M. In Advances in Bacterial Pathogen Biology (ed. Poole R. K.) 65, 1–62 (Academic Press, 2014). [DOI] [PubMed] [Google Scholar]
- Lane N., Allen J. F. & Martin W. How did LUCA make a living? Chemiosmosis in the origin of life. BioEssays 32, 271–280 (2010). [DOI] [PubMed] [Google Scholar]
- Weiss M. C. et al. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 1, 16116 (2016). [DOI] [PubMed] [Google Scholar]
- Lubitz W., Ogata H., Rüdiger O. & Reijerse E. Hydrogenases. Chem. Rev. 114, 4081–4148 (2014). [DOI] [PubMed] [Google Scholar]
- Peters J. W. et al. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta - Mol. Cell Res. 1853, 1350–1369 (2014). [DOI] [PubMed] [Google Scholar]
- Carbonero F., Benefiel A. C. & Gaskins H. R. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat Rev Gastroenterol Hepatol 9, 504–518 (2012). [DOI] [PubMed] [Google Scholar]
- Greening C. et al. Atmospheric hydrogen scavenging: from enzymes to ecosystems. Appl. Environ. Microbiol. 81, 1190–1199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D. B., Pitt L. & Love M. Biohydrogen production: prospects and limitations to practical application. Int. J. Hydrogen Energy 29, 173–185 (2004). [Google Scholar]
- Cracknell J. A., Vincent K. A. & Armstrong F. A. Enzymes as working or inspirational catalysts for fuel cells and electrolysis. Chem. Rev. 108, 2439–2461 (2008). [DOI] [PubMed] [Google Scholar]
- Shima S. et al. The crystal structure of [Fe]-Hydrogenase reveals the geometry of the active site. Science 321, 572–575 (2008). [DOI] [PubMed] [Google Scholar]
- Lenz O. & Friedrich B. A novel multicomponent regulatory system mediates H2 sensing in Alcaligenes eutrophus. Proc. Natl. Acad. Sci. USA 95, 12474–12479 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening C., Berney M., Hards K., Cook G. M. & Conrad R. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc. Natl. Acad. Sci. USA 111, 4257–4261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchmann K. & Müller V. A bacterial electron-bifurcating hydrogenase. J. Biol. Chem. 287, 31165–31171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais P. M., Billoud B. & Meyer J. Classification and phylogeny of hydrogenases. Fems Microbiol. Rev. 25, 455–501 (2001). [DOI] [PubMed] [Google Scholar]
- Calusinska M., Happe T., Joris B. & Wilmotte A. The surprising diversity of clostridial hydrogenases: a comparative genomic perspective. Microbiology 156, 1575–1588 (2010). [DOI] [PubMed] [Google Scholar]
- Atkinson H. J., Morris J. H., Ferrin T. E. & Babbitt P. C. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. Plos One 4, e4345 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed F. H. et al. Sequence-structure-function classification of a catalytically diverse oxidoreductase superfamily in mycobacteria. J. Mol. Biol. 427, 3554–3571 (2015). [DOI] [PubMed] [Google Scholar]
- Ney B. et al. The methanogenic redox cofactor F420 is widely synthesized by aerobic soil bacteria. Isme J., 10.1038/ismej.2016.100 (2016). [DOI] [PMC free article] [PubMed]
- Stetter K. O. Archaeoglobus fulgidus gen. nov., sp. nov.: a new taxon of extremely thermophilic archaebacteria. Syst. Appl. Microbiol. 10, 172–173 (1988). [Google Scholar]
- Deppenmeier U. & Blaut M. Analysis of the vhoGAC and vhtGAC operons from Methanosarcina mazei strain Gö1, both encoding a membrane-bound hydrogenase and a cytochrome b. Eur. J. Biochem. 269, 261–269 (1995). [DOI] [PubMed] [Google Scholar]
- Kim Y. J. et al. Formate-driven growth coupled with H2 production. Nature 467, 352–355 (2010). [DOI] [PubMed] [Google Scholar]
- McTernan P. M. et al. Intact functional fourteen-subunit respiratory membrane-bound [NiFe]-hydrogenase complex of the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 289, 19364–19372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie T. J. et al. Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc. Natl. Acad. Sci. USA 109, 15473–15478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auernik K. S. & Kelly R. M. Physiological versatility of the extremely thermoacidophilic archaeon Metallosphaera sedula supported by transcriptomic analysis of heterotrophic, autotrophic, and mixotrophic growth. Appl. Environ. Microbiol. 76, 931–935 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaveno M. A., Urbieta M. S., Ulloa J. R., González Toril E. & Donati E. R. Physiologic versatility and growth flexibility as the Main characteristics of a novel thermoacidophilic Acidianus strain isolated from Copahue geothermal area in Argentina. Microb. Ecol. 65, 336–346 (2012). [DOI] [PubMed] [Google Scholar]
- Poudel S. et al. Unification of [FeFe]-hydrogenases into three structural and functional groups. Biochim. Biophys. Acta (BBA)-General Subj., 10.1016/j.bbagen.2016.05.034 (2016). [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A. & Bryant S. H. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney M., Greening C., Hards K., Collins D. & Cook G. M. Three different [NiFe] hydrogenases confer metabolic flexibility in the obligate aerobe Mycobacterium smegmatis. Environ. Microbiol. 16, 318–330 (2014). [DOI] [PubMed] [Google Scholar]
- Greening C. et al. Persistence of the dominant soil phylum Acidobacteria by trace gas scavenging. Proc. Natl. Acad. Sci. 112, 10497–10502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. et al. Phaeodactylibacter xiamenensis gen. nov., sp. nov., a member of the family Saprospiraceae isolated from the marine alga Phaeodactylum tricornutum. Int. J. Syst. Evol. Microbiol. 64, 3496–3502 (2014). [DOI] [PubMed] [Google Scholar]
- Koch H. et al. Growth of nitrite-oxidizing bacteria by aerobic hydrogen oxidation. Science 345, 1052–1054 (2014). [DOI] [PubMed] [Google Scholar]
- Carere C. R. et al. Growth and persistence of methanotrophic bacteria by aerobic hydrogen respiration. Proc. Natl. Acad. Sci. USA (2016). [Google Scholar]
- Haroon M. F. et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500, 567–570 (2013). [DOI] [PubMed] [Google Scholar]
- Evans P. N. et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438 (2015). [DOI] [PubMed] [Google Scholar]
- Brown C. T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015). [DOI] [PubMed] [Google Scholar]
- Spang A. et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eloe-Fadrosh E. A. et al. Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat Commun 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. C. et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62 (2014). [DOI] [PubMed] [Google Scholar]
- Greening C. & Cook G. M. Integration of hydrogenase expression and hydrogen sensing in bacterial cell physiology. Curr. Opin. Microbiol. 18, 30–38 (2014). [DOI] [PubMed] [Google Scholar]
- Greening C. et al. Physiology, biochemistry, and applications of F420- and Fo-dependent redox reactions. Microbiol. Mol. Biol. Rev. 80, 451–493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz V. M. et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Research 40, D115–D122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover T. & Hart P. Nearest neighbor pattern classification. Ieee Trans. Inf. Theory 13 (1967). [Google Scholar]
- Constant P., Chowdhury S. P., Pratscher J. & Conrad R. Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high-affinity [NiFe]-hydrogenase. Environ. Microbiol. 12, 821–829 (2010). [DOI] [PubMed] [Google Scholar]
- Hamann E. et al. Environmental Breviatea harbour mutualistic Arcobacter epibionts. Nature 534, 254–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa F. L., Neukirchen S., Allen J. F., Lane N. & Martin W. F. Lokiarchaeon is hydrogen dependent. Nat. Microbiol. 1, 16034 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.