

ABSTRACT
With the rapid growth of biopharmaceutical product development, knowledge of therapeutic protein stability has become increasingly important. We evaluated assays that measure solution-mediated interactions and key molecular characteristics of 9 formulated monoclonal antibody (mAb) therapeutics, to predict their stability behavior. Colloidal interactions, self-association propensity and conformational stability were measured using effective surface charge via zeta potential, diffusion interaction parameter (kD) and differential scanning calorimetry (DSC), respectively. The molecular features of all 9 mAbs were compared to their stability at accelerated (25°C and 40°C) and long-term storage conditions (2–8°C) as measured by size exclusion chromatography. At accelerated storage conditions, the majority of the mAbs in this study degraded via fragmentation rather than aggregation. Our results show that colloidal stability, self-association propensity and conformational characteristics (exposed tryptophan) provide reasonable prediction of accelerated stability, with limited predictive value at 2–8°C stability. While no correlations to stability behavior were observed with onset-of-melting temperatures or domain unfolding temperatures, by DSC, melting of the Fab domain with the CH2 domain suggests lower stability at stressed conditions. The relevance of identifying appropriate biophysical assays based on the primary degradation pathways is discussed.
KEYWORDS: Aggregation, colloidal stability, diffusion interaction parameter, fragmentation, Immunoglobulin, monoclonal antibody, thermal stability, zeta potential
Abbreviations
- IgG
Immunoglobulin G
- mAb
Monoclonal antibody
- HP-SEC
High-performance Liquid Size Exclusion Chromatography
- LMW
Low Molecular Weight
- HMW
High Molecular Weight
- DSC
Differential Scanning Calorimetry
- CH
Constant heavy
- IEX
Ion Exchange Chromatography
- DLS
Dynamic Light Scattering
Introduction
Monoclonal antibodies (mAbs) are highly complex macromolecules and their stability in biotherapeutic applications is governed by multiple factors including chemical, structural, and colloidal degradation pathways. The primary goal of drug product and process development is to identify and limit these degradation pathways to enable safe and efficacious usage with commercially viable shelf life. More than 30 antibody-based therapeutics are marketed worldwide, and many more are in development.1-3 The competition in mAb development has resulted in pressure to reduce development time. Therefore, there is strong need for analytical tools that can reliably predict and differentiate between suitable candidates (in discovery) and unstable and stable formulations (in development).4-7
Product liabilities are commonly identified during development via biochemical and biophysical characterization techniques that identify molecular properties like aggregation propensity and unfolding temperature, respectively. Knowledge of these properties helps to define study parameters that expose the mAb to short-term, stressed conditions including elevated temperatures, extremes in pH, and processing conditions (e.g, high shear or agitation).6,8 Alternatively, in early development, developability and molecular profiling assessments using higher throughput technologies may also be used to identify potential liabilities and possible technical challenges associated with the protein. The output of these screening studies leads to the selection of optimum pH, buffer, and stabilizing excipients.
Given the importance of selecting an optimal drug product formulation, multiple formulations may be staged on stability following ICH-recommended conditions (2–8°C and ambient relative humidity, “25H” – 25°C and 60% relative humidity, “RH4” – 40°C and 75% relative humidity) after screening activities are completed. Elevated temperature is often used as a metric to rank-order the multiple formulations, and aid in the selection of the lead/final product formulation.9,10 However, there is very little data showing a direct correlation between the assays used during screening and stability and their ability to predict stability at the intended storage condition (2–8°C) or stability at elevated temperature conditions.
In this study, we evaluated whether commonly characterized biophysical properties provide insight into the mAb stability behavior. Nine mAbs in their lead formulation (i.e., optimally formulated) were characterized for properties such as conformational hydrophobicity, aggregation propensity, thermal stability, and measured net surface charge/ zeta potential (Table 1). Here, conformational hydrophobicity refers to the structural placement of the hydrophobic residues in the native folded mAb, aggregation propensity is defined as the tendency to form higher-order structures as detected by size-exclusion chromatography (SEC), and thermal stability reflects the temperature-induced domain unfolding monitored by differential scanning calorimetry (DSC). These characteristics were then correlated to SEC data from accelerated stability (25H and RH4) and long term stability (2–8°C).
Figure 1.
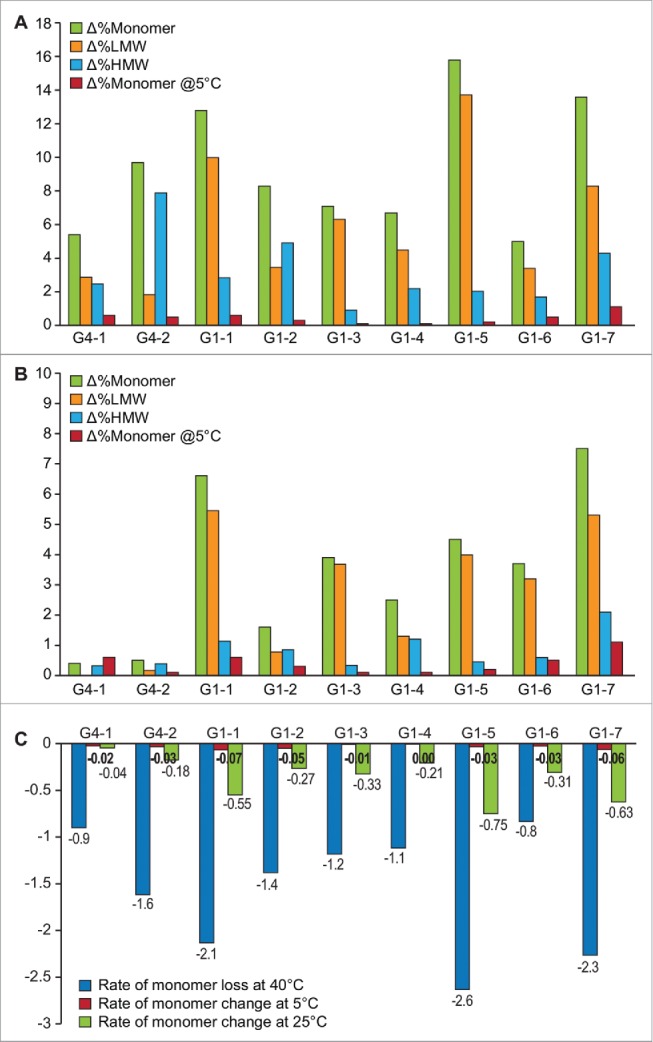
Change in % Monomer, %HMW, %LMW by SEC after 6 months of storage at 40°C along with the corresponding loss in monomer at long-term 5°C storage (A) and after storage at 25°C (B). Green bars = Δ%Monomer, orange bars = %ΔLMW, blue bars = %ΔHMW, red bars = Δ%Monomer at 5°C (C) Rate of monomer loss at 40°C, 25°C and 5°C storage conditions.
We observed differential behavior among the 9 mAbs at 40°C, and the predictive power of biophysical attributes was evaluated at this condition. Note that the comparison of stability for the optimally formulated mAbs to their molecular properties results in a data set that focuses on the predictive potential of the biophysical tools, and not how these assays were used during formulation screening activities.
Our results show that zeta potential, kD, and to an extent, exposed Trp residues may be reflective of behavior under accelerated conditions. Interestingly, thermal stability measured by DSC on the formulated mAbs did not prove useful, as minimal correlations were observed between T-onset or Tmvalues with loss of monomer. Some noteworthy points in the current discussion include relevance of: 1) SEC alone as a quality assessment tool; 2) onset-of-melting temperatures, domain-unfolding temperatures and associated apparent enthalpies by DSC; 3) inclusion of accelerated stress condition on stability and its comparison to long-term storage temperatures; and 4) preliminary critical quality attribute (pCQA) analysis to identify primary degradation mechanism.
Results
SEC
Formation of high molecular weight (HMW) species (i.e., aggregation) or low molecular weight (LMW) species (i.e., fragmentation) on stability was evaluated through SEC. The SEC results showing change in monomer content, % LMW and % HMW of all mAbs over stability when stored at 40°C and 25°C in relation to 2–8°C, are shown in Fig. 1.
When stored at 40°C for 6 months, G1–5 showed the greatest loss in monomer content (∼16%); followed by G1–7 (∼14%), and G1–1 (∼13%). Interestingly, all the 3 molecules showed higher tendency to fragment (%LMW) than aggregate. The molecules with the least amount of monomer loss were G1–6 and G4–1 (∼5%) with all others mAbs having monomer loss between ∼5–16% (Fig. 1A). In this study, most mAbs showed increased propensity to fragment (G1–1, G1–3, G1–4, G1–5, G1–6, G1–7), and we believe the fragmentation follows the classic behavior observed for mAbs involving cleavage at the hinge region.11 G4–2 and G1–2 were the only cases when HMW species formation was greater than LMW species formation. (Fig. 1A).
At 25°C storage condition, generally the least stable mAbs at 40°C were the least stable at this condition, though a different order was observed. At this condition, G1–7 showed the highest loss in monomer content (∼7.5%); followed by G1–1 (∼7%) and G1–5 (∼4.5%). To account for variations in the duration of available stability data between molecules, ( e.g., G1–5, G1–2 had only 6 months storage data while G1–1 and G1–7 had 12 months of data), the rate of monomer loss was calculated to account for storage time and expressed as rate per month, to enable a meaningful comparison (Fig. 1C).
In the current data set, except for the rapidly degrading molecules (e.g., G1–1, G1–5 and G1–7), the qualitative trends of degradation were different at 40°C and 25°C for both IgG1 and IgG4 isotypes, with this lack of correlation to storage temperature being more pronounced in the IgG4 subtype (Table 2, Fig. 1). Interestingly, despite the rapid loss of monomer seen at accelerated and stressed conditions, degradation was minimal when stored at 2–8°C for all mAbs studied.
Table 2.
Results of SEC, thermal unfolding properties by DSC, intrinsic tryptophan fluorescence and net surface charge (zeta potential) and diffusion interaction parameter (kD) of all test proteins.
| DSC |
SEC$–40C |
SEC$-5C | Trp FI | DLS |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CODE | ΔHapp (1st) Kcal/mol | ΔHapp (2nd) Kcal/mol | ΔHapp (total) Kcal/mol | Tonset (°C) | Tm1 (°C) | Tm2 (°C) | Δ %Monomer | Δ %LMW | Δ %HMW | Δ %Monomer | λmax (nm) | kD (L/g) | ZP (mV) |
| G4–1 | 246.7 | 797.6 | 1044.3 | 55.0 | 61.0 | 73.0 | 5.4 | 2.9 | 2.5 | 0.6 | 333 | 6.8 | 10.0 |
| G4–2 | 181.8 | 755.0 | 936.8 | 56.0 | 61.3 | 72.9 | 9.7 | 1.8 | 7.9 | 0.4 | 346 | 4.9 | 2.9 |
| G1–1 | 853.1 | 146.4 | 999.6 | 66.0 | 74.6 | 85.3 | 12.8 | 10.0 | 2.8 | 0.6 | 346 | 15.9 | 4.0 |
| G1–2 | 834.1 | 106.9 | 941.0 | 62.1 | 72.0 | 82.5 | 8.3 | 3.5 | 4.9 | 0.3 | 339 | −8.6 | 2.7 |
| G1–3 | 189.2 | 870.7 | 1059.8 | 62.0 | 71.1 | 83.6 | 7.1 | 6.3 | 0.9 | 0.1 | 335 | ND | 5.8 |
| G1–4 | 263.0 | 907.8 | 1170.7 | 60.3 | 69.6 | 83.0 | 6.7 | 4.5 | 2.2 | 0.1 | 336 | 17.2 | 7.8 |
| G1–5 | 271.9 | 1487.0 | 1758.9 | 60.2 | 67.6 | 80.9 | 15.8 | 13.7 | 2.0 | 0.2 | 335 | 24.0 | 6.1 |
| G1–6* | 611.3 | 347.6 | 1010.3 | 58.4 | 68.1 | 75.2 | 5.0 | 3.4 | 1.7 | 0.5 | 353 | ND | 2.0a |
| G1–7 | 1052.2 | 107.4 | 1159.6 | 58.4 | 71.6 | 81.5 | 13.6 | 8.3 | 4.3 | 1.1 | 340 | −3.9 | 3.3a |
ZP = Zeta Potential
third transition observed at 83.2°C with ΔHapp of 51.5 Kcal/mol
Δ=change in content between final and initial timepoints
diluted in 10 mM histidine buffer
DSC
The onset-of-melting temperatures (Tonset), thermal unfolding temperatures (Tm) of the domains, as well as apparent enthalpies associated with each unfolding transition, are shown in Table 2. All the mAbs tested in this study had an onset-of-melting temperature of >50°C. As expected, the IgG4 framework mAbs had an earlier onset of melting compared to the IgG1 molecules,12 even at the same pH conditions.
Assignments of CH2, CH3 and Fab domains in this study are based on reported literature and not specifically confirmed with all the test proteins. Typically, 2 distinct melting profiles are observed for mAbs, historically identified as either the unfolding of the Fab+CH2 domain or Fab+CH3 domain;13 the Fab transition is associated with a higher apparent enthalpy, given its larger size and therefore higher energy input required to cause unfolding.
We observed both instances where the Fab domain unfolded with the CH2 domain or with the CH3 domain. Among the 9 mAbs studied, G4–1, G4–2, G1–3, G1–4 and G1–5 displayed only CH2 unfolding first, followed by the Fab/CH3 (Fig. 2A). In contrast, G1–1, G1–2, G1–6 and G1–7 displayed the higher-energy Fab/CH2 unfolding prior to the CH3 domain (Fig. 2B). Interestingly, the former category of Fab/CH3 unfolding mAbs generally had lower monomer loss at 40°C, with G1–5 being an exception as it showed the highest monomer loss of 15.8%. The latter category of Fab/CH2 unfolding mAbs were all associated with higher monomer loss at 40°C; G1–6 was an exception in this set with only ∼5% monomer loss at 40°C (Table 2).
Figure 2.

(A) DSC thermograms showing the unfolding of individual domains with the CH2 unfolding preceding Fab/CH3 domain and (B) vice versa with Fab CH2 unfolding. (C) Scatter plot showing lack of correlation between T-onset or Tm1 or Tm2 and monomer loss at 40°C.
Table 2 shows the domain-associated apparent enthalpy values; all the proteins tested had an apparent enthalpy of ∼1000 kCal/ mol, which is typical for mAbs.13 We also observed that some mAbs have much higher ΔHapp; G1–5 had the highest ΔHapp of >1700 kCal/mol, with the Fab/CH3 contribution being almost 1500 kCal/mol. G1–4 and G1–7 were also high, with the ΔHapp being 1170 and 1159 kCal/mol, respectively. However, there is poor correlation of the total ΔHapp with stability at the 40°C condition; some of the test proteins with highest ΔHapp also showed the highest rate of monomer loss (e.g., G1–5, G1–7). Similar formulation matrix (G4–1, G4–2 and G1–5) with comparable domain unfolding profiles, displayed variable aggregation tendencies. G1–5 fragmented, G4–2 aggregated and G4–1 was relatively stable with low HMW or LMW. Another case in point is G1–6 and G1–7, in identical formulation, have slightly variable unfolding pattern where the former shows less cooperative unfolding with 3 transitions.
Plots of T-onset temperatures and unfolding temperatures (Tm1 and Tm2) for all test proteins in relation to the loss of monomer at 40°C storage condition are shown in Fig. 2C. We did not observe a correlation between onset (T-onset) and unfolding temperatures (Tm1 and Tm2) with loss in monomer content. For example, G1–1, G1–5 and G1–7 all had relatively high T-onset, Tm1, and Tm2 values, but also showed higher degradation rates at 40°C. In contrast, G4–1, which has a lower T-onset, Tm1 and Tm2 value, had a lower rate of monomer loss.
Intrinsic tryptophan fluorescence
Intrinsic fluorescence was used to compare the local environment around tryptophan (Trp) residues between all 9 mAbs. Due to the inherent principles of the technique, fluorescent intensity, quantum yield, and wavelength of maximum fluorescence emission of Trp are influenced by several factors, such as solvent conditions, proximity of acidic amino acids, number and location of trp residues. Trp residues that are buried in the hydrophobic core of proteins can have spectra that are blue shifted by 10 to 20 nm compared to exposed Trps on the surface of the protein.14
Fig. 3A shows the fluorescence emission spectra for all mAbs measured at the initial time point. As expected, the fluorescence emission spectra are mAb and formulation dependent, with differences observed in intensity and peak maxima. As can be seen from Table 2, the emission maxima (λmax) ranged between 333 nm in G4–1 and G1–3, representing a well-buried Trp environment, to 353 nm in G1–6, representing a highly solvent-exposed Trp environment. Intrinsic Trp fluorescence showed some correlation between λmax of Trp emission to loss of monomer, where the extent of red shift in the Trp fluorescence was suggestive of higher propensity to degrade. For example, G4–1 and G1–3, which contain buried Trp (λmax ∼ 330 nm), had lower loss of monomer under accelerated conditions while G4–2 and G1–1, which contain exposed Trp (λmax ∼ 346 nm) had higher rates of monomer loss.
Figure 3.
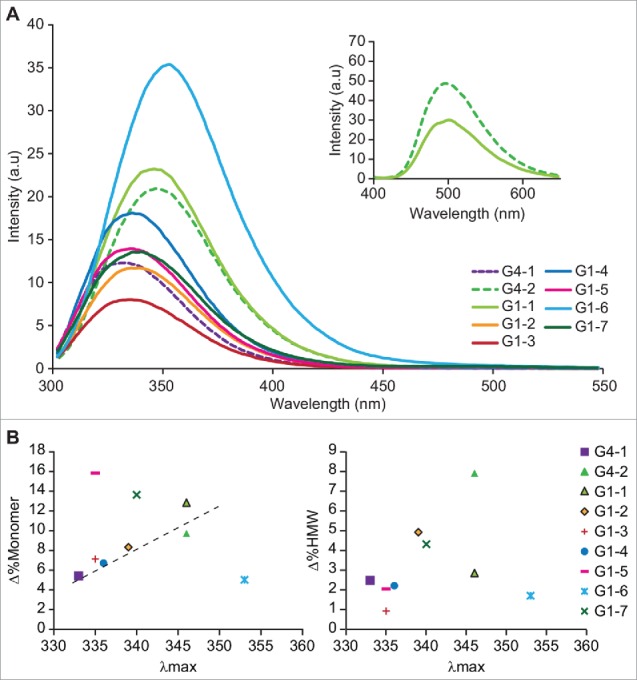
(A) Tryptophan emission spectra of all 9 mAbs, with excitation wavelength set to 295 nm. Inset shows the bis-ANS emission spectra of G4–2 (dashed line) and G1–1 (solid line); triplicate measurements of Bis-ANS fluorescence emission were recorded from 400–650 nm with excitation at 385 nm, final concentration of bis-ANS being 5 µM. All other parameters were same as used for intrinsic fluorescence. (B) Correlation of tryptophan λmax of emission with loss of monomer and gain in %HMW at 40°C storage condition
Zeta potential and kD measurements
Inherent molecular characteristics such as net surface charge and self-association propensity were also studied to evaluate their contributions to mAb stability. Two orthogonal approaches for calculating colloidal stability of various mAbs were employed, diffusion interaction parameter (kD) using dynamic light scattering and effective surface charge potential (zeta potential) using electrophoretic light scattering.
Based on published literature and application notes from Malvern Biosciences, zeta potential values greater than 5 mV were considered to be colloidally stable.15,16 In this study, G4–1, G1–3, G1–4 and G1–5 all had high zeta potential values suggestive of higher colloidal stability, whereas G4–2, G1–1, G1–2, G1–6 and G1–7 all showed zeta potential values of less than 5 mV (Fig. 4A, Table 2). Values reported for G1–6 and G1–7 are from measurements made in 10 mM histidine buffer, due to the high ionic strength of the native formulation buffers. When correlated with the loss of monomer at the 40°C storage condition, most of the mAbs that had a lower net effective surface charge (zeta potential) also had increased loss of monomer. G1–6 formulation contained high amounts of salt due to which the zeta potential measurements were made in 10 mM histidine buffer. This value thus may not reflect the net surface charge in the formulation.
Figure 4.
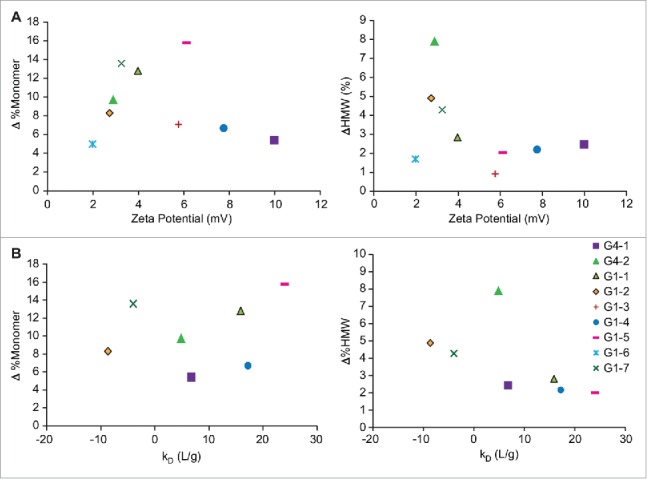
(A) Effect of zeta potential on change in monomer at 40°C, as measured by SEC (B) Effect of diffusion interaction parameter (kD) on change in monomer at 40°C, as measured by SEC.
Comparison of kD values of 7 test proteins (G1–3 and G1–6 were not tested due to material limitations) showed G1–2 and G1–7 to have negative kD suggestive of increased self-association propensity. At 40°C storage, after 6 months, the monomer content decreased by ∼14% for G1–7 and about 8% for G1–2, indicating good correlation with behavior at stressed conditions. G4–1, G4–2, G1–1, G1–4 and G1–5 had positive kDvalues of varying magnitudes, indicative of repulsive interactions, and there appears to be reasonable agreement with these results and the behavior at 40°C, when the mechanism of fragmentation or aggregation is also considered (Table 2, Fig. 4B).
Discussion
In recent years, there have been increasing reports on the sensitivity and selection of appropriate analytical and biophysical tools to assess stability of mAb product formulations.17-21 Lin et al recently demonstrated that limited information is provided by far-UV circular dichroism and Fourier transform infrared spectroscopy compared with SEC and IEX read outs in comparability studies with degraded mAb samples.22 In an effort to determine the ability of other biophysical assays to predict product stability at different conditions, we retrospectively compared the initial molecular properties of 9 mAbs in their lead (i.e., optimized) formulations with their stability behavior. Both refrigerated and elevated temperature storage conditions were studied with assays routinely used to characterize conformation and solution specific properties, such as hydrophobicity, unfolding enthalpies, kD and zeta potential. In this study, product stability was related to total monomer content as measured by SEC. Aggregates and fragments were not further classified as soluble or insoluble. All mAbs tested consisted of high monomer content (≥∼98%) at the initial characterization time point. The outcome and observed correlations of this study can inform the use of specific biophysical attributes to evaluate during early candidate assessment and formulation screening activities.
With the exception of intrinsic Trp fluorescence, analyses of the stability data for all 9 mAbs indicate limited to no correlation and predictive potential of the various biophysical assays with monomer content at elevated temperature conditions. The limited predictive potential of the biophysical assays is likely due to the lack of specificity associated with monomer loss. As measured by SEC, monomer loss may be due to either fragmentation (i.e., LMW species) or aggregation (i.e., HMW species). When evaluating the subset of mAbs that degrade substantially through aggregation (i.e., G4–2, G1–2, G1–7), some correlations are observed. Biophysical assays used to measure solution mediated properties like kD (self-association) and zeta potential (colloidal stability) show a correlation with propensity to aggregate at 40°C. Models using colloidal systems suggest charge-charge interactions play a crucial role in particle flocculation and aggregation.23 Saluja et al showed a positive correlation between the second virial coefficient (B2) and kD values in predicting solution conditions that promote heat- and agitation-induced aggregation for an IgG2 mAb.24 We observe this dependency for mAbs with low net effective surface charge (i.e., less colloidally stable), which tend to aggregate at 40°C (Fig. 4A). An increased potential for aggregation is also reflected in the kD values for G4–2, G1–2, and G1–7. For these mAbs, the neutral/negative kD values indicate less repulsive interactions or an increased likelihood for self-association, compared to the other mAbs studied (Fig. 4B). In the case of G4–2, the kD value was positive, but less than that of G1–2 and G1–7. The lower kD and low zeta potential (2.9 mV) suggest high aggregation tendency.
We observed that the Trp environment was able to inform about overall loss of monomer. In contrast, when the environment of Trp was studied in the context of aggregation tendency, a poor correlation was observed (Fig. 3B). This is not unexpected, given that conformational hydrophobicity is not the only interaction leading to mAb instability, especially when fragmentation is involved. The use of additional assays, such as extrinsic fluorescence, can provide valuable insight into the mechanism of aggregation. The two mAbs that lacked a correlation between Trp localization and stability behavior were G1–5 (well-buried Trp environment but higher fragmentation) and G1–6 (exposed Trp environment and lower degradation rate). Extrinsic 4–4-bis-1-phenylamino-8-naphthalene sulfonate (bis-ANS) fluorescence showed both these molecules to have increased solvent-exposed hydrophobic patches, which is in agreement with their tendency to form subvisible particulates (data not shown), thus suggesting that all forms of aggregates need to be monitored and not just the soluble forms detected by SEC.
Further comparison of extrinsic fluorescence of G4–2 and G1–1, where both mAbs having exposed Trp residues, revealed G4–2 to have a higher extrinsic fluorescence emission than G1–1. This indicates the presence of more surface-exposed hydrophobic residues in the former, which is suggestive of a hydrophobically mediated native state aggregation (Fig 3A inset). In addition, while comparable kD values were obtained for both G4–2 and G1–1, G4–2 displayed lower colloidal stability (ZP <5 mV), which supports an instability manifested as higher aggregation at 40°C (Fig. 3A inset). This data suggests that aggregation mediated by hydrophobic interactions can be assessed using both intrinsic and extrinsic fluorescence.
The influence of pH and ion effects on protein-protein interactions has been described previously with positive correlations to aggregation tendency.24-26 In the current study, biophysical assays used to measure solution mediated properties were useful in predicting stability of G4–2, G1–2, and G1–7 at 40°C and emphasize the importance of maintaining colloidal stability in minimizing aggregation. However, these biophysical tools did not predict the stability for mAbs that degrade primarily through fragmentation. For example, G1–1, G1–3 and G1–5 all degrade primarily through fragmentation, but do not show consistent trends associated with zeta potential or kD. This observation may be an inherent result of the nature of the biophysical assays, which focus on measuring association/aggregation tendencies.
Interestingly, DSC data also did not correlate with mAb stability independent of degradation mechanism. DSC is commonly used as an initial screening tool to measure conformational stability with the assumption that lower unfolding temperatures or lower unfolding enthalpies correspond to reduced mAb stability. From our data, DSC unfolding temperatures and unfolding enthalpies alone were not predictive of stability behavior (i.e., loss of monomer content) at accelerated temperature conditions. As shown in Fig. 2C, no correlations are observed between Tonset, Tm1, or Tm2, and no correlations are observed between ΔHtotal, ΔH1, and ΔH2 with monomer loss (Table 2). However, earlier unfolding of the Fab domain with the CH2 domain led to poor stability, at 40°C. This information could be useful during candidate selection or preformulation stages when the domain melting profile on the DSC thermogram is more relevant than the actual unfolding temperatures.
Our results on the relevance of CH2 domain corroborate previous reports by Latypov et al, who discuss the importance of CH2 domain in controlling Fc stability and aggregation propensity under low pH stress.27 In addition, the stabilizing role of the Fab domain has been proposed where the Fab domain unfolding triggered a pH- and salt-dependent aggregation of an IgG1.28 Recently, Brader et al also recommended increasing the Fab transition temperature as a strategy for improving product stability.29 However, these previous studies addressed instability as ‘aggregation’ of the mAbs, while we studied domain unfolding in relation to overall monomer loss, including fragmentation. Therefore, the nature of the stress condition (e.g., pH vs temperature) and mAb-specific propensities could be causes for these differing observations.30
The degradation mechanism at elevated temperatures for the majority of the 9 mAbs evaluated in this study was through fragmentation, and not aggregation (Fig. 1). While there are reports in the literature related to mAb stability and aggregation propensity (see references 31-34), there is very limited information on the possible consequences of fragmentation on product stability. Several questions have been raised about potential immunogenicity related to overall purity of biologic products, including fragments. 35,36 In one instance, the mechanism of metal-induced mAb fragmentation was studied where Cu(2+)-mediated fragmentation was determined to occur predominantly through a hydrolytic pathway in solution.11 In our study, the observed higher tendency of fragmentation may be a result of selection bias during formulation optimization, wherein aggregation is considered as a more significant quality attribute than fragmentation, primarily due to immunogenicity concerns.37-39 While our results suggest that aggregation-driven degradation may be predictive when using conventional biophysical characterization techniques, prediction of fragmentation was not well correlated to stability data. This observation emphasizes the importance and need to develop or utilize additional analytical tools specific for fragmentation degradation (e.g., intact and reduced LC-MS, CE-SDS).
Finally, it is also important to note that while we have been analyzing the predictive potential of various biophysical assays for mAbs stored at 40°C, there were no stability concerns when these mAbs were stored at 5°C relative to monomer content (>95% after 24 months of storage). This lack of correlation between accelerated stability and long-term low temperature stability may be a function of the degradation mechanism for the mAbs, and was discussed earlier by Brummit et al.40 Although there are several advantages of accelerated storage conditions, such as: 1) screening and elimination of poor candidates at the early stages of development; 2) supporting temperature excursions; and 3) confirming the stability-indicating nature of analytical methods, data presented here revisits the question of whether accelerated temperature conditions need to be routinely performed as part of the stability programs for biological products. The ICH guidelines on stability testing of biotechnological/ biological products state that, although accelerated conditions are useful for understanding degradation profile, identifying stability indicating assays, product integrity upon excursions/ accidental exposure, such stressed and accelerated conditions may not be appropriate for biologics.41
In summary, we characterized 9 mAbs in their lead (i.e., optimized) formulation using standard biophysical assays that measure conformational stability and solution mediated interactions, and we attempted to correlate the biophysical data with long-term and accelerated stability trends. In our evaluation, solution-mediated interaction properties (e.g., zeta potential and kD measurements) provided some prediction of stability at elevated temperatures when aggregation was a primary mechanism for degradation. Our results support the expectation that formulation optimization, through evaluation of buffer, pH, and salt levels, can help overcome aggregation propensity. However, additional biophysical characterization tools are needed to better understand fragmentation propensity. For example, the evaluation of structural stability by chemical denaturation could complement DSC results to gain a better understanding of global stability (i.e., monomer loss) as a function of temperature stress. In addition, fragmentation propensity should also be considered when choosing an optimal formulation.
It is important to note that in spite of variable mechanisms of degradation at 40°C, all mAbs showed good stability (monomer content >95%) for extended periods of time at the recommended storage condition of 2–8°C. During early formulation screening, accelerated conditions are utilized to expedite the degradation and aid in selecting the ‘optimal’ formulation. Regardless, if most mAbs show minimal degradation at 2–8°C (recommended storage condition), a phase-appropriate investment in formulation development with a balance between risk-based ‘fast to clinic’ approaches versus the generation of more relevant long-term stability data could be considered.
Our results highlight the importance of selecting the appropriate analytical tools when attempting to correlate biophysical characteristics with stability. If the primary degradation is through aggregation, common biophysical assays are available. However, if the degradation is through fragmentation, other analytical tools may be needed to inform on stability at elevated temperatures. The lack of correlation observed here may be due to the fact that these mAbs are already optimally formulated with the appropriate pH, buffers, and excipients. Additional studies to evaluate the predictive ability of biophysical assays during formulation screening are currently ongoing.
Material and methodss
This study describes 9 mAbs, all purified and formulated in their lead formulation, which contain surfactants, sugars and in some cases salts (Table 1). The lead formulation was selected after rigorous screening of pH, salts, buffers and excipients. Among the 9 mAbs, 7 were IgG1 (labeled G1–1 to G1–7) and 2 were IgG4 (labeled G4–1 and G4–2). The concentration of the formulated mAbs were as follows: 25 mg/mL (G4–1, G1–6, G1–7), 40 mg/ml (G1–3), 50 mg/mL (G4–2, G1–1, G1–2, G1–5), and 100 mg/mL (G1–4). Most of these formulations contain the same matrix consisting of histidine, sucrose and Polysorbate-80, with pH between 5.5 and 6.0.
Table 1.
Description of test proteins included in the study.
| mAb ID | IgG | pH | Conc (mg/ml) | Formulation |
|---|---|---|---|---|
| G4–1 | 4 | 5.5 | 25 | Histidine, Sucrose, PS80 |
| G4–2 | 4 | 5.5 | 50 | Histidine, Sucrose, PS80 |
| G1–1 | 1 | 6.0 | 50 | Histidine, Sucrose, PS80 |
| G1–2 | 1 | 5.5 | 50 | Phosphate, Citrate, Mannitol, PS80 |
| G1–3 | 1 | 5.5 | 40 | Histidine, Sucrose, PS80 |
| G1–4 | 1 | 6.0 | 100 | Histidine, Sucrose, PS80 |
| G1–5 | 1 | 5.5 | 50 | Histidine, Sucrose, PS80 |
| G1–6 | 1 | 6.0 | 25 | Citrate, NaCl, DTPA, PS80 |
| G1–7 | 1 | 6.0 | 25 | Citrate, NaCl, DTPA, PS80 |
Stability Data: Stability data at 2–8°C (ambient humidity), 25°C (60% relative humidity) and 40°C (75% relative humidity) from representative formulations in the liquid image were compiled and compared with the biophysical characterization assays described in this article. All batches were within acceptance criteria for critical quality attributes such as purity, content, and potency. Stability data for at least 6 months is available for all temperature conditions for all batches. Twelve months of stability data is available for mAbs G4–2, G1–1, G1–3, G1–4, G1–6 and G1–7 at 25°C. Twenty-four months of data is available for mAbs G4–1 and G1–4 at 5°C.
High-performance size-exclusion chromatography
To quantitatively detect the relative amounts of monomer, aggregates and fragments, HP-SEC was performed with a Waters 2695 Alliance system equipped with a 2487 dual wavelength detector (Waters, Milford Massachusetts, USA). Ten micrograms of sample was injected in a YMC-Diol 200 column at ambient temperature and separation achieved using a mobile phase of 50 mM phosphate, 200 mM sodium chloride, pH 7.0 at a flow rate of 0.5 ml/min. UV detection was performed at 214 nm. The integration of each sample was performed using Waters Empower 2 Software. Integrations were performed manually using baseline-to baseline, baseline-to-valley or tangential skimming, when applicable. The monomer peak for each mAb had been previously identified by on fractionation and isolation techniques. The categorization of HMW is performed using the “timed groups” function in empower and summarizes all peaks eluting prior to the monomer peak. Similarly, categorization of LMW is done using the “timed groups” function to summarize all peaks eluting after the monomer peak. The percent of monomer, HMW species and LMW species was calculated using the pre-defined functions in the Empower 2 reporting software, based on the area under the curves relative to the initial non-stressed sample
Rate of monomer loss was calculated by determining the average rate of the reaction by measuring the change in monomer concentration over time (Δt) as shown in the following equation:
A similar approach has been described earlier by Roberts et al.42
Differential scanning calorimetry
DSC was performed on a MicroCal VP-DSC (Malvern Instruments Ltd., Worcestershire, UK). Duplicate samples of each protein were diluted in appropriate formulation placebo to 1 mg/ml and heated from 20°C to 90°C at a constant rate of 1.5°C/min. Baseline correction, Tm and enthalpy values were obtained using Origin 8.5.
Fluorescence spectroscopy
Intrinsic Trp fluorescence was recorded on a Varian Cary Eclipse Spectrofluorimeter. Samples were diluted to 0.5 mg/ml in respective buffer, and placed in a 1-cm path length cuvette. Intrinsic Trp fluorescence was measured at 20°C by exciting at 295 nm and monitoring emission from 300–550 nm, with both excitation and emission slit width set at 5 nm. A scan speed of 120 nm/min and data pitch of 1 nm with 0.5 seconds averaging time was used. Duplicate measurements of each sample were made and the average reported after buffer subtraction. A 5-point Savitzky Golay smoothing function was applied to all spectra.
Zeta Potential and kD measurements
Zeta potential was measured using the Malvern Nano Zetasizer Nano-ZS (Malvern Instruments Ltd., Worcestershire, UK). Method parameters were first optimized using bovine serum albumin (BSA) to obtain values that have been previously reported.43 Based on the data from BSA, a concentration of 10 mg/ml was selected for experiments using formulated mAbs. For samples that contained high salt (e.g., G1–6 and G1–7), to overcome saturation of the current due to high ionic strength, dilutions were performed in 10 mM histidine buffer, pH 6.0. For data processing, the Smoluchowski model, which utilizes dispersant viscosity as sample viscosity, was employed. Ten measurements per sample were performed at 20°C with 120 seconds equilibration time using disposable folded capillary cells, DTS1070 (Malvern Instruments Ltd., Worcestershire, UK), with the duration per acquisition set to automatic. For kD measurements, sample dilutions were prepared between the concentration range of 2 mg/ml to 25 mg/ml in surfactant-free placebo, using mAb in buffer-only (i.e., unformulated).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr. Moumita Bhattacharya and Dr. Perumal Siva for helpful discussions. Jose K. James acknowledged the support provided by the National Institutes of Health under Ruth L. Kirschstein National research Service Award T32 GM8339 from the NIGMS.
References
- 1.Ecker DM, Jones SD, Levine HL. The Therapeutic Monoclonal Antibody Market. mAbs 2015; 7:9-14; PMID:25529996; http://dx.doi.org/ 10.4161/19420862.2015.989042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013; 341:1192; PMID:24031011; http://dx.doi.org/ 10.1126/science.1241145 [DOI] [PubMed] [Google Scholar]
- 3.Schroeder HW Jr , Cavacini L. Structure and Function of Immunoglobulins. J Allergy Clin Immunol. 2010; 125:S41-S52; PMID:20176268; http://dx.doi.org/ 10.1016/j.jaci.2009.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zurdo J, Arnell A, Obrezanova O, Smith N, Gómez de la Cuesta R, Gallagher TR, Michael R, Stallwood Y, Ekblad C, Abrahmsén L, Höidén-Guthenberg I. Early implementation of QbD in biopharmaceutical development: a practical example. Biomed Res Int. 2015; 2015:1-19; PMID:26075248; http://dx.doi.org/19880308 10.1155/2015/605427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, Gunawan R, McCampbell A, Beaumont M, Meininger D, Richardson D, Ambrogelly A. Developability studies before initiation of process development: improving manufacturability of monoclonal antibodies. MAbs, 2013; 5:787-794; PMID:23883920; http://dx.doi.org/19880308 10.4161/mabs.25269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizzo JM, Shi S, Li Y, Semple A, Esposito JJ, Yu S, Richardson D, Antochshuk V, Shameem M. Application of a high-throughput relative chemical stability assay to screen therapeutic protein formulations by assessment of conformational stability and correlation to aggregation propensity. J Pharm Sci, 2015; 104:1632-40; PMID:25757872; http://dx.doi.org/19880308 10.1002/jps.24408 [DOI] [PubMed] [Google Scholar]
- 7.Zurdo J. Developability assessment as an early de-risking tool for biopharmaceutical development. Pharmaceutical Bioprocessing 2013; 1:29-50; PMID:20489145; http://dx.doi.org/19880308 10.4155/pbp.13.3 [DOI] [Google Scholar]
- 8.Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009; 20:708-714; PMID:19880308; http://dx.doi.org/ 10.1016/j.copbio.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 9.Weiss WF, Young TM, Roberts CJ. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci. 2009; 98:1246-77; PMID:18683878; http://dx.doi.org/ 10.1002/jps.21521 [DOI] [PubMed] [Google Scholar]
- 10.Roberts CJ, Darrington RT, Whitley MB. Irreversible aggregation of recombinant bovine granulocyte-colony stimulating factor (bG-CSF) and implications for predicting protein shelf life. J Pharm Sci. 2003; 92:1095-111; PMID:12712430; http://dx.doi.org/ 10.1002/jps.10377 [DOI] [PubMed] [Google Scholar]
- 11.Glover ZK, Basa L, Moore B, Laurence JS, Sreedhara A. Metal ion interactions with mAbs: Part 1. MAbs 2015; 7:901-11; PMID:26121230; http://dx.doi.org/ 10.1080/19420862.2015.1062193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heads JT, Adams R, D'Hooghe LE, Page MJT, Humphreys DP, Popplewell AG, Lawson AD, Henry AJ. Relative stabilities of IgG1 and IgG4 Fab domains: Influence of the light–heavy interchain disulfide bond architecture. Protein Science 2012; 21:1315-22; PMID:22761163; http://dx.doi.org/ 10.1002/pro.2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of Variable Domains to the Stability of Humanized IgG1 Monoclonal Antibodies, J Pharm. Sci. 2008; 97:1414-26; PMID:17721938; http://dx.doi.org/ 10.1002/jps.21104 [DOI] [PubMed] [Google Scholar]
- 14.Eftink MR. Intrinsic Fluorescence of Proteins. In: Lakowicz JR, ed. Topics in Fluorescence Spectroscopy, Mew York: Kluwer Academic Publishers, 2000:1-15 [Google Scholar]
- 15.Malvern Instruments Ltd Developing A Bioformulation Stability Profile: Light Scattering and Micro- Capillary Viscometry. 2013, 7 [Google Scholar]
- 16.Yadav S, Liu J, Shire SJ, Kalonia DS. Specific interactions in high concentration antibody solutions resulting in high viscosity. J Pharm Sci. 2010; 99:1152-68; PMID:19705420; http://dx.doi.org/ 10.1002/jps.21898 [DOI] [PubMed] [Google Scholar]
- 17.Valente JJ, Payne RW, Manning MC, Wilson WW, Henry CS. Colloidal behavior of proteins: Effects of the second virial coefficient on solubility, crystallization and aggregation of proteins in aqueous solution. Curr Pharm Biotechnol 2005; 6:427-36; PMID:16375727; http://dx.doi.org/ 10.2174/138920105775159313 [DOI] [PubMed] [Google Scholar]
- 18.Banks DD, Latypov RF, Ketchem RR, Woodard J, Scavezze JL, Siska CC, Razinkov VI. Native-State Solubility and Transfer Free Energy as Predictive Tools for Selecting Excipients to Include in Protein Formulation Development Studies. J. Pharm. Sci. 2012; 101:2720-32; PMID:22648863; http://dx.doi.org/ 10.1002/jps.23219 [DOI] [PubMed] [Google Scholar]
- 19.Cheng W, Joshi SB, He F, Brems DN, He B, Kerwin BA, Volkin DB, Middaugh CR. Comparison of High-Throughput Biophysical Methods to Identify Stabilizing Excipients for a Model IgG2 Monoclonal Antibody: Conformational Stability and Kinetic Aggregation Measurements. J. Pharm. Sci. 2012; 101:1701-20; PMID:22323186; http://dx.doi.org/ 10.1002/jps.23076 [DOI] [PubMed] [Google Scholar]
- 20.Sukumar M, Doyle BL, Combs JL, Pekar AH. Opalescent appearance of an IgG1 antibody at high concentrations and its relationship to noncovalent association. Pharm Res. 2004; 21:1087-93; PMID:15290846; http://dx.doi.org/ 10.1023/B:PHAM.0000032993.98705.73 [DOI] [PubMed] [Google Scholar]
- 21.Fesinmeyer RM, Hogan S, Saluja A, Brych SR, Kras E, Narhi LO, Brems DN, Gokarn YR. Effect of Ions on Agitation and Temperature-Induced Aggregation Reactions of Antibodies. Pharm. Res. 2009; 26:903-13; PMID:19104916; http://dx.doi.org/ 10.1007/s11095-008-9792-z [DOI] [PubMed] [Google Scholar]
- 22.Lin JC, Glover ZK, Sreedhara A. Assessing the Utility of Circular Dichroism and FTIR Spectroscopy in Monoclonal-Antibody Comparability Studies. J Pharm Sci 2015; 104:4459-66; PMID:26505267; http://dx.doi.org/ 10.1002/jps.24683 [DOI] [PubMed] [Google Scholar]
- 23.Wu W, Giese RF Jr, van Oss CJ. Linkage between ζ-potential and electron donicity of charged polar surfaces. Implications for the mechanism of flocculation of particle suspensions with plurivalent counterions. Colloids and Surfaces A: Physicochemical and Engineering Aspects 1994; 89:241-52; http://dx.doi.org/ 10.1016/0927-7757(94)80122-3 [DOI] [Google Scholar]
- 24.Saluja A, Fesinmeyer RM, Hogan S, Brems DN and Gokarn YR. Diffusion and Sedimentation Interaction parameters for measuring the second virial coefficient and their utility as predictors of protein aggregation. Biophys J 2010; 99:2657-65; PMID:20959107; http://dx.doi.org/ 10.1016/j.bpj.2010.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnan S, Chi EY, Webb JN, Chang BS, Shan D, Goldenberg M, Manning MC, Randolph TW, Carpenter JF. Aggregation of granulocyte colony stimulating factor under physiological conditions: characterization and thermodynamic inhibition. Biochemistry. 2002; 41:6422-31; PMID:12009905; http://dx.doi.org/ 10.1021/bi012006m [DOI] [PubMed] [Google Scholar]
- 26.Barnett GV, Razinkov VI, Kerwin BA, Laue TM, Woodka AH, Butler PD, Perevozchikova T, Roberts CJ. Specific-ion effects on the aggregation mechanisms and protein-protein interactions for anti-streptavidin immunoglobulin gamma-1. J Phys Chem B. 2015; 119:5793-804; PMID:25885209; http://dx.doi.org/ 10.1021/acs.jpcb.5b01881 [DOI] [PubMed] [Google Scholar]
- 27.Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of Acid-induced Unfolding and Aggregation of Human Immunoglobulin IgG1 and IgG2 Fc. J Biol Chem 2012; 287:1381-96; PMID:22084250; http://dx.doi.org/ 10.1074/jbc.M111.297697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim N, Remmele RL, Liu D, Razinkov VI, Fernandez EJ, Roberts CJ. Aggregation of Anti-Streptavidin Immunoglobulin gamma-1 Involves Fab Unfolding and Competing Growth Pathways Mediated by pH and Salt Concentration. Biophys. Chem. 2013; 172:26-36; PMID:23334430; http://dx.doi.org/ 10.1016/j.bpc.2012.12.004 [DOI] [PubMed] [Google Scholar]
- 29.Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of Thermal Unfolding and Aggregation Profiles of a Series of Developable Therapeutic Monoclonal Antibodies. Mol. Pharmaceutics 2015; 12:1005-7; PMID:25687223; http://dx.doi.org/22477068 10.1021/mp400666b [DOI] [PubMed] [Google Scholar]
- 30.Paul R, Graff-Meyer A, Stahlberg H, Lauer ME, Rufer AC, Beck H, Briguet A, Schnaible V, Buckel T, Boeckle S. Structure and Function of Purified Monoclonal Antibody Dimers Induced by Different Stress Conditions. Pharm. Res. 2012; 29:2047-59; PMID:22477068; http://dx.doi.org/ 10.1007/s11095-012-0732-6 [DOI] [PubMed] [Google Scholar]
- 31.Narhi LO, Schmit J, Bechtold-Peters K, Sharma D. Classification of Protein Aggregates. J Pharm Sci 2012; 101:493-98; PMID:21989781; http://dx.doi.org/ 10.1002/jps.22790 [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Roberts CJ. Aggregation of Therapeutic Proteins; Hoboken, New Jersey: John Wiley & Sons, 2010 [Google Scholar]
- 33.Brummitt RK, Nesta DP, Chang L, Chase SF, Laue TM, Roberts CJ. Nonnative aggregation of an IgG1 antibody in acidic conditions: part 1. Unfolding, colloidal interactions, and formation of high-molecular-weight aggregates. J Pharm Sci. 2011; 100:2087-103; PMID:21213308; http://dx.doi.org/ 10.1002/jps.22448 [DOI] [PubMed] [Google Scholar]
- 34.Brummitt RK, Nesta DP, Chang L, Kroetsch AM, Roberts CJ. Nonnative aggregation of an IgG1 antibody in acidic conditions, part 2: Nucleation and growth kinetics with competing growth mechanisms. J Pharm Sci 2011; 100:2104-19; PMID:21213307; http://dx.doi.org/ 10.1002/jps.22447 [DOI] [PubMed] [Google Scholar]
- 35.Shi S. Biologics: An Update and challenge of their Pharmacokinetics. Curr Drug Metab. 2014; 15:271-90; PMID:24745789; http://dx.doi.org/ 10.2174/138920021503140412212905 [DOI] [PubMed] [Google Scholar]
- 36.Mok CC, Tsai WC, Chen DY, Wei JCC. Immunogenicity of anti-TNF biologic agents in the treatment of rheumatoid arthritis. Exp Opinion on Biol Ther. 2016; 16:201-11; PMID:26560845; http://dx.doi.org/25630815 10.1517/14712598.2016.1118457 [DOI] [PubMed] [Google Scholar]
- 37.Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, Kiialainen A, Koulov A, Boll B, Weiser T, Singer T, Rolink AG, Iglesias A. The Immunogenicity of Antibody Aggregates in a Novel Transgenic Mouse Model. Pharm. Res. 2015; 32:2344-59; PMID:25630815; http://dx.doi.org/ 10.1007/s11095-015-1627-0 [DOI] [PubMed] [Google Scholar]
- 38.Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of Protein Therapeutics. Self Nonself 2010; 1:314-22; PMID:21487506; http://dx.doi.org/ 10.4161/self.1.4.13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of Therapeutic Proteins: Influence of Aggregation. J. Immunotoxicol. 2014; 11:99-109; PMID:23919460; http://dx.doi.org/ 10.3109/1547691X.2013.821564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brummitt RK, Nesta DP, Roberts CJ. Predicting accelerated aggregation rates for monoclonal antibody formulations, and challenges for low-temperature predictions. J Pharm Sci. 2011; 100:4234-43; PMID:21671226; http://dx.doi.org/ 10.1002/jps.22633 [DOI] [PubMed] [Google Scholar]
- 41.ICH harmonised tripartite guideline, Stability testing of biotechnological/biological products Q5C. Current Step 4 version dated 30 November 1995 [Google Scholar]
- 42.Roberts CJ, Das TK, Sahin E. Predicting solution aggregation rates for therapeutic proteins: Approaches and challenges. International J. Pharmaceutics 2011; 418:318-33; PMID:2149188; http://dx.doi.org/ 10.1016/j.ijpharm.2011.03.064 [DOI] [PubMed] [Google Scholar]
- 43.Corbett JCW, Jack RO. Measuring protein mobility using modern microelectrophoresis. Colloids and Surfaces A: Physiochemical and Engineering Aspects. 2011; 376:31-41; http://dx.doi.org/ 10.1016/j.colsurfa.2010.10.029 [DOI] [Google Scholar]