

ABSTRACT
Vibrio cholerae is the etiological agent of the acute intestinal disorder cholera. The toxin-coregulated pilus (TCP), a type IVb pilus, is an essential virulence factor of V. cholerae. Recent work has shown that TcpB is a large minor pilin encoded within the tcp operon. TcpB contributes to efficient pilus formation and is essential for all TCP functions. Here, we have initiated a detailed targeted mutagenesis approach to further characterize this salient TCP component. We have identified (thus far) 20 residues of TcpB which affect either the steady-state level of TcpB or alter one or more TCP functions. This study provides a solid framework for further understanding of the complex role of TcpB and will be of use upon determination of the crystal structure of TcpB or related minor pilin orthologs of type IVb pilus systems.
IMPORTANCE Type IV pili, such as the toxin-coregulated pilus (TCP) in V. cholerae, are bacterial appendages that often act as essential virulence factors. Minor pilins, like TcpB, of these pili systems often play integral roles in pilus assembly and function. In this study, we have generated mutations in tcpB to determine residues of importance for TCP stability and function. Combined with a predicted tertiary structure, characterization of these mutants allows us to better understand critical residues in TcpB and the role they may play in the mechanisms underlying minor pilin functions.
INTRODUCTION
Vibrio cholerae O1 is a Gram-negative pathogenic bacterium and the etiologic agent of the acute intestinal disorder cholera (for a review, see the work of Kaper et al. [1]). Cholera remains a major health concern and economic burden in developing nations. As the source of cholera infection is typically fecal contamination of potable water sources, developing nations and nations devastated by natural disasters are particularly vulnerable to cholera epidemics. Cholera is readily treatable with oral rehydration therapy, but left untreated, it can quickly lead to severe dehydration, shock, and death in less than 24 h postinfection (2).
Essential to V. cholerae pathogenesis are two virulence factors, cholera toxin (CT) (reviewed by Sandkvist [3]) and the toxin-coregulated pilus (TCP) (4, 5). CT is secreted via the extracellular protein secretion (EPS) type II secretion system (T2SS) and internalized via receptor-mediated endocytosis by epithelial cells of the host small intestine (3). There, CT initiates a biochemical cascade of events leading to a massive efflux of Na+, K+, Cl−, and HCO3−, followed by H2O, giving rise to the hallmark symptom of profuse watery diarrhea. TCP, a type IV pilus, is essential for pathogenesis in both the infant mouse cholera model (4) and infant rabbit model (6), as well as in humans (5). TCP is essential for secretion of the colonization factor TcpF (7), is the receptor for the lysogenic bacteriophage (CTXϕ) carrying the CT genes (ctxAB) (8), and mediates bacterial colonization of the human small intestine (5, 9).
Type IV pili are flexible surface appendages found to be important in the environmental lifestyle and are essential for pathogenesis of a number of bacterial species. Type IV pilus structures have been shown to be essential for cellular adhesion (10), natural transformation (11), motility (12), biofilm formation (13), and virulence (4, 5, 14–18). Characteristics of the system's major pilin protein, which forms the pilus structure, along with the organization of the genes encoding components of the pilus biogenesis apparatus, are used to classify type IV pili into two distinct subtypes, IVa and IVb (19, 20).
The V. cholerae TCP, along with the colonization factor antigen III (CFA/III) and longus pili of enterotoxigenic Escherichia coli (ETEC) and the colonization factor Citrobacter (CFC) pilus of Citrobacter rodentium (21), fall under the type IVb subtype. The major pilin proteins in these systems contain longer leader sequences (25 to 30 amino acids), compared to the 6- to 8-amino-acid-long leader sequences of the type IVa pilins (20). The type IVb pilus machinery is also simpler, in that the dozen or so genes encoding the assembly apparatus proteins are arranged in the same operon, whereas the type IVa assembly apparatus often requires 40 or more proteins encoded by genes distributed throughout the organism's genome (22). All type IV pilus systems also contain minor pilins, characterized by their hydrophobic α-helices, which are also highly conserved in all major pilins. Minor pilins are much less abundant than major pilins. Most minor pilins in the type IVa systems are similar in size to their major pilins and have been demonstrated to be incorporated into the pilus structure to mediate pilus functions (23–25). In contrast, the minor pilins of the type IVb systems are much larger than their major pilin counterparts, and their precise roles in pilus assembly and function are less well understood.
TcpB is characterized as the minor pilin of TCP. The tcpB gene is found in the tcp operon adjacent to the major pilin gene tcpA, and the protein it encodes contains the highly conserved pilin-like hydrophobic N-terminal α-helix. Similar to minor pilins of the CFA/III and longus type IVb systems in ETEC, TcpB has a short (7-amino-acid) type IVa pilin-like leader sequence and is significantly larger than the TcpA pilin (423 amino acids [aa] and 199 aa, respectively). TcpB is essential for all known TCP functions (7); however, unpublished data from our laboratory show that the formation of pilus structures can still occur in the absence of TcpB. A ΔtcpB mutant strain is unable to autoagglutinate (an in vitro phenotype that correlates with colonization), deficient in secretion of the colonization factor TcpF, unable to be transduced by CTX-Kmϕ, and unable to colonize the infant mouse (7, 26). In addition, the pili produced by a ΔtcpB mutant strain are only found as small thin twisted bundles in a lower abundance than the wild type and are incapable of forming the large supertwists seen for wild-type strains and essential for autoagglutination and colonization (27), which may explain their inability to carry out any TCP-associated functions.
Recently, the crystal structure of the ETEC CFA/III minor pilin CofB has been determined (28, 29). CofB contains a pilin-like N-terminal region and two discrete C-terminal domains connected by flexible linkers. It was also revealed that CofB is required for and likely initiates CFA/III pilus assembly. The minor pilins CofB and TcpB share many similarities. Although their sequences are only homologous in the N-terminal α-helix region, they are both substantially larger than their pilin counterparts and have multiple cysteines and other shared residues. The similarities between CofB and TcpB allow for reliable structural modeling of TcpB using the solved CofB structure. Based on the similarities, it could also be surmised that TcpB is likely required for efficient TCP assembly. A finer understanding of specific residues of TcpB is needed to decipher the role the protein plays in the TCP machinery. Such information could also provide insights into the mechanism of action of all type IVb minor pilins. In this study, we initiated a targeted mutagenesis approach to determine residues that are important for TcpB stability and function. We hypothesized that by identifying residues that are important for some, but not all, TCP-associated functions, we could gain insight into the roles played by different regions of the protein, with the ultimate goal of understanding the possible mechanisms by which TcpB mediates the functions of TCP.
MATERIALS AND METHODS
Bacterial strains and growth media.
The bacterial strains and plasmids used in this study are described in Table S1 in the supplemental material. All strains were maintained at −80°C in lysogeny broth (LB) containing 20% (vol/vol) glycerol. V. cholerae strains grown under TCP-expressing conditions were grown in LB, with a starting pH of 6.5 and aeration at 30°C for 12 to 16 h, as previously described (4, 30).
When appropriate, strains were grown with antibiotics at the following final concentrations: 100 μg/ml ampicillin, 30 μg/ml gentamicin, 45 μg/ml kanamycin, and 100 μg/ml or 1,000 μg/ml streptomycin. All DNA manipulations were performed using standard molecular and genetic techniques (31). Strains containing plasmid pBAD22, pTK85, pTRNS101, or their derivatives were grown in LB containing ampicillin, and expression of the fusion protein was induced with 0.01% (wt/vol) arabinose as needed.
Plasmid and strain construction.
Plasmid pJMA32 was constructed via the QuikChange PCR-based mutagenesis system (Stratagene). Complementary mutagenic primers ABN75 (5′-GGCACAACCCAAAAGGATGGTAGCGCGGGACCGCATGCG-3′) and ABN76 (5′-CCGGATAGAAGCGCATGCGGTCCCGCGCTACCATCCTTTTGGG-3′) were used for inverse PCR to amplify the entire pTK85 (26) template construct, incorporating the mutation. The resulting plasmid (pJMA32) was then transformed into S17-1 λpir via electroporation, isolated via a Qiagen miniprep kit, using the manufacturer's directions, and verified by sequencing. Following verification, the plasmid was introduced in a V. cholerae O395 ΔtcpB strain, RT4368, via electroporation. All additional plasmids used in the mutagenesis screen were constructed similarly, with the exception of plasmids pJMA46, pJMA70, pJMA57, and pJMA71. These plasmids contain fortuitous additional mutations generated during the QuikChange PCR process, which were discovered during sequencing verification. Plasmid descriptions are found in Table 1.
TABLE 1.
Phenotypic screening of strains with plasmid-carried tcpB
| Construct | Alteration(s)a | TcpB stabilityb | Autoagglutinationb | Transduction efficiencyc | TcpF secretionb |
|---|---|---|---|---|---|
| pTK85 | WT TcpB | + | + | ++ | + |
| pBAD22 | Empty vector | − | − | − | − |
| pJMA82 | E5V | + | + | ++ | − |
| pJMA83 | R26E | + | + | + | + |
| pJMA86 | W79A | + | + | ++ | +/− |
| pJMA85 | K81A | + | + | ++ | + |
| pJMA84 | E83R | + | + | ++ | + |
| pJMA37 | C85A | − | − | ++ | +/− |
| pJMA72 | C85S | − | − | − | − |
| pJMA54 | C107A | − | − | + | + |
| pJMA73 | C107S | + | − | + | − |
| pJMA45 | LCWD249-252AAAA | +/− | − | +/− | − |
| pCHG016 | L249A | + | + | ++ | +/− |
| pJMA38 | C250A | + | − | − | +/− |
| pJMA75 | C250S | − | − | − | − |
| pCHG017 | W251A | + | − | ++ | − |
| pJMA58 | D252A | +/− | + | ++ | + |
| pJMA39 | C261A | + | − | +/− | +/− |
| pJMA76 | C261S | + | − | +/− | − |
| pJMA52 | K272A | + | + | ++ | + |
| pJMA62 | E274A | + | + | ++ | + |
| pJMA61 | D276A | + | + | ++ | + |
| pJMA49 | K278A | + | + | ++ | +/− |
| pJMA51 | D281A | +/− | + | ++ | +/− |
| pJMA60 | K286A | + | + | ++ | + |
| pJMA48 | K295A | + | + | ++ | + |
| pJMA53 | K297A | + | + | ++ | + |
| pJMA69 | F307A | + | − | + | +/− |
| pJMA47 | D309A | + | + | ++ | + |
| pJMA50 | K314A | +/− | ++ | ++ | + |
| pJMA40 | C321A | − | − | − | +/− |
| pJMA74 | C321S | +/− | − | ++ | − |
| pJMA41 | R328A | + | + | ++ | + |
| pJMA68 | SS334-335AA | + | − | + | +/− |
| pJMA42 | E348A | + | + | ++ | + |
| pJMA34 | KD351-2AA | +/− | − | ++ | − |
| pJMA46 | L262P, KD351-2AA | − | +/− | ++ | − |
| pJMA67 | S353A | + | +/− | + | +/− |
| pJMA35 | KD359-60AA | + | + | ++ | + |
| pJMA66 | S362A | + | +/− | ++ | + |
| pJMA32 | K363A | + | + | ++ | + |
| pJMA59 | H366A | + | + | ++ | +/− |
| pJMA65 | L369A | + | + | ++ | + |
| pJMA64 | S370A | + | + | ++ | + |
| pJMA43 | K381A | + | + | ++ | + |
| pJMA63 | W383A | + | − | + | +/− |
| pJMA33 | D384A | + | + | ++ | + |
| pJMA55 | E392A | + | +/− | ++ | + |
| pJMA70 | E392A, D402A | +/− | + | + | +/− |
| pJMA56 | D402A | + | + | ++ | + |
| pJMA36 | RNPK408-11AAAA | + | − | − | +/− |
| pJMA57 | RNPK408-11ANPA | + | + | ++ | + |
| pJMA71 | Δ392-401, RNPK408-11AKNA | +/− | − | − | + |
| pCHG005 | W420A | − | − | − | − |
| pCHG003 | C421S | − | − | − | − |
Alterations in bold type were selected for further study as chromosomal mutations.
+, wild type like; −, ΔtcpB mutant like; +/−, a decrease in function compared to the wild type but not complete loss; ++, exceeds wild-type levels.
Level of transduction efficiency: ++, approximately wild-type level; +, approximately 1 to 2 log below wild-type level; +/− approximately 3 to 5 log below wild-type level; −, no Kmr bacteria were recovered and transduction efficiency is therefore below the limit of detection, equivalent to >7 to 8 log below the wild-type level.
Plasmid pTRNS101 was constructed as follows. Briefly, an approximately 2,800-bp region of V. cholerae O395 containing tcpB was cloned into the allelic exchange vector pKAS32 (32). The resulting plasmid (pTRNS101) was then verified by sequencing and used as a template construct for QuikChange (Stratagene) or Phusion (New England BioLabs) PCR-based mutagenesis (as described above for pJMA32). The resulting constructs were used to introduce chromosomal tcpB point mutations via allelic exchange into strain RT4368 (Table 2), as previously described (30). The resulting strains, RT4634 to RT4656, RT4658, RT4659, and RT4660, were then verified by sequencing to ensure no unintended mutations occurred during the procedure.
TABLE 2.
Summary of TCP functional assays for tcpB chromosomal missense mutants
| Strain | Alteration(s) | TcpB stabilitya | Autoagglutinationa | Transduction efficiencyb | TcpF secretiona | Pilus phenotype |
|---|---|---|---|---|---|---|
| V. cholerae O395 | None (WT) | + | + | ++ | + | WT |
| RT4368 | ΔtcpB | NA | − | − | − | ΔtcpB mutant |
| RT4634 | E5V | + | +/− | + | − | WT |
| RT4635 | R26E | + | + | + | + | WT (looser twists) |
| RT4637 | E83R | + | + | ++ | + | WT |
| RT4638 | C85A | +/− | − | +/− | − | ΔtcpB mutant |
| RT4639 | C85S | +/− | − | +/− | − | ΔtcpB mutant |
| RT4640 | C107A | +/− | − | +/− | − | ΔtcpB mutant |
| RT4641 | C107S | +/− | − | +/− | − | ΔtcpB mutant |
| RT4644 | C250A | + | − | − | − | ΔtcpB mutant |
| RT4645 | C250S | + | − | − | − | ΔtcpB mutant |
| RT4646 | W251A | − | − | − | − | ΔtcpB mutant |
| RT4648 | C261A | + | − | − | − | ΔtcpB mutant |
| RT4649 | C261S | + | − | − | − | ΔtcpB mutant |
| RT4650 | K278A | + | + | ++ | +/− | WT |
| RT4651 | D281A | + | + | + | +/− | WT |
| RT4652 | F307A | + | − | +/− | − | WT (many singular pilus fragments but also a few larger WT-like supertwists; some twists appear to be looser than normal) |
| RT4653 | K314A | + | + | + | + | WT |
| RT4654 | C321A | − | − | − | − | ΔtcpB mutant (occasional large twists but no supertwists) |
| RT4655 | C321S | − | − | − | − | ΔtcpB mutant |
| RT4656 | KD351-2AA | − | − | − | − | ΔtcpB mutant |
| RT4657 | S353A | + | + | ++ | +/− | WT (intermediate in size) |
| RT4658 | H366A | + | + | ++ | − | WT |
| RT4659 | W383A | + | − | +/− | − | WT (very loose strands, fraying) |
| RT4660 | Δ392-401, RNPK408-411AKNA | + | − | − | − | ΔtcpB mutant |
| RT4662 | C421S | − | − | − | − | ΔtcpB mutant |
+, wild type like; − ΔtcpB mutant like; +/−, a decrease in function compared to the wild type but not complete loss; NA, not applicable.
Level of transduction efficiency: ++, approximately wild-type level; +, approximately 1 to 2 log below wild-type level; +/−, approximately 3 to 5 log below wild-type level; −, no Kmr bacteria were recovered and transduction efficiency is therefore below the limit of detection, equivalent to >7 to 8 log below the wild-type level.
Strains RT4657 and RT4662 were constructed using the following approach due to difficulty generating these mutations. Primer pairs were used to amplify ∼500-bp chromosomal regions flanking the intended mutation site. Each primer pair was designed to include a common restriction site to join the two products and a unique restriction site for vector incorporation. The upstream primer pair was additionally designed to incorporate the intended mutation. The resulting PCR products were then cloned into pKAS32, resulting in plasmids pTRNS132 and pTRNS133. The plasmids were verified by sequencing and the mutations then introduced into RT4368 using previously described methods (30).
Plasmid pTRNS120 was constructed by gene splicing by overlap extension (gene SOEing) (33). Briefly, (i) pJMA71, containing the desired tcpB mutant allele, was used as a template for PCR amplification. An ∼600-bp region encompassing the 3′ end of tcpB was PCR amplified. (ii) O395 wild-type (WT) chromosomal DNA both upstream and downstream of the amplified region in step 1 was PCR amplified. The reverse primer of the upstream region and the forward primer of the downstream region were designed to incorporate segments overlapping the PCR amplified region from step 1. (iii) The three PCR-amplified regions produced in steps 1 and 2 were subjected to two additional rounds of PCR amplification using the outside-most primers to generate a single PCR product of ∼2,700 bp. This final PCR product was ligated into the allelic exchange vector pKAS32 and propagated in S17-1 λpir. The desired mutation was then introduced into the O395 WT strain via allelic exchange and verified by sequencing.
Autoagglutination assay.
Cultures were inoculated for each strain from an individual colony and grown under TCP-expressing conditions, as described above, with antibiotics and/or arabinose as required. The autoagglutination phenotype was scored visually after allowing cultures to stand at room temperature for 15 min, and 1 ml of each culture was removed from the top of each culture tube and the optical density at 550 nm (OD550) measured.
CTX-Kmϕ transduction assay.
Strains were grown as described for the autoagglutination assay. The CTX-Kmϕ transduction assay was performed as previously described (8, 9). Briefly, equal volumes of CTX-Kmϕ-containing supernatants and bacterial cultures were mixed and incubated in a water bath at 37°C for 45 min. Following incubation, dilutions of each sample were plated on LB agar containing kanamycin. Additionally, dilutions of bacterial cultures were plated to determine input bacteria. Transduction efficiency is calculated as the ratio of kanamycin-resistant (Kmr) test strain transductants to the number of input CFU divided by the ratio of Kmr wild-type O395 transductants (or ΔtcpB/pTK85 transductants for a positive control) to the number of wild-type O395 (or ΔtcpB/pTK85) input CFU.
Western immunoblotting.
Whole-cell lysates were assayed for total protein concentrations using a bicinchoninic acid protein assay kit (Pierce). Equal amounts of total protein for each sample were resuspended in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis buffer. Culture supernatant fractions were resuspended in 6× buffer due to their lower protein concentrations and were loaded at equal volumes of sample. All samples were boiled for 10 min prior to being loaded on 16% precast Tris-glycine gels (Invitrogen). Proteins were electroblotted onto a nitrocellulose membrane via an iBlot dry blotting system (Invitrogen). The membrane was blocked with 3% bovine serum albumin in 1× Tris-buffered saline with 0.1% Tween (TBST) overnight. The primary antisera used included lab collections of polyclonal antipeptide or monoclonal antibodies against TcpB and TcpF.
Transmission electron microscopy.
Strains were grown under TCP-expressing conditions. A Formvar-coated copper grid was inverted and suspended on top of a 50-μl drop from an overnight culture on Parafilm for 10 min. Grids were wicked dry with Whatman filter paper, negatively stained with 0.5% phosphotungstic acid (pH 6.5) for 2 min, and stored in a desiccated chamber until viewing. Grids were viewed using a Jeol 100CX electron microscope at 100 kV. Each strain was observed at magnifications of up to ×25,000.
Modeling of residues altered in this study.
Predicted secondary and tertiary structures were generated by Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2) using the mature TcpB amino acid sequence. Phyre2 generates a multisequence alignment by scanning the query sequence against a curated protein sequence database and uses the Hidden Markov method (HMM) to construct a tertiary model based on a database of HMMs of proteins of known structure (34). Phyre analysis revealed CofB to be the primary structural match to TcpB, with 99% of TcpB residues modeled at 100% confidence in homology models to the CofB crystal structure. The predicted tertiary structure of TcpB was generated using PyMOL (http://www.pymol.org/).
RESULTS
Construction and analysis of plasmid-borne tcpB mutant alleles.
In an effort to better understand the role of TcpB in TCP functions, we initiated an extensive genetic analysis of TcpB by targeting tcpB codons for mutagenesis. The corresponding residues we selected included all cysteine residues, residues conserved among TcpB orthologs, and additional charged residues, with emphasis on those closer to the C terminus, as previous work has shown the C-terminal region of the pilin TcpA is important for pilin function (9), and TcpB has a similar pilin-like domain. Selected residues were changed to Ala or a specific alternative amino acid based on previous studies involving pilin proteins. In addition, each Cys residue was changed to a Ser to investigate the importance of disulfide bond formation, as changing the -SH group to an -OH group alters the bonding capabilities of the side chain while maintaining the overall conformation of the amino acid. Additional mutations that arose during the PCR process, which were identified during the sequence verification step, were analyzed as well, bringing the total to 53 different alleles of tcpB.
To simplify the process of identifying important residues of TcpB, we initially screened these mutations by generating each allele on a pBAD22 expression construct using a PCR-based mutagenesis system. Each plasmid-borne allele was then introduced into an O395 ΔtcpB strain via electroporation. Strains were grown under TCP-expressing conditions and assessed for TcpB steady-state level, along with any effect on TCP functions using in vitro assays to assess autoagglutination (an in vitro phenotype that correlates with the ability to colonize the small intestine [9, 27]), ability to secrete TcpF, and CTX-Kmϕ transduction frequency. The results for each assay were compared to those for an O395 ΔtcpB/pTK85 strain and an O395 ΔtcpB/pBAD22 strain as positive and negative controls, respectively. A summary of the results relative to the control strains can be found in Table 1. A panel of mutations affecting one or more, but not all, TCP functions (highlighted in bold in Table 1) were then selected for further study by generating equivalent chromosomal mutations in tcpB. Additionally, residues corresponding to those shown to be important in previous studies of pilin TcpA function were also generated in tcpB to determine if they play a role in the function of this minor pilin as well (35, 36).
Construction of chromosomal tcpB mutant alleles.
The results of our screening study identified a number of alleles that exhibited an effect on either the TcpB steady-state level or one or more TCP functions (Table 2). The 24 selected alleles were regenerated using either a PCR-based mutagenesis method or other standard molecular cloning methods in an allelic exchange vector, as described in Materials and Methods. The mutations were then recombined into the chromosome via allelic exchange. By introducing the mutations into the chromosome, the tcpB alleles are expressed using their native promoter, thereby eliminating any consequences of TcpB overexpression which may have occurred in the initial plasmid-based screen. Each allele was verified by sequencing, and each resulting strain was tested for the allele's effect on TcpB steady-state level, pilus formation visually assessed using transmission electron microscopy (TEM), autoagglutination, TcpF secretion, and CTX-Kmϕ transduction frequency. A subset of altered residues was labeled on the predicted secondary and tertiary structures of TcpB (see Fig. 5), which may provide context to better understand the subsequent phenotypic results.
FIG 5.

Selected residues mapped onto predicted TcpB structural models. Residues that do not affect the stability of TcpB but impact the phenotypic profile of TCP, as well as the cysteine residues, are mapped onto the predicted secondary structure (A) and tertiary structure (B). (A) Cysteines (blue), E5 (orange), residues that primarily affect TcpF secretion (red), and residues that have an intermediate effect (green) are highlighted on the mature TcpB amino acid sequence. The predicted secondary structure of the protein generated using Phyre2 (33) is shown below the amino acid sequence. Green coils represent α-helices, blue arrows represent β-sheets, and gray lines indicate coils. (B) Theoretical structural cartoon model of TcpB created using Phyre2. The predicted structure was modeled largely on the crystal structure of CofB; however, the N-terminal region was modeled based on the full-length PAK pilin. Residues of interest are highlighted using the same color code as in panel A. The image was generated in PyMOL (http://www.pymol.org/).
TcpB steady-state levels of tcpB chromosomal missense mutants.
Strains containing chromosomal mutations in tcpB were grown overnight under TCP-expressing conditions with whole-cell extracts (WCE) assayed via Western immunoblotting for the presence of TcpB using anti-TcpB antiserum (Fig. 1, arrow). TcpB was detected in each strain, with the exception of mutants RT4646 (TcpBW251A), RT4654 (TcpBC321A), RT4655 (TcpBC321S), RT4656 (TcpBKD351-2AA), RT4662 (TcpBC421S), and the O395 ΔtcpB strain. Notably, TcpB was detectable in strains encoding mutations of C250 and C261 but not in C321 or C421 mutants. Both C85 and C107 mutants also yielded detectable TcpB, albeit the protein levels were severely reduced. In addition, an unidentified cross-reactive protein was observed at ∼180 kDa only in the TcpBC250A mutant strain and was consistently seen on the immunoblot even with increased levels of dithiothreitol (DTT) added to the protein sample (data not shown). This higher-molecular-weight band was also detected in strains expressing either a plasmid-borne allele that encodes TcpBC250A or a chromosomal allele encoding TcpBC250A constructed from an independent allelic exchange experiment (data not shown), suggesting that this band is not the result of any technical errors made during the construction of the mutant. A nonspecific band observed on the immunoblot served as a loading control.
FIG 1.
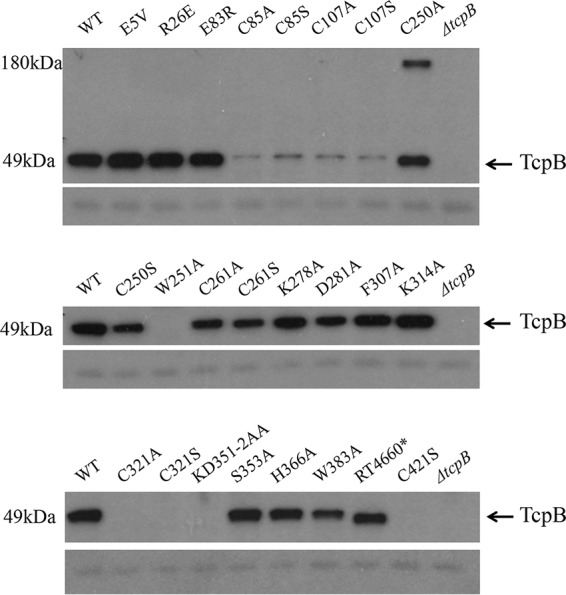
Steady-state level of tcpB chromosomal missense mutants. Western immunoblot of whole-cell lysates of O395 WT, ΔtcpB, and tcpB chromosomal mutants grown under TCP-expressing conditions and probed with anti-TcpB antiserum. A nonspecific band on the immunoblot beneath TcpB serves as the loading control. RT4660* refers to the TcpB alteration Δ392-401, RNPK408-411AKNA.
Pilus formation by the tcpB chromosomal missense mutants.
To ensure the effects on TCP function observed in the tcpB missense mutants are attributable to alterations of the amino acid(s) on the function of TcpB and not the result of an inability of the strain to elaborate TCP, each strain was visually assessed via TEM to confirm that a pilus structure was produced. This analysis also permitted the opportunity to characterize the pili produced by each strain and categorize them based on our previously proposed model of the hierarchy of pilus bundling (27).
Figure 2 shows representative images of pili from O395 WT, ΔtcpB mutant, and tcpB missense mutants. We previously designated the small bundles of pili produced by the ΔtcpB strain as “twists” and the large bundles of pili observed in the O395 WT strain as “supertwists” (27). Our results show O395 WT forming supertwists, while only twists were observed for the ΔtcpB mutant strain. Strains RT4634 (TcpBE5V), RT4637 (TcpBE83R), RT4650 (TcpBK278A), RT4651 (TcpBD281A), and RT4658 (TcpBH366A) appear capable of forming supertwists similar to those observed for the O395 WT strain. Strains RT4638 (TcpBC85A), RT4639 (TcpBC85S), RT4640 (TcpBC107A), RT4641 (TcpBC107S), RT4645 (TcpBC250S), RT4646 (TcpBW251A), RT4648 (TcpBC261A), RT4649 (TcpBC261S), RT4660 (TcpBΔ392–401, RNPK408–411AKNA), and RT4662 (TcpBC421S) produced pili bundles that were similar in size to the twists produced by the ΔtcpB mutant strain, and the supertwists seen in the O395 WT strain were not observed. Interestingly, an intermediate level of bundling (neither completely WT nor ΔtcpB mutant-like in nature) was observed for the RT4635 (TcpBR26E), RT4652 (TcpBF307A), RT4657 (TcpBS353A), and RT4659 (TcpBW383A) strains, where they readily form supertwists but the bundles appear much looser than those of the wild type.
FIG 2.

Pilus morphology of tcpB chromosomal missense mutants. Representative images of mutants under TCP-inducing growth conditions. Overnight cultures were negatively stained with phosphotungstic acid and viewed using TEM. The black arrow in the WT panel indicates pili, and white arrows indicate flagella. All images were taken at a magnification of ×25,000.
Autoagglutination phenotype of tcpB chromosomal missense mutants.
Next, we characterized the autoagglutination phenotype of the tcpB missense mutants. When grown under TCP-expressing conditions, the O395 WT strain forms visible aggregates that rapidly precipitate out of a suspension upon removal of the culture from the rotator. To determine whether mutations in tcpB altered the autoagglutination phenotype, strains were grown under TCP-expressing conditions as described in the Materials and Methods and scored both visually and by a spectrophotometer (Fig. 3). The OD550 values shown in Fig. 3 represent an average of three replicates. A ΔtcpB mutant strain was used as a negative control and showed no autoagglutination, consistent with previous studies (7, 26).
FIG 3.

Autoagglutination phenotype of tcpB chromosomal missense mutants. Strains were grown under TCP-expressing conditions, followed by 15 min of standing at room temperature (RT). Images show the various levels of autoagglutination of the different tcpB mutants. Numbers below the images indicate average OD550 from three separate cultures for each strain.
Seven of the mutant strains retained the ability to autoagglutinate at WT levels. The TcpBE5V mutant consistently resulted in reduced levels of autoagglutination, exhibiting a visible pellet on the bottom of the culture tube and a turbid supernatant, while 16 of the mutations (including all strains encoding Cys alterations) resulted in the inability of the strain to form aggregates, appearing similar to the ΔtcpB mutant strain. The autoagglutination phenotypes appeared to be consistent with the formation of supertwists (Table 2).
TcpF secretion by tcpB chromosomal missense mutants.
TcpB is essential for secretion of the colonization factor TcpF (7, 26). Whole-cell extracts (WCE) and filtered culture supernatants (Supe) were assayed for the presence of TcpF via immunoblotting with anti-TcpF antiserum (Fig. 4). Only strains encoding mutants R26E, E83R, and K314A (RT4635, RT4637, and RT4653, respectively) retained the ability to secrete TcpF at WT levels, where all of the TcpF is secreted into the culture supernatant and none remains in the WCE. Strains encoding the mutations K278A, D281A, and S353A (RT4650, RT4651, and RT4657, respectively) showed an intermediate phenotype, as a band corresponding to TcpF was observed in the supernatant fraction, but TcpF was still detectable in the WCE. A ΔtcpF mutant strain was used as a negative antibody control.
FIG 4.
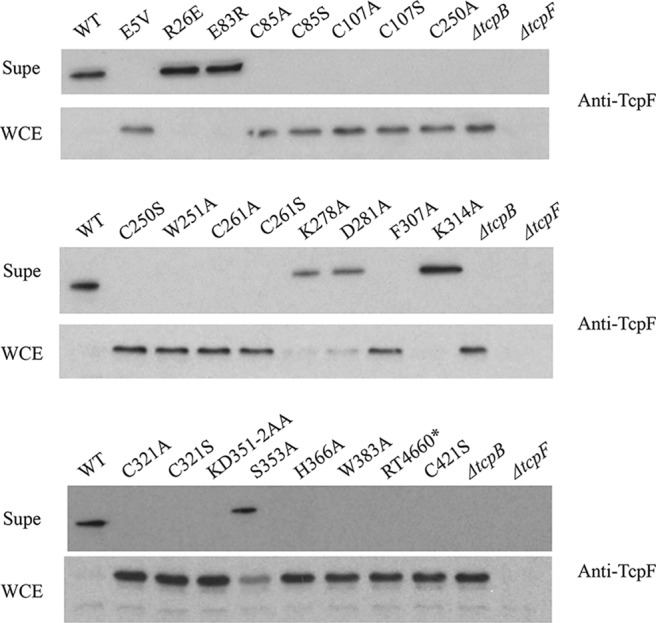
TcpF secretion by tcpB chromosomal missense mutants. Western immunoblots of filter-sterilized culture supernatant (Supe) and whole-cell extract (WCE) samples from O395 WT, ΔtcpB mutant, and tcpB chromosomal mutant strains grown under TCP-expressing conditions and probed with anti-TcpF antiserum. The ΔtcpF mutant served as an antiserum control.
CTX-Kmϕ transduction frequency of tcpB chromosomal missense mutants.
An O395 ΔtcpB strain is deficient for CTX-Kmϕ transduction (7, 26). To determine whether the tcpB missense mutations altered the transduction frequency by CTX-Kmϕ, each strain was tested as described in Materials and Methods, with the results shown in Table 3. O395 WT and ΔtcpB mutant strains were used as positive and negative controls, respectively. Transduction efficiency was determined by dividing the transduction frequency of the experimental strain by WT.
TABLE 3.
CTX-Kmϕ transduction of tcpB chromosomal missense mutants
| Strain | Alteration(s) | Transduction frequency | Transduction efficiency |
|---|---|---|---|
| WT | 1.02E−02 | 1 | |
| ΔtcpB mutant | <4.50E−10 | <4.41E−08 | |
| RT4634 | E5V | 1.42E−04 | 1.39E−02 |
| RT4635 | R26E | 2.49E−04 | 2.44E−02 |
| RT4637 | E83R | 2.97E−02 | 2.91 |
| RT4638 | C85A | 3.48E−07 | 3.42E−05 |
| RT4639 | C85S | 5.87E−07 | 5.75E−05 |
| RT4640 | C107A | 1.47E−06 | 1.44E−04 |
| RT4641 | C107S | 6.67E−07 | 6.54E−05 |
| RT4644 | C250A | <2.18−10 | <2.14E−08 |
| RT4645 | C250S | <2.18E−09 | <2.14E−07 |
| RT4646 | W251A | <1.27E−10 | <1.25E−08 |
| RT4648 | C261A | <1.16E−09 | <1.14E−07 |
| RT4649 | C261S | <1.43E−09 | <1.40E−07 |
| RT4650 | K278A | 2.66E−03 | 0.261 |
| RT4651 | D281A | 4.47E−04 | 4.38E−02 |
| RT4652 | F307A | 9.95E−07 | 9.75E−05 |
| RT4653 | K314A | 4.08E−04 | 4.00E−02 |
| RT4654 | C321A | <6.20E−10 | <6.08E−08 |
| RT4655 | C321S | <3.20E−10 | <3.14E−08 |
| RT4656 | KD351-2AA | <1.14E−09 | <1.12E−07 |
| RT4657 | S353A | 3.92E−03 | 0.384 |
| RT4658 | H366A | 1.66E−03 | 0.163 |
| RT4659 | W383A | 2.56E−06 | 2.51E−04 |
| RT4660 | Δ392-401, RNPK408-11AKNA | <3.17E−10 | <3.11E−08 |
| RT4662 | C421A | <5.91E−10 | <5.79E−08 |
Four tcpB mutant trains resulted in WT levels of transduction efficiencies. Four mutants resulted in transduction efficiencies of approximately 1 to 2 log below the levels of the WT strain. Six mutants resulted in transduction efficiency 4 to 5 log below WT levels and are considered to be severely defective in CTXϕ transduction. The remaining 10 tcpB mutant strains were greater than 7 log below WT efficiencies and were considered to be transduction negative.
DISCUSSION
The V. cholerae O395 minor pilin TcpB is required for efficient TCP formation, although some pili are still produced by a tcpB mutant. However, TcpB is essential for all known TCP functions both in vitro and in vivo. To further characterize this important protein and better understand how TcpB mediates the various TCP functions, we initiated a targeted mutagenesis study to identify those residues of TcpB important for TCP function.
Through the analysis of mutants that were unaffected in TcpB stability but altered in one or more TCP-associated functions, we found that changes in autoagglutination and phage transduction phenotypes are correlated. If a mutant is able to autoagglutinate to a wild-type level when grown under TCP-inducing conditions, it also has a wild-type level of phage transduction efficiency. If a mutant is unable to autoagglutinate, it is also either severely defective in phage transduction or unable to be transduced by phage at a level detectable by our assay. However, this phenotypic correlation does not extend to TcpF secretion, as there are two mutants (RT4634 and RT4658) that have wild-type, or near-wild-type, levels of autoagglutination and phage transduction efficiency but are unable to secrete TcpF (Table 2). This finding may reflect a disruption in the interaction between the pilus and its secreted protein, i.e., a potential TcpB-TcpF interaction.
The recent crystallization of enterotoxigenic E. coli minor pilin CofB (28, 29) may provide new insights into the significance of the TcpB point mutants generated in this study, as 99% of the TcpB residues modeled at 100% confidence in homology models with the CofB crystal structure (which was crystalized without the N-terminal α-helix) when analyzed using the Phyre2 modeling server (28). CofB consists of a pilin domain, a linker connecting the pilin domain to a β-repeat region containing two β-repeats, and a second connective linker, followed by a C-terminal β-sandwich domain (28). The Phyre2 protein folding recognition software generated secondary and tertiary structural models for TcpB that we used to map our selected amino acids of interest from the mutagenesis study (Fig. 5). These residues include all the cysteines as well as amino acids that, when altered, do not affect the stability of TcpB but impact the phenotypic profile of TCP. Given the homology to CofB, it appears that TcpB consists of a pilin domain, one β-repeat region, and the β-sandwich domain.
When viewing the tertiary predicted model for TcpB, of first interest were the Cys missense mutations (Fig. 5B, blue). In type IV pilins, the disulfide bridge forms a loop structure, termed the D-region (37), shown in TCP pilin TcpA to contain residues important for TCP function. TcpB contains 6 Cys residues, making it difficult to predict a priori which ones might be critical for TcpB stability. Some of the Cys residue alterations resulted in a loss of detectable TcpB in the WCE of strains grown under TCP-expressing conditions, suggesting that these mutations decrease the stability of TcpB. None of the Cys mutant strains showed the ability to autoagglutinate or secrete TcpF, and all were markedly deficient compared to O395 WT in phage transduction frequency.
In CofB, two disulfide bonds are present within a β-repeat region, where each links a pair of β-strands (28, 29). In the Phyre2-predicted model, the homologous cysteine residues within TcpB would be C250 and C261, which could be a disulfide bonded pair within the corresponding β-repeat region that links the predicted pilin domain to the β-sandwich domain in TcpB. Of note, the TcpB steady-state level was not affected in strains carrying these mutations (RT4645 and RT4649, respectively), indicating that alterations at C250 or C261 do not affect the overall stability of TcpB. These cysteine mutations may prevent proper folding of a domain of the protein, thereby preventing full TcpB function, perhaps due to the disruption of a disulfide bond. In TcpB, C321 and C421 are homologous to a pair of cysteines in CofB that form a disulfide bond within the β-sandwich region (25), suggesting that a similar disulfide bond occurs in TcpB as well. Changing these residues (in RT4655 and RT4662, respectively) completely abolished TcpB stability and TCP functions, suggesting that these cysteines, and potential disulfide bond, are critical for TcpB. The C85 and C107 residues are located in the N-terminal pilin domain region and, as in TcpA, are most likely also disulfide bonded and are critical for TcpB structure and function. Although altering either of the cysteines did not completely render the protein unstable, as for TcpA (9), these two Cys residues are partially responsible for TcpB stability and the ability of TcpB to contribute to all TCP-associated functions.
In type IV pilins, the glutamic acid residue at position 5 of the mature pilin is highly conserved and thought to aid in subunit incorporation into the growing filament, where it neutralizes the N-terminal positive charge so that pilins may assemble (35, 38, 39, 40). A mutation corresponding to E5V in the gene encoding the P. aeruginosa major pilin PilA resulted in a loss of piliation in this strain (35). An E5A mutant in V. cholerae TcpA also disrupted efficient pilus formation (39). Strain RT4634 (TcpBE5V) produced a stable TcpB protein but showed intermediate phenotypes for autoagglutination (pellet at bottom of culture tube with turbid supernatant) and phage transduction efficiency, and it had a severe deficiency in its ability to secrete TcpF. Since this mutation is located in the N-terminal helical region (Fig. 5A, orange), it likely does not disrupt the overall structure of TcpB, hence the normal steady-state level of TcpB, but it could disrupt the possible ability of TcpB to be incorporated into the pilus. It is possible that proper TcpB incorporation into a growing pilus is particularly critical for TcpF secretion.
This mutagenesis study has identified four additional TcpB residues (K278, D281, S353, and H366) that appear important primarily for TcpF secretion. Altering the K278, D281, and S353 residues (strains RT4650, RT4651, and RT4657, respectively) reduced the secretion of TcpF, as some TcpF remained in the WCE, while the H366A (strain RT4658) alteration completely abolished TcpF secretion. Based on the predicted TcpB structure, these residues are located on one side of the β-sandwich region (Fig. 5B [all four fall within the top plane of this region]), which may be an important interaction domain for TcpB with TcpF, TcpB with TcpB, or between TcpB and other components of the pilus biogenesis apparatus that enable TcpF secretion.
Other notable alterations include F307A and W383A (strains RT4652 and RT4659, respectively). Both mutants produced stable TcpB and WT-like pili when viewed under TEM, but do not autoagglutinate or secrete TcpF, and are deficient in phage transduction. Although they produce WT-like pilus supertwists, these mutants also produce many singular pilus fragments and some looser-appearing supertwists. Based on the predicted TcpB structural model, F307 is located near the C-terminal end of the β-sandwich region, and W383 is also located on the same side of the β-sandwich region, falling within a β-sheet (Fig. 5B). Since some supertwists appeared loosely packed, the F307 and W383 residues may be located in a region of TcpB that is important for assisting pili in tightly and efficiently supertwisting (although the single point mutations may not be sufficient to completely abolish these tight twists, as some were still observed), and these tight supertwists may be necessary for the other TCP functions (TcpF secretion, phage transduction, and autoagglutination). It is possible that this region of TcpB also stabilizes pili in a way that allows the tight supertwists to form.
The individual residues (K278, D281, S353, H366, W383, and F307) within the β-sandwich region appear to be of particular importance to TcpB function, as additional selected residues that were screened in our original plasmid mutagenesis (Table 1) were located in this region as well and did not affect TcpB stability or function. In the initial screen, we selected many more residues in the C-terminal region because of their conservation in TcpB orthologs and their polar or charged properties. Among the residues tested, many are located near the four residues that affected only TcpF secretion (K278, D281, S353, and H366), and these mutant proteins all exhibited characteristics identical to those of the wild type. Similarly, the F307 and W383 residues that are important for tight supertwist formation are also surrounded by amino acids with biochemical properties that potentially play a role in protein-protein interaction or pilus stabilization, but from our plasmid mutagenesis screen, none of them seem to contribute to TcpB stability or function (Table 1). The set of six mutations we tested on the chromosome appear to be uniquely critical for TCP function, as surrounding mutations (on the plasmid) did not share the same functional effects. Future immunoprecipitation or two-hybrid assays using these mutants will allow us to study TcpB interaction with TcpF and other proteins in the TCP biogenesis apparatus, providing more insight into the mechanisms underlying TcpB function.
Through this study, we have identified 20 residues of TcpB which affect either the steady-state level of TcpB or one or more in vitro functions of TCP. Now that the structure of CofB has been deduced (28, 29), the model for TcpB will aid in determining potential residues of interest for further studies in TcpB interaction and function. In CofB, the flexible C-terminal region is required for CofB to initiate pilus assembly, likely by recruiting the initial major pilin CofA to the assembly apparatus (28). Recently, it has also been proposed that CofB may form a homotrimer that acts as a tip complex to initiate pilus formation (29). The proposed model suggests that the CofB complex forms largely through the hydrophobic interactions of the β-repeat and β-sandwich domains. This new model further illustrates the importance of the C-terminal domains of these minor pilins and provides insight into the potential mechanism that TcpB may adopt for its role in TCP functions.
We propose that TcpB is necessary for efficient TCP formation, as a ΔtcpB mutant strain and many of the mutants tested still produce a pilus, but it is nonfunctional. It is possible that TcpB helps to stabilize the major pilin TcpA for assembly, contributing to the ability to form supertwists and perform associated pilus functions. Additionally, current efforts to determine the crystal structure of TcpB will provide the opportunity to map these important residues onto the true structure of TcpB and might further allow us to explore the role of TcpB in V. cholerae pathogenesis, while providing new insights into minor pilin functions in other pilus systems.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Howard and the Rippel Electron Microscope Facility of Dartmouth College for the assistance in collecting microscopy data. We also thank T. Kirn for plasmid pTK85.
Funding was provided by the National Institutes of Health grant AI025096 to R.K.T. Additional funding was provided for J.M.M. and C.A.H. by the National Institutes of Health training grants AI007363 and AI007519.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00309-16.
REFERENCES
- 1.Kaper JB, Morris JG Jr, Levine MM. 1995. Cholera. Clin Microbiol Rev 8:48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barua D. 1992. History of cholera, p 1–36. In Barua D, Greenbough WB III (ed), Cholera. Plenum Publishing Corp., New York, NY. [Google Scholar]
- 3.Sandkvist M. 2001. Biology of type II secretion. Mol Microbiol 40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- 4.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritchie JM, Rui H, Bronson RT, Waldor MK. 2010. Back to the future: studying cholera pathogenesis using infant rabbits. mBio 1(1):e00047-10. doi: 10.1128/mBio.00047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirn TJ, Bose N, Taylor RK. 2003. Secretion of a soluble colonization factor by the TCP type 4 pilus biogenesis pathway in Vibrio cholerae. Mol Microbiol 49:81–92. doi: 10.1046/j.1365-2958.2003.03546.x. [DOI] [PubMed] [Google Scholar]
- 8.Waldor MK, Mekalanos JJ. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 9.Kirn TJ, Lafferty MJ, Sandoe CM, Taylor RK. 2000. Delineation of pilin domains required for bacterial association into microcolonies and intestinal colonization by Vibrio cholerae. Mol Microbiol 35:896–910. doi: 10.1046/j.1365-2958.2000.01764.x. [DOI] [PubMed] [Google Scholar]
- 10.Lee KK, Sheth HB, Wong WY, Sherburne R, Paranchych W, Hodges RS, Lingwood CA, Krivan H, Irvin RT. 1994. The binding of Pseudomonas aeruginosa pili to glycosphingolipids is a tip-associated event involving the C-terminal region of the structural pilin subunit. Mol Microbiol 11:705–713. doi: 10.1111/j.1365-2958.1994.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 11.Cehovin A, Simpson PJ, McDowell MA, Brown DR, Noschese R, Pallett M, Brady J, Baldwin GS, Lea SM, Matthews SJ, Pelicic V. 2013. Specific DNA recognition mediated by a type IV pilin. Proc Natl Acad Sci U S A 110:3065–3070. doi: 10.1073/pnas.1218832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi W, Sun H. 2002. Type IV pilus-dependent motility and its possible role in bacterial pathogenesis. Infect Immun 70:1–4. doi: 10.1128/IAI.70.1.1-4.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 14.Girón JA, Ho AS, Schoolnik GK. 1991. An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science 254:710–713. doi: 10.1126/science.1683004. [DOI] [PubMed] [Google Scholar]
- 15.Strom MS, Nunn DN, Lory S. 1993. A single bifunctional enzyme, PilD, catalyzes cleavage and N-methylation of proteins belonging to the type IV pilin family. Proc Natl Acad Sci U S A 90:2404–2408. doi: 10.1073/pnas.90.6.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobbs M, Mattick JS. 1993. Common components in the assembly of type 4 fimbriae, DNA transfer systems, filamentous phage and protein-secretion apparatus: a general system for the formation of surface-associated protein complexes. Mol Microbiol 10:233–243. doi: 10.1111/j.1365-2958.1993.tb01949.x. [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi T, Fujino Y, Yamamoto K, Miwatani T, Honda T. 1995. Sequencing of the gene encoding the major pilin of pilus colonization factor antigen III (CFA/III) of human enterotoxigenic Escherichia coli and evidence that CFA/III is related to type IV pili. Infect Immun 63:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XL, Tsui IS, Yip CM, Fung AW, Wong DK, Dai X, Yang Y, Hackett J, Morris C. 2000. Salmonella enterica serovar Typhi uses type IVB pili to enter human intestinal epithelial cells. Infect Immun 68:3067–3073. doi: 10.1128/IAI.68.6.3067-3073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strom MS, Lory S. 1993. Structure-function and biogenesis of the type IV pili. Annu Rev Microbiol 47:565–596. doi: 10.1146/annurev.mi.47.100193.003025. [DOI] [PubMed] [Google Scholar]
- 20.Craig L, Pique ME, Tainer JA. 2004. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 21.Mundy R, Pickard D, Wilson RK, Simmons CP, Dougan G, Frankel G. 2003. Identification of a novel type IV pilus gene cluster required for gastrointestinal colonization of Citrobacter rodentium. Mol Microbiol 48:795–809. doi: 10.1046/j.1365-2958.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 22.Pelicic V. 2008. Type IV pili: e pluribus unum? Mol Microbiol 68:827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- 23.Helaine S, Dyer DH, Nassif X, Pelicic V, Forest KT. 2007. 3D structure/function analysis of PilX reveals how minor pilins can modulate the virulence properties of type IV pili. Proc Natl Acad Sci U S A 104:15888–15893. doi: 10.1073/pnas.0707581104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giltner CL, Habash M, Burrows LL. 2010. Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J Mol Biol 398:444–461. doi: 10.1016/j.jmb.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 25.Brown DR, Helaine S, Carbonnelle E, Pelicic V. 2010. Systematic functional analysis reveals that a set of seven genes is involved in fine-tuning of the multiple functions mediated by type IV pili in Neisseria meningitidis. Infect Immun 78:3053–3063. doi: 10.1128/IAI.00099-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marles JM. 2012. Characterization of type IVb pilus components essential for functional TCP of Vibrio cholerae O395, including implications on the hierarchy of microcolony formation and virulence. Ph.D. dissertation. Dartmouth College, Hanover, NH. [Google Scholar]
- 27.Jude BA, Taylor RK. 2011. The physical basis of type 4 pilus-mediated microcolony formation by Vibrio cholerae O1. J Struct Biol 175:1–9. doi: 10.1016/j.jsb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolappan S, Ng D, Yang G, Harn T, Craig L. 2015. Crystal structure of the minor pilin CofB, the initiator of CFA/III pilus assembly in enterotoxigenic Escherichia coli. J Biol Chem 290:25805–25818. doi: 10.1074/jbc.M115.676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawahara K, Oki H, Fukakusa S, Yoshida T, Imai T, Maruno T, Kobayashi Y, Motooka D, Iida T, Ohkubo T, Nakamura S. 2016. Homo-trimeric structure of the type IVb minor pilin CofB suggests mechanism of CFA/III pilus assembly in human enterotoxigenic Escherichia coli. J Mol Biol 428:1209–1226. doi: 10.1016/j.jmb.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Martinez RM, Megli CJ, Taylor RK. 2010. Growth and laboratory maintenance of Vibrio cholerae. Curr Protoc Microbiol 6:Unit-6A.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maniatis T, Fritsch EF, Sambrook J. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 32.Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 33.Horton RM. 1995. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol 3:93–99. [DOI] [PubMed] [Google Scholar]
- 34.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 Web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strom MS, Lory S. 1991. Amino acid substitutions in pilin of Pseudomonas aeruginosa. Effect on leader peptide cleavage, amino-terminal methylation, and pilus assembly. J Biol Chem 266:1656–1664. [PubMed] [Google Scholar]
- 36.Li J, Lim MS, Li S, Brock M, Pique ME, Woods VL Jr, Craig L. 2008. Vibrio cholerae toxin-coregulated pilus structure analyzed by hydrogen/deuterium exchange mass spectrometry. Structure 16:137–148. doi: 10.1016/j.str.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim MS, Ng D, Zong Z, Arvai AS, Taylor RK, Tainer JA, Craig L. 2010. Vibrio cholerae El Tor TcpA crystal structure and mechanism for pilus-mediated microcolony formation. Mol Microbiol 77:755–770. doi: 10.1111/j.1365-2958.2010.07244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macdonald DL, Pasloske BL, Paranchych W. 1993. Mutations in the fifth-position glutamate in Pseudomonas aeruginosa pilin affect the transmethylation of the N-terminal phenylalanine. Can J Microbiol 39:500–505. doi: 10.1139/m93-071. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Egelman EH, Craig L. 2012. Structure of the Vibrio cholerae type IVb pilus and stability comparison with the Neisseria gonorrhoeae type IVa pilus. J Mol Biol 418:47–64. doi: 10.1016/j.jmb.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA. 2006. Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol Cell 23:651–662. doi: 10.1016/j.molcel.2006.07.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.