

Abstract
Oridonin belongs to ent-kaurane tetracyclic diterpenoid and was first isolated from Isodon species. It exhibits inhibitory activities against a variety of tumor cells, and pharmacological study shows that oridonin could inhibit cell proliferation, DNA, RNA and protein synthesis of cancer cells, induce apoptosis and exhibit an antimutagenic effect. In addition, the large amount of the commercially-available supply is also very important for the natural lead oridonin. Moreover, the good stability, suitable molecular weight and drug-like property guarantee its further generation of a natural-like compound library. Oridonin has become the hot molecule in recent years, and from the year 2010, more than 200 publications can be found. In this review, we summarize the synthetic medicinal chemistry work of oridonin from the first publication 40 years ago and share our research experience of oridonin for about 10 years, which may provide useful information to those who are interested in this research field.
Keywords: oridonin, ent-kaurane, medicinal chemistry, structural modification, biological activity
1. Introduction
Oridonin (1, Figure 1) is an ent-kaurane diterpenoid isolated from Isodon of the Labiatae family, the structure and absolute configuration of which were first confirmed in the year 1970 [1]. Since then, hundreds of research articles, mainly in the antitumor field, have been published. Only from the year 2010, more than 200 papers can be found using Web of Science (searching oridonin as the topic), and obviously, it has become a molecule in focus from a natural source for the treatment of cancer. Oridonin is also a good lead in the field of medicinal chemistry: (a) it shows antitumor activities against many tumor-related cells [2,3] (for example, IC50 values were 21.48 μM against human esophageal squamous EC9706 cells [4], 5 μM against leukemia-derived Jurkat cells [5], 2.5 μM against human umbilical vascular endothelial cells [6], 15.6 μM against gastric cancer SGC-7901 cells [7], 19.32 μM against human pancreatic cancer BxPC-3 cells [8], 37.90 μM against human liver carcinoma HepG2 cells [9], 3.1 and 6.1 μM against uveal melanoma OCM-1 and MUM2B cells for a treatment of 24 h [10], 54.2 μM against human lung cancer A549 cells [11], 41.8 μM against leukemia HPB cells [12], 63.7 μM against human fibrosarcoma HT1080 cells [13], 15.18 μM against human epidermoid carcinoma A431 cells [14], 37.1 μM against human laryngeal cancer HEp-2 cells [15,16], 7.4 μM against melanoma K1735M2 cells [17], 3.88 μM against human colon tumor SW620 cells [18], 3.74 μM against bone marrow tumor K562 cells [18], 5.12 μM against breast tumor MCF7 cells [18], and so on) and relatively low toxicity (18.26 μM against human liver L-02 cells and 7.48 μM against human liver carcinoma Bel-7402 cells for a treatment of 72 h [19]); (b) the well-studied multitargeting properties of the antitumor activity [20,21] through specific chemical modifications enable the development of natural product (NP)-based novel drugs; (c) oridonin possessed good stability in stock solutions (1 mg/mL of oridonin and the internal standard were over 99% of the nominal concentrations compared with freshly prepared solutions after storage at −20 °C for 30 days), rat plasma (the starting concentrations were 12.3, 246 and 984 ng/mL; the measured concentrations were 12.0, 248 and 973 ng/mL after 24 h at room temperature and 12.1, 252 and 976 ng/mL at −20 °C for 30 days, respectively) and even after three freeze–thaw cycles (freezing at −20 °C and thawing at room temperature on three consecutive days) with concentrations of 11.8, 253 and 978 ng/mL [22,23]; (d) oridonin meets the criteria of Lipinski’s rule of five and is a lead-like, fragment-like and drug-like molecule [24,25]; the molecular weight (Mw) of oridonin is 362.2, which makes it suitable for further optimization, because the Mw value always increases during the process from lead to drug-like candidate [26]; (e) there are many functional groups (double bond, carbonyl group and hydroxyl groups in different chemical environments), which provide good synthetic accessibility to efficiently generate libraries of derivatives; and (f) last, but not least, for all natural products, it is commercially available for large-scale compound supply. Despite these promising profiles as an anticancer lead, there are also some shortcomings to be overcome during structural modification processes, such as relatively low aqueous solubility and bioavailability, moderate potency and undefined mechanisms of action. On the basis of the above, oridonin was widely used as a lead compound, especially to develop novel antitumor natural product-derived agents, by many medicinal chemistry researchers, including our research group [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41]. With its molecular mechanisms being gradually clarified, more scientists will join this field to discover promising anti-neoplastic drug from oridonin, as well as tetracyclic diterpenoids. Although there are several reviews concerning the biological activities [20,21,42,43,44] and medicinal chemistry [45] (structural modification and biological evaluation) of oridonin, herein, we would like to review the synthetic work of the lead oridonin from the first publication 40 years ago and to share our research experience of oridonin for about 10 years, which not only provides the best convenience to whomever wants to join this research field, but also a good example for lead optimization research from natural sources.
Figure 1.
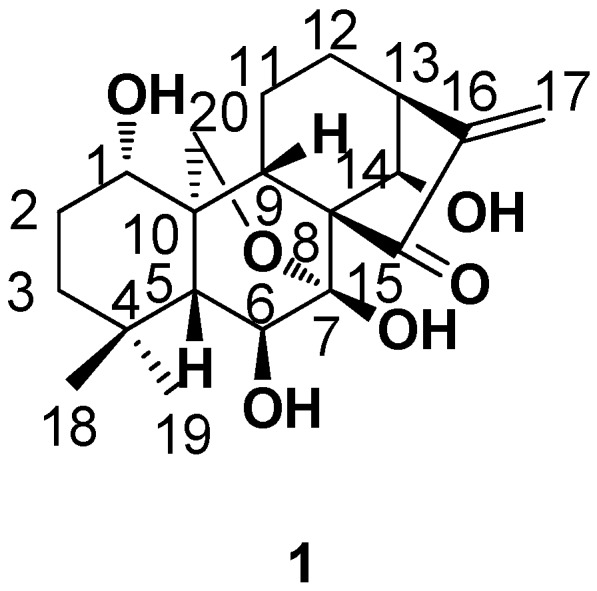
The structure and numbering of oridonin.
2. Structure Modification on the Core Structure of Oridonin
In the year 1976, biomimetic reactions of oridonin and alkane thiols were done by Fujita et al. under mild conditions to give alkane thiol adducts (2) at C-17 [46]. The dihydro-derivative of oridonin (3) was obtained by the reduction reaction of 2 with Raney Ni (Figure 2). Compound 3 showed no antitumor activity against Ehrlich ascites carcinoma cells in mouse and antibacterial activity against 19 selected bacteria. Therefore, the α-methylene-cyclopentanone system in the structure of oridonin was first considered as an important active center.
Figure 2.

The biomimetic products (2) and dihydro-derivative (3) of oridonin.
Five years later (in 1981), Fujita et al. selectively synthesized the first series of derivatives of 1 by acylation at 6-O (4) or/and 14-O (5, Figure 3) [47]. The antitumor activity of synthetic derivatives against Ehrlich ascites carcinoma cells in mouse were evaluated, and the structure activity relationship (SAR) showed that the activity increased with the increase of the acyl carbon chain length. The importance of the hydrogen bond and the ester side chain for the antitumor activity was also demonstrated in the derivatives of oridonin; while the 1,14-diacetate derivative of oridonin could be obtained by the treatment of oridonin with acetic anhydride in pyridine [48].
Figure 3.

Selective acylated products of oridonin at 6-O (4) or 14-O (5).
In 1990, eriocalyxin B (6) and its analogues were obtained by Zhou et al. from oridonin [49]. The overall yields of 6 and 14-hydroxyeriocalyxin B (7) were 11% and 57% in six and four steps, correspondingly (Figure 4).
Figure 4.

Synthesis of eriocalyxin B (6) and 14-hydroxyeriocalyxin B (7) from oridonin.
Oridonin-6-O-α-d-glucopyranoside (8) was synthesized by Liu’s group from 1 by five steps in a total yield of 23% (Figure 5). This method could improve the water solubility of oridonin derivatives [50].
Figure 5.

Synthesis of oridonin-6-O-α-d-glucopyranoside (8).
1-OAc oridonin was also a natural product, which was called lasiokaurin (11). In 2006, Liu’s group synthesized 11 from 1 in a 69% overall yield via three steps by selective acetonide protection (9), acetylation (10) and deprotection (Figure 6). The antiprotozoan activity of lasiokaurin and oridonin was tested, and the median lethal concentration was 25 and 50 μM, respectively, which indicated that 1-hydroxyl had some benefits on the antiprotozoan activity [51]. These reactions were used extensively in the further modification of oridonin.
Figure 6.

Synthesis routine of lasiokaurin (11). (a) 2,2-Dimethoxypropane, acetone, TsOH, 56 °C; (b) Ac2O, TEA (triethylamine), DMAP (4-dimethylaminopyridine), rt (room temperature); (c) 10% HCl, THF (tetrahydrofuran), rt.
In the next year, a series of aromatic amine derivatives (12, Figure 7) of oridonin was designed and obtained by Liu’s group [52]. The antiproliferative activity against oral epithelial KB cells was evaluated and all of the derivatives showed cytotoxicity to some extent, which was similar to or stronger than oridonin. When R4 was carboxyl, Compound 13 exhibited the strongest activity with an inhibition rate of 38.0% at a concentration of 0.8 mg/mL.
Figure 7.

Aromatic amine derivatives (12 and 13) of oridonin.
In the year 2008, our research group published our first work in the field of the structural modification of oridonin [41]. Some 1-O and 14-O-derivatives of oridonin were synthesized (Figure 8) and biologically evaluated against six cancer cell lines (SW-480, BGC-7901, HL-60, A549, Bel-7402 and B16). All of the derivatives exhibited stronger cytotoxicity than oridonin in vitro, and three of them were further evaluated in vivo. Derivatives 16 and 17 showed the most potent antiproliferative activity against HL-60 and Bel-7402 cell lines with the IC50 values of 0.84 and 1.00 μM, respectively. The preliminary SAR suggested that the introduction of both terminal carboxylic acid and the ester side chain of the lipophilicity moiety to the 14-O position of oridonin appeared to increase the antiproliferative activity. The cytotoxicity of the derivatives with the substituent of acetyl at the 1-O position was better than those with the propylsulfonyl group and the 1-hydroxyl oxidated derivatives. Compounds 16 and 17 also had stronger anti-tumor activity in mice bearing H22 liver tumor than oridonin (45.9%) with the inhibition ratios of 69.9% and 61.2%, correspondingly.
Figure 8.

Synthesis of 1-O and 14-O-derivatives (14–17) of oridonin.
Nitric oxide (NO) is a key mediator involved in many physiological and pathological processes. Li et al. synthesized several series of furoxan/oridonin hybrids (Figure 9) and evaluated the antiproliferative activity against four cancer cell lines (Bel-7402, CaEs-17, MGC-803, and K562) [38]. All of the target compounds released high levels of NO (more than 15 μM) at the time point of 60 min in vitro and exhibited stronger antiproliferative activity than the parent oridonin. Higher levels of NO-releasing capacity could be beneficial to cytotoxicity. In each series of the target synthetic hybrids, the derivative (18) with R10 of o-C6H4 and R11 of (CH2)3 exhibited the strongest antiproliferative activity among the designed hybrids with IC50 values in (sub)micromolar ranges. The linkages between the NO donor and drug molecule always affect the NO-releasing ability and biological activity.
Figure 9.

Synthesis of furoxan/oridonin NO-releasing hybrids (18).
In 2013, Zhou’s group developed an efficient and concise synthetic approach to install the azide functional group at the C-1, C-2 or C-3 positions of the A-ring of oridonin rapidly and diversely with highly-controlled regio- and stereo-selectivity (Figure 10) [53]. These azides were further functionalized through click chemistry to yield triazole derivatives. The antiproliferative activity of representative 1,2,3-triazolesubstituted derivatives (19 and 20) against breast cancer cell lines MCF-7 and MDA-MB-231 was tested. These derivatives with 1,2,3-triazole installed in the A-ring exhibited significantly improved antiproliferative activity compared to oridonin. Among them, Compound 21 showed the strongest potency with IC50 of 0.38 and 0.48 μM, respectively. This work provided access to an expanded potential anticancer natural scaffold-based compound library from the lead oridonin.
Figure 10.

Installation of azides and 1,2,3-triazole at the C-1, -2, or -3 position derivatives (19–21) of oridonin.
In the same year, Zhou’s group synthesized a series of oridonin derivatives (22) with thiazole fused in the A-ring through a protecting group-free synthetic strategy (Figure 11) [54]. Most of the derivatives exhibited potent antiproliferative effects against selected pancreatic, breast and prostate cancer cells with low (sub)micromolar IC50 values and enhanced aqueous solubility. These molecules not only showed enhanced growth inhibitory effects against MCF-7 cells, but also on the other oridonin insensitive cancer cells, including the highly invasive triple-negative MDA-MB-231 cell line with low IC50 values. Particularly, the most potent derivative (23) with an N-allyl substituted thiazole moiety exhibited IC50 values of 0.2 μM against both MDA-MB-231 and MCF-7 cells, which are approximately 147-fold and 33-fold more potent than oridonin, respectively. It was also found to induce the apoptosis of MCF-7/ADR and MDA-MB-231 cells in a concentration-dependent manner through similar multiple pathways as oridonin. The above derivative significantly suppressed MDA-MB-231 xenograft tumor growth at 5 mg/kg and was more efficacious than oridonin. Furthermore, one analogue (24, R14 = H, R15 = n-butane), which significantly inhibited HCC1806 and HCC1937 triple-negative breast cancer cells’ proliferation, was selected for an intensive mechanism study. The results showed that it could induce apoptosis and cell cycle arrest at the G2/M phase of HCC1806 and HCC1937 cells. The inhibitory potency would be caused by the expression of death receptor 5 (DR5), p21 and pERK and downregulations of cyclin D1, XIAP, FLIPL, pSTAT3 and pAKT. Besides, the suppression of HCC1806 xenograft tumor growth at 5 mg/kg in nude mice without the loss of body weight also guaranteed its further development as a drug candidate [55].
Figure 11.

The thiazole-fused A-ring modified oridonin derivatives (22–24).
In late 2013, a series of dienone derivatives of oridonin with an additional α,β-unsaturated ketone system installed in the A-ring was synthesized by Zhou’s group (Figure 12) [56]. Regioselective enone construction strategies were established. These derivatives significantly induced apoptosis and exhibited superior antitumor effects to oridonin against drug-resistant and aggressive breast cancer cells in vitro and in vivo (26 suppressed MDA-MB-231 xenograft tumor growth at 5.0 mg/kg) and also exhibited low toxicity to normal human mammary epithelial cells. The preliminary mechanism studies revealed that selected dienone analogues (25, 26) were found to induce the apoptosis of MDA-MB-231 cells in a concentration-dependent manner through regulation of a series of apoptotic-related proteins.
Figure 12.

The dienone derivatives (25–28) of oridonin with the α,β-unsaturated ketone system in the A-ring.
It was also reported that oridonin [57,58,59] and its derivatives [60,61] exhibited anti-inflammatory activity, such as the ability to treat hepatic fibrosis. The antifibrogenic effects of Compounds 27 [60] and 25 [61] were investigated on the activated rat HSC-T6 and human LX-2 stellate cell lines. These two derivatives could inhibit the proliferation of HSC-T6 and LX-2 cells in a dose- and time-dependent manner. However, for the human hepatocyte cell line C3A, no significant antiproliferative effects were observed. These two derivatives also induced apoptosis and cycle arrest at the S phase in the LX-2 cell line. The apoptosis-related property of Compound 27 was associated with the activation of p53, p21 and cleaved caspase-3, while Compound 25 could activate cleaved PARP, p21 and p53 and decrease cyclin-B1. Both of them could markedly downregulate major ECM proteins type I collagen and fibronectin and the myofibroblast marker protein α-smooth muscle actin in a time- and dose-dependent fashion and blocked transforming growth factor-β-induced type I collagen and fibronectin production. Oridonin analogues 25 and 27 would have great potential to act as promising antifibrogenic agents to treat hepatic fibrosis.
In 2014, a series of oridonin and nitrogen mustard hybrids were designed and synthesized by Xu et al. to find more efficacious and less toxic antitumor agents (Figure 13) [30]. The antiproliferative activity of the hybrids was more potent than oridonin and the clinically used nitrogen mustards against four selected human cancer cell lines (Bel-7402, MCF-7, K562 and MGC-803). Some representative derivatives exhibited antiproliferative activities against the multidrug-resistant cell lines (NCI-H460/MX20 and SW620/AD300). The most effective compound (29) of this series showed strong inhibitory activity with an IC50 value (0.67 μM) 21-fold lower than that of oridonin (14.60 μM) against MCF-7 cells and also exhibited selective cytotoxicity toward different cancer cells. It was demonstrated to affect cell cycle progression and significantly induce apoptosis in human hepatoma Bel-7402 cells.
Figure 13.

The hybrid of oridonin and nitrogen mustard (29).
In the same year, a series of dihydropyran-fused oridonin derivatives was designed and synthesized as potential anticancer agents by Zhou’s group (Figure 14) [62]. 3,4-dihydro-2H-pyran moiety was introduced into the A-ring of oridonin by an optimized IED HDA (inverse electron demand hetero-Diels–Alder) reaction in a mild and concise approach. The inhibitory effects of these derivatives were evaluated against MCF-7, MDAMB-231, MDA-MB-468 and MCF-7/ADR cell lines. Among them, Compound 30 was the most potent one, which showed submicromolar IC50 values in MCF-7 (IC50 = 0.44 μM), MDA-MB-231 (IC50 = 0.54 μM) and MDA-MB-468 (IC50 = 0.52 μM) cells and an improved capability to overcome chemoresistance against the MCF-7/ADR cell line (IC50 = 1.6 μM).
Figure 14.

The dihydropyran-fused oridonin 30.
From 2014–2016, the antibacterial activity of 1- or/and 14-position modified oridonin derivatives (31) was evaluated (Figure 15) [29,31,32]. Some derivatives were screened against Mycobacterium marinum, Mycobacterium smegmatis and Mycobacterium phlei. Among them, the compounds containing the trans-cinnamic acid moiety showed the most potent inhibitory activity against M. phlei with MICs of 0.5 μg/mL, and the SARs were analyzed. Five compounds were tested against Mycobacterium tuberculosis H37Rv based on the preliminary screening results. Among them, Compound 32 showed an IC50 value of 17.1 μg/mL. The antibacterial activity of some derivatives against Escherichia coli, Staphylococcus aureus, Bacillus subtilis and Monilia albicans was evaluated for the first time. Most of them showed good antibacterial activity against Gram-positive bacteria B. subtilis and S. aureus. Additionally, no obvious inhibitory activity was observed against Gram-negative bacterium E. coli and fungus M. albicans (MIC > 100 μg/mL).
Figure 15.

The 1- or/and 14-position modified oridonin derivatives (31 and 32) with antibacterial activity.
Early in this year, a series of oridonin fluorescent probes linked with coumarin moieties were designed and synthesized to explore the anticancer mechanism by our research group (Figure 16) [27]. Most of the probe molecules displayed optimal antiproliferative activity and a fluorescent property. Fluorescence microscopy and confocal imaging studies were examined by using typical probes. The results indicated that oridonin fluorescent probe 33 could be rapidly taken up into tumor cells, and the mitochondrion was the main site where it accumulated. Moreover, we confirmed that the α,β-unsaturated ketone group is the active moiety, which is crucial to its cytotoxicity, localization and uptake. The studies on mitochondrial physiology suggested that the mitochondrion-related pathway was involved in oridonin-induced apoptosis, and cytochrome c played an important role. These results provide new insights into the cellar mechanism of oridonin and may be useful to study the insight of the anticancer action and the targets of other ent-kaurane diterpenoids.
Figure 16.

The oridonin fluorescent probe 33.
3. Oridonin as the Lead to Synthesize ent-Kaurane Diterpenoid Derivatives of Other Types
ent-Kaurane diterpenoid derivatives with diverse unique chemical skeletons are generally quite complex, incorporating numbers of intricate ring systems and stereogenic centers, and exhibit promising biological activities [63,64,65,66,67,68,69]. The preparation of ent-kaurane diterpenoid libraries inevitably involved rather laborious and complex synthetic sequences, especially total synthesis [70,71,72]. Therefore, the therapeutic development of these compounds was significantly impeded by the problem of large-scale compound supply. It is found that oridonin is a good relevant commercially-available lead to semi-synthesize novel ent-kaurane diterpenoid derivatives with different scaffolds bypassing the de novo synthetic strategy.
3.1. 15,16-seco-ent-Kaurane Diterpenoid Derivatives
In the year 2011, Zhang et al. synthesized rubescensin S (34) from oridonin by two steps and revised its stereochemistry (Figure 17) [73]. An effective two-step transformation from oridonin to the 15,16-seco-ent-kaurane skeleton (rubescensin S) was achieved. This key compound provided a good building block for further construction of a natural product-like 15,16-seco-ent-kaurane compound library. They also revised its structure of the 13S configuration instead of the reported 13R.
Figure 17.

The synthesis of 15,16-seco-ent-kaurane rubescensin S (34) from oridonin.
3.2. 6,7-seco-ent-Kaurane Diterpenoid Derivatives
6,7-seco-ent-Kaurane diterpenoid derivatives (including enmein- and spirolactone-type 6,7-seco-ent-kaurane diterpenoids) showed stronger cytotoxicity than oridonin. These compounds also have more complex structures (Figure 18) and are more difficult to isolate from natural sources [74,75,76,77,78,79]. We used oridonin as the starting material to semi-synthesize enmein- and spirolactone-type core structures by the ring-opening reaction between C-6 and C-7. A compound library containing more than 100 enmein- and spirolactone-type diterpenoid derivatives was built up mainly by our research group for further medicinal study.
Figure 18.

Natural spirolactone- and enmein-type 6,7-seco-kaurane diterpenoids.
3.2.1. Enmein-Type 6,7-seco-ent-Kaurane Diterpenoid Derivatives
Several series of ester derivatives (35) of enmein-type diterpenoid at 14-O were synthesized from commercially-available oridonin by efficient and practical synthetic methods (Figure 19) [33,36]. The antiproliferative activity was evaluated against a set of human cancer cell lines. Some derivatives showed even smaller IC50 values than positive control paclitaxel. The apoptotic properties of the selected Compound 36 (IC50 = 0.71 μM) in human hepatocarcinoma Bel-7402 cells were evaluated. It caused cell-cycle arrest at the G2/M phase and induced apoptosis. Moreover, Compound 36 exhibited potent antitumor activity in vivo in MGC-803 mice. These results warranted further preclinical investigations of these enmein-type diterpenoid derivatives as potential anticancer agents.
Figure 19.

Enmein-type 6,7-seco-kaurane diterpenoid derivatives 35 and 36.
The antimycobacterial activity of some enmein-type 6,7-seco-kaurane diterpenoid derivatives (35) was also evaluated [31,32]. Most of the derivatives showed antimycobacterial activity against M. phlei, of which Compounds 37–39 (Figure 19) exhibited the strongest activity with MIC of 0.5 μg/mL and were 15-fold stronger than that of oridonin (8 μg/mL). The trans-cinnamic acid moiety benefitted the antimycobacterial activity. Compounds 38 and 40 (Figure 20) also showed moderate antitubercular activity against M. tuberculosis H37Rv with MICs of 28.8 and 24.0 μg/mL, respectively. These findings could provide new insights into the development of novel antitubercular agents from enmein-type 6,7-seco-kaurane diterpenoid derivatives.
Figure 20.

Antimycobacterial enmein-type 6,7-seco-kaurane diterpenoid derivatives 37–40.
In this year, a series of NO-donating enmein-type diterpenoid derivatives (41) was designed and synthesized (Figure 21) [19]. The target derivatives showed potent antibacterial activity against Gram-positive bacteria S. aureus and B. subtilis with the most promising MICs of 4 and 2 μg/mL, respectively, while the MICs of oridonin were both 32 μg/mL. The antiproliferative activity against human tumor and human normal cells was also tested. Most of these NO-releasing molecules showed good cytotoxic selectivity and released high levels (above 20 μmol/L) of NO at the time point of 60 min. Compound 42 was the most promising one with IC50 values of 1.68, 1.11, 3.60 and 0.72 μM against K562, MGC-803, CaEs-17 and Bel-7402 cells and 18.80 μM against normal liver cell line L-02. The selectivity index (SI) of 42 between tumor and normal liver cells was about 26.1, while the SI of oridonin was only 2.4. Compound 42 also induced apoptosis by the mitochondria-related pathway and arrested Bel-7402 cell cycle at the S phase.
Figure 21.

NO-releasing enmein-type 6,7-seco-kaurane diterpenoid derivatives 41 and 42.
3.2.2. Spirolactone-Type 6,7-seco-ent-Kaurane Diterpenoid Derivatives
The spirolactone-type 6,7-seco-ent-kaurane diterpenoid derivatives (43) were also obtained from commercial-available oridonin (Figure 22) [34,37]. These derivatives showed improved antiproliferative activity against a panel of human cancer cell lines, and some of them were more potent than the positive control Taxol. For example, the most potent Compound 44 with chloro substitution at the ortho-position of the benzene ring showed IC50 values of 0.39, 1.28, 0.60 and 1.39 μM against K562, MGC-803, CaEs-17 and Bel-7402 cells, which were 11.2-, 3.4-, 17.4- and 4.3-fold stronger than oridonin, correspondingly. The cellular mechanisms showed that Compound 44 could induce apoptosis at low micromolar concentrations in human hepatoma Bel-7402 cells.
Figure 22.

Spirolactone-type 6,7-seco-kaurane diterpenoid derivatives 43 and 44.
3.3. ent-Kaurane Diterpenoid Dimers
Many ent-kaurane diterpenoid dimers with diverse structures were isolated from natural sources [80,81,82]. It was found that oridonin could be a good lead to semi-synthesize ent-kaurane diterpenoid dimer derivatives to achieve the final drug candidate. A 3,4-dihydro-2H-pyran ring was firstly constructed in the A-ring of oridonin using an optimized IED HDA (inverse electron demand hetero-Diels–Alder) reaction. ent-Kaurane diterpenoid dimers were synthesized through a homo-HDA reaction by a self-dimerization of the exocyclic enone in the A-ring (Figure 23) [62]. The antiproliferative effects were evaluated against four breast cancer cell lines, MCF-7, MDA-MB-468, MDAMB-231 and MCF-7/ADR, by the MTT method, and the IC50 values were in the submicromolar range with a significantly improved capability to overcome chemoresistance.
Figure 23.

ent-Kaurane diterpenoid dimer 45.
4. Conclusions
In summary, there is an increasing number of studies advocating the medicinal chemistry field of the lead oridonin especially in the treatment of cancer. We propose ent-kaurane tetracyclic diterpenoids as natural multi-target agents represent promising therapeutic agents. However, further studies are required to elucidate the detailed molecular mechanisms of their actions. Structural modification should focus on the enhancement of potency and activity spectrum and, thereby, counter resistance mechanisms. Meanwhile, improving water solubility, the reduction of toxicity and increasing metabolic stability will continue to make significant contributions to the drug development of oridonin. Furthermore, the synthesis of some bioactive natural rare ent-kaurane diterpenoid analogues will be another key research direction for oridonin, which will provide structurally simpler compounds with retained bioactivities. It is also an economic strategy for the development of natural product-derived drugs.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81373280, 21502121), the Project Funded by the China Post-Doctoral Science Foundation (2015M570258), the General Scientific Research Projects of the Department of Education in Liaoning Province (L2014382), the Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources (Guangxi Normal University), the Ministry of the Education of China (CMEMR2015-B07), the Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy (KF-XY-201401) and the Career Development Support Plan for Young and Middle-aged Teachers in Shenyang Pharmaceutical University.
Abbreviations
| Mw | molecular weight |
| NP | natural product |
| SAR | structure activity relationship |
| DMAP | 4-dimethylaminopyridine |
| NO | nitric oxide |
| rt | room temperature |
| THF | tetrahydrofuran |
| TEA | triethylamine |
| Ac | acetyl group |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MIC | minimal inhibitory concentration |
| DR5 | death receptor 5 |
| IED | inverse electron demand |
| HDA | hetero-Diels–Alder |
| SI | selectivity index |
Author Contributions
Dahong Li, Shengtao Xu, Huiming Hua and Jinyi Xu designed the review. Dahong Li, Tong Han, Kangtao Tian, Xiaoke Gu and Keguang Cheng collected the literature. Dahong Li, Jie Liao, Xu Hu, Zhanlin Li and Jinyi Xu wrote and corrected the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Fujita E., Fujita T., Katayama H., Shibuya M., Shingu T. Terpenoids. Part XV. Structure and absolute configuration oridonin isolated from Isodon japonicus and Isodon trichocarpus. J. Chem. Soc. C. 1970;21:1674–1681. doi: 10.1039/j39700001674. [DOI] [Google Scholar]
- 2.Fujita E., Nagao Y., Node M., Kaneko K., Nakazawa S., Kuroda H. Antitumor activity of the isodon diterpenoids: Structural requirements for the activity. Cell. Mol. Life Sci. 1976;32:203–206. doi: 10.1007/BF01937766. [DOI] [PubMed] [Google Scholar]
- 3.Weng H., Huang H., Dong B., Zhao P., Zhou H., Qu L. Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res. 2014;74:4409–4419. doi: 10.1158/0008-5472.CAN-13-1748. [DOI] [PubMed] [Google Scholar]
- 4.Liu J.B., Yue J.Y. Preliminary study on the mechanism of oridonin-induced apoptosis in human squamous cell oesophageal carcinoma cell line EC9706. J. Int. Med. Res. 2014;42:984–992. doi: 10.1177/0300060513507389. [DOI] [PubMed] [Google Scholar]
- 5.Dal Piaz F., Cotugno R., Lepore L., Vassallo A., Malafronte N., Lauro G., Bifulco G., Belisario M.A., de Tommasi N. Chemical proteomics reveals HSP70 1A as a target for the anticancer diterpene oridonin in Jurkat cells. J. Proteom. 2013;82:14–26. doi: 10.1016/j.jprot.2013.01.030. [DOI] [PubMed] [Google Scholar]
- 6.Dong Y., Zhang T., Li J., Deng H., Song Y., Zhai D., Peng Y., Lu X., Liu M., Zhao Y., et al. Oridonin inhibits tumor growth and metastasis through anti-angiogenesis by blocking the Notch signaling. PLoS ONE. 2014;9:1395. doi: 10.1371/journal.pone.0113830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao S.Y., Li J., Qu X.Y., Zhu N., Ji Y.B. Downregulation of Cdk1 and cyclinB1 expression contributes to oridonin-induced cell cycle arrest at G2/M phase and growth inhibition in SGC-7901 gastric cancer cells. Asian Pac. J. Cancer Prev. 2014;15:6437–6441. doi: 10.7314/APJCP.2014.15.15.6437. [DOI] [PubMed] [Google Scholar]
- 8.Chen R.Y., Xu B., Chen S.F., Chen S.S., Zhang T., Ren J., Xu J. Effect of oridonin-mediated hallmark changes on inflammatory pathways in human pancreatic cancer (BxPC-3) cells. World J. Gastroenterol. 2014;20:14895–14903. doi: 10.3748/wjg.v20.i40.14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H., Ye Y., Yu Z.L. Proteomic and functional analyses demonstrate the involvement of oxidative stress in the anticancer activities of oridonin in HepG2 cells. Oncol. Rep. 2014;31:2165–2172. doi: 10.3892/or.2014.3081. [DOI] [PubMed] [Google Scholar]
- 10.Gu Z., Wang X., Qi R., Wei L., Huo Y., Ma Y., Shi L., Chang Y., Li G., Zhou L. Oridonin induces apoptosis in uveal melanoma cells by upregulation of Bim and downregulation of fatty acid synthase. Biochem. Biophys. Res. Commun. 2015;457:187–193. doi: 10.1016/j.bbrc.2014.12.086. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y., Liu J.H., Chai K., Tashiro S., Onodera S., Ikejima T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J. Pharm. Pharmacol. 2013;65:1622–1642. doi: 10.1111/jphp.12140. [DOI] [PubMed] [Google Scholar]
- 12.Liu J.J., Huang R.W., Lin D.J., Wu X.Y., Peng J., Pan X.L., Lin Q., Hou M., Zhang M.H., Chen F. Antiproliferation effects of oridonin on HPB-ALL cells and its mechanisms of action. Am. J. Hematol. 2006;81:86–94. doi: 10.1002/ajh.20524. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Wu Y., Wu D., Tashiro S., Onodera S., Ikejima T. NF-κB facilitates oridonin-induced apoptosis and autophagy in HT1080 cells through a p53-mediated pathway. Arch. Biochem. Biophys. 2009;489:25–33. doi: 10.1016/j.abb.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y., Fan S.M., Song J.K., Tashiro S., Onodera S., Ikejima T. Hydroxyl radical (·OH) played a pivotal role in oridonin-induced apoptosis and autophagy in human epidermoid carcinoma A431 cells. Biol. Pharm. Bull. 2012;35:2148–2159. doi: 10.1248/bpb.b12-00405. [DOI] [PubMed] [Google Scholar]
- 15.Kang N., Zhang J.H., Qiu F., Tashiro S., Onodera S., Ikejima T. Inhibition of EGFR signaling augments oridonin-induced apoptosis in human laryngeal cancer cells via enhancing oxidative stress coincident with activation of both the intrinsic and extrinsic apoptotic pathways. Cancer Lett. 2010;294:147–158. doi: 10.1016/j.canlet.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Kang N., Zhang J.H., Qiu F., Chen S., Tashiro S., Onodera S., Ikejima T. Induction of G2/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. J. Nat. Prod. 2010;73:1058–1063. doi: 10.1021/np9008199. [DOI] [PubMed] [Google Scholar]
- 17.Ren K.K., Wang H.Z., Xie L.P., Chen D.W., Liu X., Sun J., Nie Y.C., Zhang R.Q. The effects of oridonin on cell growth, cell cycle, cell migration and differentiation in melanoma cells. J. Ethnopharmacol. 2006;103:176–180. doi: 10.1016/j.jep.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 18.Ji Z., Tang Q., Zhang J., Yang Y., Liu Y., Pan Y. Oridonin-induced apoptosis in SW620 human colorectal adenocarcinoma cells. Oncol. Lett. 2011;2:1303–1307. doi: 10.3892/ol.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li D., Hu X., Han T., Xu S., Zhou T., Wang Z., Cheng K., Li Z., Hua H., Xiao W., et al. Synthesis, biological activity, and apoptotic properties of NO-Donor/enmein-type ent-kauranoid hybrids. Int. J. Mol. Sci. 2016;17:747. doi: 10.3390/ijms17060747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z., Ouyang L., Peng H., Zhang W.Z. Oridonin: Targeting programmed cell death pathways as an anti-tumour agent. Cell Prolif. 2012;45:499–507. doi: 10.1111/j.1365-2184.2012.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C.Y., Wang E.Q., Cheng Y., Bao J.K. Oridonin: An active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int. J. Biochem. Cell Biol. 2011;43:701–704. doi: 10.1016/j.biocel.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Mei Y.H., Xu J., Zhao J.H., Feng N.P., Liu Y., Wei L. Method for determination of oridonin in rabbits using isopsoralen as an internal standard and its application to pharmacokinetic studies for oridonin-loaded nanoparticles. J. Chromatogr. B. 2008;869:138–141. doi: 10.1016/j.jchromb.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Du Y.F., Liu P.W., Shi X.W., Jin Y.R., Wang Q., Zhang X.W. A novel analysis method for diterpenoids in rat plasma by liquid chromatography–electrospray ionization mass spectrometry. Anal. Biochem. 2010;407:111–119. doi: 10.1016/j.ab.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 25.Manly C.J., Chandrasekhar J., Ochterski J.W., Hammer J.D., Warfield B.B. Strategies and tactics for optimizing the Hit-to-Lead process and beyond—a computational chemistry perspective. Drug Discov. Today. 2008;13:99–109. doi: 10.1016/j.drudis.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Hann M.M., Oprea T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Xu S., Luo S., Yao H., Cai H., Miao X., Wu F., Yang D.H., Wu X., Xie W., Yao H., et al. Probing the anticancer action of oridonin with fluorescent analogues: Visualizing subcellular localization to mitochondria. J. Med. Chem. 2016;59:5022–5034. doi: 10.1021/acs.jmedchem.6b00408. [DOI] [PubMed] [Google Scholar]
- 28.Xu S., Wang G., Lin Y., Zhang Y., Pei L., Yao H., Hu M., Qiu Y., Huang Z., Zhang Y., et al. Novel anticancer oridonin derivatives possessing a diazen-1-ium-1,2-diolate nitric oxide donor moiety: Design, synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2016;26:2795–2800. doi: 10.1016/j.bmcl.2016.04.068. [DOI] [PubMed] [Google Scholar]
- 29.Li D., Han T., Xu S., Zhou T., Tian K., Hu X., Cheng K., Li Z., Hua H., Xu J. Antitumor and antibacterial derivatives of oridonin: A main composition of Dong-Ling-Cao. Molecules. 2016;21:575. doi: 10.3390/molecules21050575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S.T., Pei L.L., Wang C.Q., Zhang Y.K., Li D.H., Yao H.Q., Wu X.M., Chen Z.S., Sun Y.J., Xu J.Y. Novel hybrids of natural oridonin-bearing nitrogen mustards as potential anticancer drug candidates. ACS Med. Chem. Lett. 2014;5:797–802. doi: 10.1021/ml500141f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S., Pei L., Li D., Yao H., Cai H., Yao H., Wu X., Xu J. Synthesis and antimycobacterial evaluation of natural oridonin and its enmein-type derivatives. Fitoterapia. 2014;99:300–306. doi: 10.1016/j.fitote.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Xu S., Li D., Pei L., Yao H., Wang C., Cai H., Yao H., Wu X., Xu J. Design, synthesis and antimycobacterial activity evaluation of natural oridonin derivatives. Bioorg. Med. Chem. Lett. 2014;24:2811–2814. doi: 10.1016/j.bmcl.2014.04.119. [DOI] [PubMed] [Google Scholar]
- 33.Li D., Xu S., Cai H., Pei L., Zhang H., Wang L., Yao H., Wu X., Jiang J., Sun Y., et al. Enmein-type diterpenoid analogs from natural kaurene-type oridonin: Synthesis and their antitumor biological evaluation. Eur. J. Med. Chem. 2013;64:215–221. doi: 10.1016/j.ejmech.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Li D., Cai H., Jiang B., Liu G., Wang Y., Wang L., Yao H., Wu X., Sun Y., Xu J. Synthesis of spirolactone-type diterpenoid derivatives from kaurene-type oridonin with improved antiproliferative effects and their apoptosis-inducing activity in human hepatoma Bel-7402 cells. Eur. J. Med. Chem. 2013;59:322–328. doi: 10.1016/j.ejmech.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Li D.H., Wang L., Cai H., Jiang B.W., Zhang Y.H., Sun Y.J., Xu J.Y. Synthesis of novel furozan-based nitric oxide-releasing derivatives of 1-oxo-oridonin with anti-proliferative activity. Chin. J. Nat. Med. 2012;10:471–476. doi: 10.1016/S1875-5364(12)60089-2. [DOI] [Google Scholar]
- 36.Li D., Xu S., Cai H., Pei L., Wang L., Wu X., Yao H., Jiang J., Sun Y., Xu J. Library construction and biological evaluation of enmein-type diterpenoid analogues as potential anticancer agents. ChemMedChem. 2013;8:812–818. doi: 10.1002/cmdc.201200559. [DOI] [PubMed] [Google Scholar]
- 37.Wang L., Li D., Xu S., Cai H., Yao H., Zhang Y., Jiang J., Xu J. The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: Synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 2012;52:242–250. doi: 10.1016/j.ejmech.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 38.Li D., Wang L., Cai H., Zhang Y., Xu J. Synthesis and biological evaluation of novel furozan-based nitric oxide-releasing derivatives of oridonin as potential anti-tumor agents. Molecules. 2012;17:7556–7568. doi: 10.3390/molecules17067556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L., Li D.H., Wang C.L., Zhang Y.H., Xu J.Y. Recent progress in the development of natural ent-kaurane diterpenoids with anti-tumor activity. Mini Rev. Med. Chem. 2011;11:910–919. doi: 10.2174/138955711796575416. [DOI] [PubMed] [Google Scholar]
- 40.Wang L., Ran Q., Li D.H., Yao H.Q., Zhang Y.H., Yuan S.T., Zhang L.Y., Shen M.Q., Xu J.Y. Synthesis and anti-tumor activity of 14-O-derivatives of natural oridonin. Chin. J. Nat. Med. 2011;9:194–198. [Google Scholar]
- 41.Xu J.Y., Yang J.Y., Ran Q., Wang L., Liu J., Wang Z.X., Wu X.M., Hua W.Y., Yuan S.T., Zhang L.Y., et al. Synthesis and biological evaluation of novel 1-O- and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008;18:4741–4744. doi: 10.1016/j.bmcl.2008.06.097. [DOI] [PubMed] [Google Scholar]
- 42.Owona B.A., Schluesener H.J. Molecular insight in the multifunctional effects of oridonin. Drugs R D. 2015;15:233–244. doi: 10.1007/s40268-015-0102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Z., Chen Y. Oridonin, a promising antitumor natural product in the chemotherapy of hematological malignancies. Curr. Pharm. Biotechnol. 2014;15:1083–1092. doi: 10.2174/1389201015666141111115608. [DOI] [PubMed] [Google Scholar]
- 44.Tian W., Chen S.Y. Recent advances in the molecular basis of anti-neoplastic mechanisms of oridonin. Chin. J. Integr. Med. 2013;19:315–320. doi: 10.1007/s11655-013-1437-3. [DOI] [PubMed] [Google Scholar]
- 45.Ding Y., Ding C., Ye N., Liu Z., Wold E.A., Chen H., Wild C., Shen Q., Zhou J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016;122:102–117. doi: 10.1016/j.ejmech.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujita E., Nagao Y., Kaneko K., Nakazawa S., Kuroda H. The antitumor and antibacterial activity of isodon diterpenoid. Chem. Pharm. Bull. 1976;24:2118–2127. doi: 10.1248/cpb.24.2118. [DOI] [PubMed] [Google Scholar]
- 47.Fujita E., Nagao Y., Kohno T., Matsuda M., Ozaki M. Antitumor activity of acylated oridonin. Chem. Pharm. Bull. 1981;29:3208–3213. doi: 10.1248/cpb.29.3208. [DOI] [PubMed] [Google Scholar]
- 48.Nagao Y., Fujita E., Kohno T., Yagi M. An efficient method for selective acetylation of alcoholic hydroxyl groups. Chem. Pharm. Bull. 1981;29:3202–3207. doi: 10.1248/cpb.29.3202. [DOI] [Google Scholar]
- 49.Zhou W.S., Cheng Y.X. The chemical selective synthesis of eriocalyxin B and its analogues. Acta Chim. Sin. 1990;48:1185–1190. [Google Scholar]
- 50.Yan X.B., Lei M., Zhang J.Y., Liu H.M. Synthesis of oridonin glucopyranoside. Chin. J. Org. Chem. 2005;25:222–224. [Google Scholar]
- 51.Yan X.B., Li W., Liu H.M. Study on the synthesis and activity of lasiokaurin (in Chinese) J. Zhengzhou Univ. Eng. Sci. 2006;27:113–115. [Google Scholar]
- 52.Yan X.B., Zhang J.Y., Ke Y., Zhou X., Liu H.M. Synthesis of amino-derivatives of oridonin and their antitumor activity (in Chinese) J. Zhengzhou Univ. Med. Sci. 2007;42:39–41. [Google Scholar]
- 53.Ding C., Zhang Y., Chen H., Wild C., Wang T., White M.A., Shen Q., Zhou J. Overcoming synthetic challenges of oridonin A-ring structural diversification: Regio- and stereoselective installation of azides and 1,2,3-triazoles at the C-1, C-2, or C-3 position. Org. Lett. 2013;15:3718–3721. doi: 10.1021/ol4015865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding C., Zhang Y., Chen H., Yang Z., Wild C., Chu L., Liu H., Shen Q., Zhou J. Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: Protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem. 2013;56:5048–5058. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J., Ding Y., Chen C.H., Zhou Z., Ding C., Chen H., Zhou J., Chen C. A new oridonin analog suppresses triple-negative breast cancer cells and tumor growth via the induction of death receptor 5. Cancer Lett. 2016;380:393–402. doi: 10.1016/j.canlet.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 56.Ding C., Zhang Y., Chen H., Yang Z., Wild C., Ye N., Ester C.D., Xiong A., White M.A., Shen Q., et al. Oridonin ring A-based diverse constructions of enone functionality: Identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J. Med. Chem. 2013;56:8814–8825. doi: 10.1021/jm401248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luan J., Zhang Y., Yang S., Wang X., Yu B., Yang H. Oridonin: A small molecule inhibitor of cystic fibrosis transmembrane conductance regulator (CFTR) isolated from traditional Chinese medicine. Fitoterapia. 2015;100:88–94. doi: 10.1016/j.fitote.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 58.Bohanon F.J., Wang X., Ding C., Ding Y., Radhakrishnan G.L., Rastellini C., Zhou J., Radhakrishnan R.S. Oridonin inhibits hepatic stellate cell proliferation and fibrogenesis. J. Surg. Res. 2014;190:55–63. doi: 10.1016/j.jss.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuo L.M., Kuo C.Y., Lin C.Y., Hung M.F., Shen J.J., Hwang T.L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules. 2014;19:3327–3344. doi: 10.3390/molecules19033327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bohanon F.J., Wang X., Graham B.M., Ding C., Ding Y., Radhakrishnan G.L., Rastellini C., Zhou J., Radhakrishnan R.S. Enhanced effects of novel oridonin analog CYD0682 for hepatic fibrosis. J. Surg. Res. 2015;199:441–449. doi: 10.1016/j.jss.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bohanon F.J., Wang X., Graham B.M., Prasai A., Vasudevan S.J., Ding C., Ding Y., Radhakrishnan G.L., Rastellini C., Zhou J., et al. Enhanced anti-fibrogenic effects of novel oridonin derivative CYD0692 in hepatic stellate cells. Mol. Cell. Biochem. 2015;410:293–300. doi: 10.1007/s11010-015-2562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding C., Wang L., Chen H., Wild C., Ye N., Ding Y., Wang T., White M.A., Shen Q., Zhou J. ent-Kaurane-based regio- and stereoselective inverse electron demand hetero-Diels-Alder reactions: Synthesis of dihydropyran-fused diterpenoids. Org. Biomol. Chem. 2014;12:8442–8452. doi: 10.1039/C4OB01040J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun H.D., Huang S.X., Han Q.B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006;23:673–698. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- 64.Yang J., Wang W.G., Wu H.Y., Du X., Li X.N., Li Y., Pu J.X., Sun H.D. Bioactive enmein-type ent-kaurane diterpenoids from Isodon phyllostachys. J. Nat. Prod. 2016;79:132–140. doi: 10.1021/acs.jnatprod.5b00802. [DOI] [PubMed] [Google Scholar]
- 65.Wang W.G., Du X., Li X.N., Wu H.Y., Liu X., Shang S.Z., Zhan R., Liang C.Q., Kong L.M., Li Y., et al. New bicyclo[3.1.0]hexane unit ent-kaurane diterpene and its seco-derivative from Isodon eriocalyx var. laxiflora. Org. Lett. 2012;14:302–305. doi: 10.1021/ol203061z. [DOI] [PubMed] [Google Scholar]
- 66.Zou J., Pan L., Li Q., Zhao J., Pu J., Yao P., Gong N., Lu Y., Kondratyuk T.P., Pezzuto J.M., et al. Rubesanolides A and B: Diterpenoids from Isodon rubescens. Org. Lett. 2011;13:1406–1409. doi: 10.1021/ol200086k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brucoli F., Borrello M.T., Stapleton P., Parkinson G.N., Gibbons S. Structural characterization and antimicrobial evaluation of atractyloside, atractyligenin, and 15-didehydroatractyligenin methyl ester. J. Nat. Prod. 2012;75:1070–1075. doi: 10.1021/np300080w. [DOI] [PubMed] [Google Scholar]
- 68.Wang W.G., Li X.N., Du X., Wu H.Y., Liu X., Su J., Li Y., Pu J.X., Sun H.D. Laxiflorolides A and B, epimeric bishomoditerpene lactones from Isodon eriocalyx. J. Nat. Prod. 2012;75:1102–1107. doi: 10.1021/np300106j. [DOI] [PubMed] [Google Scholar]
- 69.Wijeratne E.M., Bashyal B.P., Liu M.X., Rocha D.D., Gunaherath G.M., U’Ren J.M., Gunatilaka M.K., Arnold A.E., Whitesell L., Gunatilaka A.A. Geopyxins A-E, ent-kaurane diterpenoids from endolichenic fungal strains Geopyxis aff. majalis and Geopyxis sp. AZ0066: Structure-activity relationships of geopyxins and their analogues. J. Nat. Prod. 2012;75:361–369. doi: 10.1021/np200769q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gong J.X., Lin G.A., Sun W.B., Li C.C., Yang Z. Total synthesis of (±) maoecrystal, V. J. Am. Chem. Soc. 2010;132:16745–16746. doi: 10.1021/ja108907x. [DOI] [PubMed] [Google Scholar]
- 71.Cha J.Y., Yeoman J.T.S., Reisman S.E. A concise total synthesis of (−)-maoecrystal, Z. J. Am. Chem. Soc. 2011;133:14964–14967. doi: 10.1021/ja2073356. [DOI] [PubMed] [Google Scholar]
- 72.Pan Z., Zheng C., Wang H., Chen Y., Li Y., Cheng B., Zhai H. Total synthesis of (±)-sculponeatin, N. Org. Lett. 2014;16:216–219. doi: 10.1021/ol403208g. [DOI] [PubMed] [Google Scholar]
- 73.Zhang M., Zhang Y., Lu W., Nan F.J. Synthesis and revision of stereochemistry of rubescensin, S. Org. Biomol. Chem. 2011;9:4436–4439. doi: 10.1039/c1ob05611e. [DOI] [PubMed] [Google Scholar]
- 74.Li S.H., Wang J., Niu X.M., Shen Y.H., Zhang H.J., Sun H.D., Li M.L., Tian Q.E., Lu Y., Cao P., et al. Maoecrystal V, cytotoxic diterpenoid with a novel C19 skeleton from Isodon eriocalyx (Dunn.) hara. Org. Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- 75.Jiang H.Y., Wang W.G., Zhou M., Wu H.Y., Zhan R., Li X.N., Du X., Li Y., Pu J.X., Sun H.D. Diterpenoids from Isodon sculponeatus. Fitoterapia. 2014;93:142–149. doi: 10.1016/j.fitote.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 76.Han Q.B., Cheung S., Tai J., Qiao C.F., Song J.Z., Tso T.F., Sun H.D., Xu H.X. Maoecrystal Z, a cytotoxic diterpene from Isodon eriocalyx with a unique skeleton. Org. Lett. 2006;8:4727–4730. doi: 10.1021/ol061757j. [DOI] [PubMed] [Google Scholar]
- 77.Li L.M., Li G.Y., Pu J.X., Xiao W.L., Ding L.S., Sun H.D. ent-Kaurane and cembrane diterpenoids from Isodon sculponeatus and their cytotoxicity. J. Nat. Prod. 2009;72:1851–1856. doi: 10.1021/np900406c. [DOI] [PubMed] [Google Scholar]
- 78.Jiang H.Y., Wang W.G., Zhou M., Wu H.Y., Zhan R., Li X.N., Du X., Li Y., Pu J.X., Sun H.D. Enmein-type 6,7-seco-ent-kauranoids from Isodon sculponeatus. J. Nat. Prod. 2009;72:1851–1856. doi: 10.1021/np400669t. [DOI] [PubMed] [Google Scholar]
- 79.He F., Xiao W.L., Pu J.X., Wu Y.L., Zhang H.B., Li X.N., Zhao Y., Yang L.B., Chen G.Q., Sun H.D. Cytotoxic ent-kaurane diterpenoids from Isodon sinuolata. Phytochemistry. 2009;70:1462–1466. doi: 10.1016/j.phytochem.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 80.Thuong P.T., Pham T.H., Le T.V., Dao T.T., Dang T.T., Nguyen Q.T., Oh W.K. Symmetric dimers of ent-kaurane diterpenoids with cytotoxic activity from Croton tonkinensis. Bioorg. Med. Chem. Lett. 2012;22:1122–1124. doi: 10.1016/j.bmcl.2011.11.116. [DOI] [PubMed] [Google Scholar]
- 81.Bai S.P., Wei Q.Y., Jin X.L., Wu Q.X., Yang L. Two novel ent-kauranoid diterpenoids from Isodon japonica leaves. Planta Med. 2005;71:764–769. doi: 10.1055/s-2005-871246. [DOI] [PubMed] [Google Scholar]
- 82.Braca A., Abdel-Razik A.F., Mendez J., Morelli I. A new kaurane diterpene dimer from Parinari campestris. Fitoterapia. 2005;76:614–619. doi: 10.1016/j.fitote.2005.05.005. [DOI] [PubMed] [Google Scholar]