

ABSTRACT
When microbes are faced with an environmental challenge or opportunity, preexisting enzymes with promiscuous secondary activities can be recruited to provide newly important functions. Mutations that increase the efficiency of a new activity often compromise the original activity, resulting in an inefficient bifunctional enzyme. We have investigated the mechanisms by which growth of Escherichia coli can be improved when fitness is limited by such an enzyme, E383A ProA (ProA*). ProA* can serve the functions of both ProA (required for synthesis of proline) and ArgC (required for synthesis of arginine), albeit poorly. We identified four genetic changes that improve the growth rate by up to 6.2-fold. Two point mutations in the promoter of the proBA* operon increase expression of the entire operon. Massive amplification of a genomic segment around the proBA* operon also increases expression of the entire operon. Finally, a synonymous point mutation in the coding region of proB creates a new promoter for proA*. This synonymous mutation increases the level of ProA* by 2-fold but increases the growth rate by 5-fold, an ultrasensitive response likely arising from competition between two substrates for the active site of the inefficient bifunctional ProA*.
IMPORTANCE The high-impact synonymous mutation we discovered in proB is remarkable for two reasons. First, most polar effects documented in the literature are detrimental. This finding demonstrates that polar effect mutations can have strongly beneficial effects, especially when an organism is facing a difficult environmental challenge for which it is poorly adapted. Furthermore, the consequence of the synonymous mutation in proB is a 2-fold increase in the level of ProA* but a disproportionately large 5.1-fold increase in growth rate. While ultrasensitive responses are often found in signaling networks and genetic circuits, an ultrasensitive response to an adaptive mutation has not been previously reported.
INTRODUCTION
Metabolic enzymes, although prodigious catalysts, are not perfectly specific for their physiological substrates. They typically possess secondary activities as a consequence of the assemblage of highly reactive functional groups, metal ions, and cofactors in their active sites. Secondary activities that are physiologically irrelevant, either because they are too inefficient to contribute to fitness or because the enzyme never encounters the substrate, are termed promiscuous activities (1).
Promiscuous activities are important from an evolutionary standpoint because they provide a reservoir of catalytic potential within a proteome that can be drawn upon when the environment changes. A promiscuous activity may become important for fitness when a new source of carbon, nitrogen, or phosphorous appears in the environment or when a previously available compound, such as an amino acid or cofactor, becomes unavailable. A promiscuous activity may also become critical when the organism is exposed to a novel toxin, such as an antibiotic or pesticide.
A newly recruited promiscuous activity is unlikely to be the optimal solution to an environmental challenge or opportunity. The evolutionary path by which fitness can be improved depends upon whether the original activity of the enzyme is still important for fitness. If it is not, the gene encoding the promiscuous enzyme is free to evolve in the absence of constraints imposed by the need to retain the original activity. Such is often the case for antibiotic resistance enzymes, which typically evolve under selective pressure for detoxification of a new antibiotic in the absence of selective pressure to maintain their former activity. The opposite end of the spectrum is a situation in which an essential enzyme harbors the newly critical secondary activity and there is a strong trade-off between the two activities such that improvements in the new activity result in a significant decrease in the original activity. The ultimate outcome of such an adaptive conflict will depend upon a number of factors, including (i) the flux through each reaction that is necessary to satisfy the needs of the cell, (ii) the frequency of duplication of the region surrounding the gene encoding the “weak-link” bifunctional enzyme, (iii) the costs and benefits of duplication and subsequent amplification, and (iv) the potential for point mutations to either increase the amount of the weak-link enzyme or improve one or both of its activities.
E. coli ProA (γ-glutamyl phosphate reductase), which catalyzes the second step in the pathway for synthesis of proline, has an inefficient promiscuous ability to catalyze the reduction of N-acetyl glutamyl phosphate (2, 3). This reaction, normally catalyzed by ArgC, is essential for biosynthesis of arginine. The chemical transformations catalyzed by ProA and ArgC are identical, although the two enzymes are not homologous. The substrates for ProA and ArgC differ only in the absence or presence of an acetyl moiety on the amino group (Fig. 1).
FIG 1.

Reactions catalyzed by ProA (γ-glutamyl phosphate reductase) and ArgC (N-acetyl glutamyl phosphate reductase).
Deletion of argC prevents E. coli from growing on glucose as the sole carbon source, demonstrating that the promiscuous activity of ProA is insufficient to support synthesis of arginine. However, a mutation that changes Glu383 to Ala in ProA allows the ΔargC strain to grow, albeit slowly, on glucose (3). We previously found that E383A ProA (ProA*) has improved activity with the substrate for ArgC but substantially poorer activity with its normal substrate (2). (For technical reasons, both reactions were assayed in the reverse direction, but decreased activity in one direction implies decreased activity in the other, as well.) Reducing the efficiency of the original catalytic activity when a secondary activity becomes important may be an important general theme in the recruitment of existing enzymes to serve new functions. When two substrates compete for the same active site, each will be a competitive inhibitor of the other. Decreasing the kcat/Km for the original activity will decrease the inhibition of the newly important reaction by the original substrate. Thus, our model system in which E383A ProA provides two essential activities exemplifies the evolutionarily challenging situation in which successful recruitment of an essential enzyme to serve a new function requires a mutation that diminishes the efficiency of its original activity.
Since ΔargC proA* E. coli grows on glucose at a fraction of the rate of the wild-type strain, there is strong selective pressure for improvement in the ability to produce arginine, proline, or both. Here we describe an exploration of the initial events that improve fitness when growth of E. coli is limited by the inefficiency of this bifunctional enzyme. We identified four high-impact genetic changes that improve the growth rate on glucose by factors of 3.2 to 6.2. In addition to two previously identified mutations in the promoter of the proBA* operon (3), we found a synonymous mutation in the coding region of proB and large segmental amplifications with more than 50 copies of the proBA* operon. The promoter mutations and segmental amplifications led to substantial overexpression of the proBA* operon. In contrast, the synonymous mutation in proB results in a 9.6-fold increase in expression of proA* alone, suggesting that the mutation has generated a new promoter within the body of proB. Thus, this mutation provides a mechanism for increasing the level of ProA* with no associated costs due to overexpression of other genes and/or maintenance of extra DNA. This is an effective strategy; the increase in growth rate resulting from the synonymous mutation in proB is equivalent to that achieved by segmental amplifications with as many as 53 copies of the proBA* operon. Furthermore, this mutation produces an ultrasensitive response in that the improvement in growth rate (5.4-fold) is larger than the increase in the level of ProA* (2-fold). Although ultrasensitivity appears to be a common mechanism for achieving sharp transitions in signaling networks and genetic circuits (4), an ultrasensitive response caused by an adaptive mutation that improves the performance of a metabolic pathway has not previously been reported.
MATERIALS AND METHODS
Primers for strain construction, PCR, qPCR, Melt-MAMA qPCR, and 5′-RACE.
Primers (see Tables S1 to S5 in the supplemental material) were ordered from IDT with standard desalting unless otherwise stated.
Strains and culture conditions.
The strains and plasmids used in this study are listed in Tables 1 and 2, respectively. E. coli cultures were routinely grown in LB medium (37) at 37°C with 20 μg/ml kanamycin (Kan) (LB/Kan), 35 μg/ml chloramphenicol (Cm), or 100 μg/ml ampicillin (Amp), as required. Cultures for adaptation or analysis of growth curves were grown in M9 minimal medium (37) containing 0.2% glucose (M9/glucose). In some cases, 5.2 mM arginine, 0.4 mM proline, or both were included in the M9/glucose. (These amino acid concentrations correspond to those in EZ-rich medium [5].)
TABLE 1.
Strains used in this study
| Strain(s) | Genotype | Note(s) | Reference |
|---|---|---|---|
| BW25113 | Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 54 | |
| GS325 | BW25113 proA* argC::kan; hemL allele encoding G277S HemL | Contains unexpected mutation in hemL | |
| JK290 | BW25113 argC::kan | 54 | |
| JK293a and JK293b | BW25113 proA* argC::kan | 2 isolates obtained from JK290 by gene gorging | This work |
| JK306 | JK293a(G259542A) | Contains M1 (−30G→A) in −35 region of proBA promoter | This work |
| JK311 | JK293a(C259567T) | Contains M2 (−5C→T) in −10 region of proBA promoter | This work |
| JK313a and JK313b | JK293a(A260655T) | 2 isolates in which M3 (1044A→T) was inserted in proB in JK293a | This work |
TABLE 2.
Plasmids used in this study
| Plasmid | Description and/or use | Source or reference |
|---|---|---|
| pACYC184 | Amplification of cat cassette | New England Biolabs |
| pSMART-HCAmp | High-copy-no. cloning vector; Ampr | Lucigen |
| pACBSR | Gene-gorging plasmid; contains gene encoding I-SceI endonuclease and λ red recombinase genes under control of the PBAD promoter; temp-sensitive origin of replication; Cmr | 55 |
| pK-HT | Mutagenesis plasmid; contains gene encoding I-SceI endonuclease under control of the Ptet promoter and λ red recombinase genes under control of the Prha promoter; Ampr | 56 |
| pKDTS | Mutagenesis plasmid; contains gene encoding I-SceI endonuclease under control of the PtetA promoter and λ red recombinase genes under control of the PBAD promoter; temp-sensitive origin of replication; Ampr | 9 |
Strain construction.
Construction of the ΔargC proA* strain and ΔargC proA* strains containing the M1, M2, and M3 point mutations is described in the supplemental material.
Adaptation of the ΔargC proA* strain on M9/glucose.
Individual colonies of JK293b obtained from LB/Kan plates were washed 3 times with sterile PBS and resuspended in 100 μl sterile phosphate-buffered saline (PBS). Aliquots of each cell suspension were diluted and plated on M9/glucose at 5,000 to 10,000 CFU per plate. Serial dilutions were also plated onto LB in triplicate for colony enumeration. The M9/glucose plates were incubated for 5 days at 37°C and then for 1 to 3 additional days at room temperature (to prevent the plates from drying out) before larger colonies were selected for analysis. Large colonies (0.5 to >1 mm in diameter) were picked from a background of smaller colonies (0.1 to 0.5 mm) and suspended in 10 μl sterile water. (Sterile water rather than PBS was used because the suspensions were subsequently used for PCR, and it was preferable to avoid introduction of extra salts.) Aliquots (0.5 to 1 μl) were streaked onto LB/Kan plates and incubated overnight at 37°C. The remainder of each suspension was diluted with 50 μl sterile water and heated to 98°C for 10 min before being stored at 4°C for the melt analysis of mismatch amplification assay (Melt-MAMA) (6) and quantitative PCR (qPCR) analysis. Individual colonies from each LB/Kan isolation plate (4 clones per mutant) were suspended in 10 μl PBS and used to inoculate individual wells of 96-well blocks containing 0.5 ml LB/Kan. After overnight growth at 37°C with shaking, 50 μl dimethyl sulfoxide (DMSO) was added to make freezer stocks.
Longer-term adaptation was carried out for four strains in which increases in the copy number of proA* had been detected in order to allow further increases in copy number. Freezer stocks of strains JK293b A1, A2, A3, and A4 were streaked onto M9/glucose plates and grown at 37°C until the colonies had grown to ∼0.5 to 1 mm in diameter (3 to 5 days). Five individual colonies of each strain were suspended in 300 μl sterile PBS and inoculated into 5 ml M9/glucose to give an initial optical density at 600 nm (OD600) of 0.0005. Cultures were incubated at 37°C with shaking at 225 rpm. Five serial transfers were performed when cultures reached mid-log phase. At each transfer, the cells were diluted into 5 ml of fresh M9/glucose to obtain an initial OD600 of 0.0005. At each transfer, an aliquot of cells was used to make freezer stocks by adding 10% DMSO, and a second aliquot (0.5 to 2 ml) was harvested, resuspended in 100 μl sterile water, and heated to 100°C for 10 min for qPCR determination of proA* copy number.
Determination of the number of cells in colonies.
An individual colony of the JK293b strain was washed 5 times and resuspended in 100 μl sterile PBS before being streaked onto plates containing M9/glucose. Plates were grown at 37°C for 6 days until the colonies were approximately 0.5 mm in diameter. Agar plugs of 5 individual colonies were suspended in 100 μl sterile PBS, and serial dilutions were spread onto LB/Kan plates to enable enumeration of colonies.
Illumina genome sequencing.
Procedures for construction of libraries, whole-genome resequencing, and sequence analysis are described in the supplemental material.
Screening for point mutations M1, M2, and M3.
Colonies containing M1, M2, and M3 were identified using the Melt-MAMA single nucleotide polymorphism (SNP) detection method (6). In this assay, the primer that anneals to the mutant sequence has an appended GC-rich tail, and the primer that anneals to the wild-type sequence does not (see Table S3 in the supplemental material). Consequently the melting temperature of amplicons containing a point mutation is higher than that of wild-type amplicons. Regions surrounding the point mutation were amplified using a common primer and a primer including either the point mutation or the corresponding wild-type base at the 3′ end. An additional mismatch was added to the latter primers at the second or third position from the 3′ end as described by Birdsell et al. (6). Primer sets were validated using PCR products containing each point mutation or the wild-type sequence as the template to ensure the absence of interference from primer-dimers, as well as good efficiency and discrimination between fragments with known point mutations.
Colonies picked from M9/glucose plates after adaptation were tested for the presence of all three point mutations. Melt-MAMA was performed in reaction mixtures containing 2 μl boiled cell suspensions, 200 nM (each) common, mutant, and wild-type primers, and 10 μl Fast SYBR green master mix (Applied Biosystems) in a total volume of 20 μl. Cycling was carried out in a Step One real-time PCR system as follows: 95°C for 20 s followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. A melt curve was performed immediately following (95°C for 15 s, 60°C for 60 s, increasing the temperature by 0.3°C to 95°C). Figure S1 in the supplemental material shows the melt curves obtained using the wild-type and mutant primers for M1, M2, and M3. The melting temperatures of each amplicon are listed in Table S3 in the supplemental material.
Amplicons from many colonies exhibited melt curves that indicated the presence of both wild-type and mutant sequences (either M1, M2, or M3, but never more than one of these), likely due to the presence of both wild-type and mutant cells in the colony or inadvertent picking of an overlapping wild-type colony. In these cases, Melt-MAMA was repeated using colonies obtained from frozen stocks of isolated subclones. In most cases, at least one subclone contained the point mutation.
Determination of proA* copy number.
The copy number of proA* was determined by qPCR using gyrB and icd, which are expected to be present in one copy each, as internal reference genes. Primer sets (see Table S4 in the supplemental material) were validated for qPCR using purified genomic DNA as well as boiled cell suspensions. The excellent correlation between copy numbers measured using boiled cell suspensions and purified genomic DNA (see Fig. S2 in the supplemental material) enabled us to screen large numbers of colonies using boiled cell suspensions. Details of the qPCR procedure are described in the supplemental material.
Determination of the boundaries of amplified regions.
The sizes of amplified regions were determined by PCR using primers oriented in divergent directions along the genome. A primer pair oriented in divergent directions will not result in amplification of a PCR fragment from genomic DNA in the absence of a duplication or amplification. However, annealing of such a primer pair in the context of a duplication/amplification will result in two primers oriented toward each other across the boundary of the amplified region and consequently will allow amplification of a PCR fragment. To diagnose the lengths of amplified regions in ΔargC proA* strains containing amplifications, primer K12-273095-F, which anneals immediately before the IS5A insertion element (right-facing blue arrowhead in Fig. 4), was used in PCRs with a series of primers (left-facing blue arrowheads in Fig. 4) oriented in the reverse direction (see Table S2 in the supplemental material). PCR mixtures contained 2 μM primer K12-273095-F, 2 μM upstream reverse primer, 0.5 μl boiled colony suspension, and 1× Q5 high-fidelity master mix (NEB) in a total volume of 10 μl. The cycling conditions were as follows: 98°C for 30 s, followed by 30 cycles of 98°C for 10 s, 67°C for 15 s, and 72°C for 2 min, and then 72°C for 5 min. PCR products were obtained using the following upstream primers: A1, primer K12-253037-R (∼1,800-bp product); A2, primer K12-259497-R (∼1,200-bp product); A3, primer K12-242970-R (∼5,500-bp product); and A4, primer K12-242970-R (∼1,000-bp product). PCRs resulting in the amplicons listed above were scaled up to a total volume of 100 μl. The PCR products were purified by agarose gel electrophoresis and extracted and cleaned up using QIAquick columns (Qiagen). Purified products were sent to Macrogen for Sanger sequencing using primer K12-274-057-F (blue arrowhead upstream of the orange arrowhead in Fig. 4).
FIG 4.

Extent of segmental amplifications in four strains derived from the ΔargC proA* strain. proB and proA* are highlighted in red. Blue arrowheads indicate sites and direction of primers used for identification of the size of amplified segments. The orange arrowhead indicates the site of the primer used to sequence PCR amplicons to identify amplification boundaries. The extent of A4 is indicated in dashed lines because the exact boundaries could not be determined.
Measurement of growth rates.
Aliquots of freezer stocks were streaked onto M9/glucose plates and grown overnight at 37°C. Single colonies were inoculated into 2 ml M9/glucose and grown to early log phase at 37°C. Cells (0.5 ml) were harvested by centrifugation at 16,000 × g for 1 min, washed 5 times with 0.5 ml PBS, and resuspended in 0.5 ml PBS at room temperature. The cells were diluted to an OD600 of 0.01 in M9/glucose, and a 10-μl aliquot was inoculated into each of three wells containing 90 μl M9/glucose in the interior of a 96-well plate to give an initial OD600 of 0.001. Two peripheral rows of wells were filled with 200 to 300 μl water to slow evaporation. This procedure resulted in only a 10% loss in volume over 7 days. The plates were incubated in a Varioskan (Thermo) plate reader at 37°C with shaking every 5 min. The absorbance at 600 nm was measured every 20 min for up to 120 h.
The data were smoothed by averaging the absorbance value over 5 time points. Outlier points for which the difference between the absorbance and the averaged absorbance was greater than the standard deviation over the averaged points were replaced by the previous averaged absorbance value. The baseline absorbance for each well (the average over several smoothed data points before growth) was subtracted from each point of the smoothed growth curve, and 0.001 (the expected initial OD600 resulting from 1:10 dilution of a culture with an OD600 of 0.01) was added to obtain the absorbance values used to fit the data. Growth parameters (maximum specific growth rate [μmax], lag time [λ], and maximum growth [Amax]) were estimated by nonlinear regression using the modified Gompertz equation (7). Nonlinear least-squares regression was performed in Excel using the Solver feature.
Analysis of transcription levels by RT-qPCR.
Growth of starter cultures from freezer stocks was carried out as described above. Culture tubes containing 4.9 ml M9/glucose were inoculated with 100 μl cells at an OD of 0.05 to give an initial OD600 of 0.001. Triplicate cultures were grown at 37°C with shaking at 225 rpm to mid-log phase (OD600 of 0.2 to 0.4, 1 to 5 days). Cells (0.5 ml) were harvested by centrifugation at 16,000 × g and 4°C for 1 min and treated with RNAprotect (Qiagen) before storage at −68°C. Frozen pellets were thawed, and the RNA was purified using RNeasy spin columns (Qiagen) according to the manufacturer's protocol. An additional DNase treatment performed using Turbo DNase (Life Technologies) according to the manufacturer's protocol was followed by purification with RNA Clean & Concentrator-5 columns (Zymo Research). RNA concentration was determined using the Qubit RNA HS assay kit with a Qubit 3.0 fluorometer (Life Technologies). Reverse transcription (RT) was performed with 300 ng RNA using Superscript VILO master mix (Invitrogen) according to the manufacturer's protocol, except that the 42°C incubation step was increased to 2 h. Aliquots of the cDNA obtained from these reactions were stored at −20°C. qPCR was carried out as described in the supplemental material.
Identification of the transcription start site for proA*.
RNA was prepared as described above. Reverse transcription, cDNA purification, and dA-tailing were performed using a 5′/3′ random amplification of cDNA ends (RACE) kit, 2nd Generation (Roche), and a High Pure PCR product purification kit (Roche). proA cDNA was generated using primer r1358. 5′ cDNA ends were amplified with 1× OneTaq Hot Start master mix and standard buffer (NEB). Primers rAM056 and rAM057 were used for the first and second rounds of amplification, respectively. After two rounds of amplification, the reaction mixture was run on a 1% agarose gel, and the expected ∼700-bp fragment was purified using the GeneJET gel extraction kit (Thermo). Purified DNA fragments were cloned into a pUC19 vector using Gibson Assembly master mix (NEB). The inserted DNA was sequenced using Sanger sequencing in order to identify the transcription start site upstream of proA*. Sequences were aligned to the proBA operon using CLC Main Workbench.
Analysis of ProA and ProB levels using mass spectrometry.
Freezer stocks of the ΔargC proA* strain (JK293a) and the constructed M3 strain (JK313a) were streaked onto LB/Kan plates. Single colonies were inoculated into 2 ml LB/Kan and grown to early log phase. Cells were harvested by centrifugation at 16,000 × g at room temperature for 1 min, washed 5 times with sterile PBS, and suspended in 0.5 ml PBS. Suspensions were diluted to an OD600 of 0.5 in M9/glucose. The suspended cells were inoculated into 30 ml M9/glucose in 150-ml flasks to give an initial OD600 of 0.001. The cultures were grown at 37°C with shaking to an OD600 of 0.2 to 0.4, at which time 10-ml aliquots were harvested by centrifugation at 5,000 × g at 4°C for 8 min. Cell pellets were frozen in liquid nitrogen and stored at −68°C prior to analysis.
For proteomic analyses, total protein was extracted from PBS-washed cell pellets (107 to ∼108 cells for ∼200 μg total protein) by a slight modification of the procedure described by Wisniewski et al. (8) using 4% (wt/vol) sodium dodecyl sulfate (SDS) in 100 mM Tris-HCl (pH 8.5). Soluble protein was treated with the reducing agent Tris(2-carboxyethyl)phosphine hydrochloride (TCEP [10 mM]) for 30 min with rocking at room temperature, followed by 10-fold dilution with 8 M urea in 100 mM Tris-HCl (pH 8.5). At that point, iodoacetamide was added to a concentration of 25 mM to alkylate cysteine residues. The reaction mixture was incubated in the dark for 30 min with rocking at room temperature. The denatured, reduced, and alkylated proteins were concentrated in an Amicon Ultra-0.5 centrifugal filter device (Millipore) with a nominal molecular weight limit (NMWL) of 30,000 and washed three times with 8 M urea in 100 mM Tris-HCl (pH 8.5) to remove SDS, TCEP, and iodoacetamide and then three times with 2 M urea in 100 mM Tris-HCl (pH 8.5) to reduce the urea concentration. The solution was incubated in the centrifugal filter device with lysyl endopeptidase (Wako) at a ratio of approximately 1:50 protein to protease with rocking for 4 h at room temperature. Trypsin (Promega) was then added at a ratio of approximately 1:50 protein to protease, and incubation with rocking at room temperature was continued overnight. The centrifugal filter device was subjected to centrifugation at 14,000 × g for 10 min at 25°C. Peptides in the filtrate were passed through a C18 spin column (Pierce) to remove urea and Tris-HCl. The peptide mixture was evaporated to dryness in a Speed-Vac and resuspended in 0.1% (vol/vol) formic acid in high-performance liquid chromatography (HPLC)-grade water. Aliquots containing 2 μg of peptides were injected onto a C18 ultraperformance liquid chromatography (UPLC) column (Waters nanoACQUITY 1.7-μm BEH130, 75 μm by 250 mm). The column was eluted with a gradient from 3% to 33% acetonitrile at 0.3 μl/minute over a period of 5 h. Eluted peptides were detected using an LTQ-Orbitrap (Thermo Scientific). Spectra were searched against an E. coli database using Mascot (Matrix Sciences) to identify peaks corresponding to peptides from ProA and ProB.
RESULTS
A synonymous mutation in proB enhances growth rate during short-term adaptation of a ΔargC proA* strain on M9/glucose.
A ΔargC::kan proA* strain of E. coli BW25113 (GS325) was constructed by replacing proA in a ΔargC strain with proA*, which encodes E383A ProA (Table 1). This strain was cultured for 6 days in 100 ml of M9/glucose containing kanamycin. Aliquots of the culture were then spread onto plates containing M9/glucose and kanamycin. The proBA* operon was amplified by PCR from colonies that were visibly larger than most. Sanger sequencing revealed one of three mutations in most of the large colonies (Fig. 2). Two point mutations in the promoter of the proBA* operon (M1 and M2) had previously been found to increase the growth rate of a ΔargC::kan proA* strain (3). A third point mutation (A1044T [M3]) was found in the 3′ end of proB (which encodes glutamyl kinase) 72 bp upstream of the start codon for proA. M3 is a synonymous change of a GCA codon to a GCU codon; both codons specify Ala. However, we detected no point mutations in the proBA* operon in several large colonies. Resequencing the genomes of two of these strains revealed massive amplifications of a large region of the genome surrounding the proBA* operon (see Fig. S3 in the supplemental material). Unfortunately, the resequencing data revealed that the GS325 ΔargC::kan proA* strain used for these initial adaptation experiments carried an unrecognized mutation in hemL. Comparison of the growth rates of GS325 and two newly constructed strains (JK293a and JK293b) in which argC had been deleted and the mutation changing Glu383 to Ala had been introduced into proA revealed that the hemL mutation had an adverse effect on growth on M9/glucose. Consequently all further adaptation experiments were carried out using the newly constructed strains.
FIG 2.
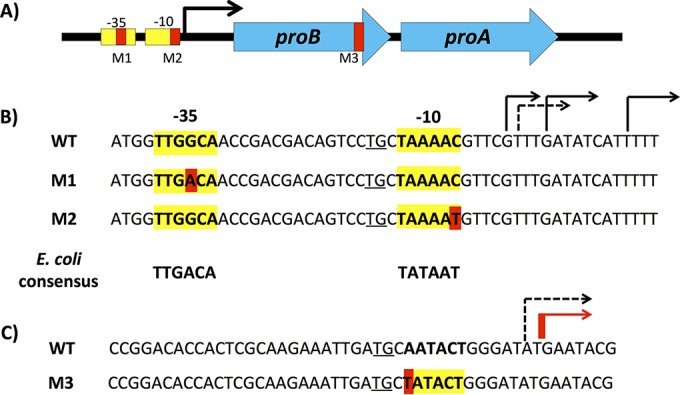
Mutations identified in unusually large colonies of E. coli ΔargC proA* cells after growth in M9/glucose for 6 days followed by plating on M9/glucose. (A) Locations of point mutations in the proBA* operon. (B) Sequences surrounding point mutations M1 and M2 in the promoter of the proBA operon. The E. coli consensus sequence was defined in references 51 and 52. The ex-10 TG element is underlined. The dashed line indicates the transcription start site identified by Thomason et al. (24), and the black lines indicate the transcription start sites identified by Cho et al. (53). WT, wild type. (C) Region in the 3′ end of proB that encodes a weak promoter for proA. The red line indicates the transcription start site identified by 5′-RACE and the dashed line indicates the transcription start site identified by Thomason et al. (24).
Effects of the point mutations M1, M2, and M3 on growth rate.
We introduced M1, M2, and M3 individually into the ΔargC proA* strain JK293a by genome editing (9) and measured the growth rate for each strain as well as the wild-type BW25113 strain and the parental ΔargC proA* strain JK293 in M9/glucose in the absence and presence of proline and arginine. M1, M2, and M3 increase the growth rate in M9/glucose by 4.7-, 3.2-, and 5.1-fold, respectively, relative to the parental strain (Fig. 3). Each strain was able to grow at the wild-type rate only when both proline and arginine were supplied in the medium, suggesting that growth is limited in each case by a poor ability to produce both proline and arginine. Notably, the ability to make arginine seems to be more impaired than the ability to make proline; addition of arginine to the medium has a greater effect on growth rate than addition of proline. This difference is less marked in the strain carrying M3.
FIG 3.
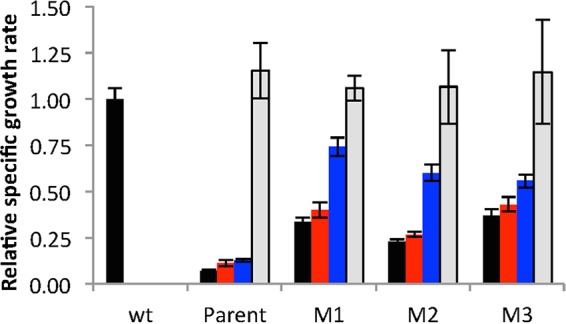
Effect of point mutations M1, M2, and M3 on the growth rate of ΔargC proA* E. coli cells. Black bars, M9/glucose; red bars, M9/glucose plus 0.4 mM proline; blue bars, M9/glucose plus 5.2 mM arginine; gray bars, M9/glucose plus 0.4 mM proline and 5.2 mM arginine. wt, wild type. Error bars represent standard deviations from 3 replicates.
Amplification of large genomic regions surrounding the proBA* operon increases growth rate.
When the ΔargC proA* strain was plated on M9/glucose, we observed a few anomalously large colonies (0.5 to >1 mm in diameter) against a background of smaller colonies. Colonies that were 0.5 mm in diameter contained (8.2 ± 1.8) × 106 cells, and thus represented approximately 23 generations of growth from a single founding cell. Given the known frequency of point mutations in E. coli (2.2 × 10−10 per nucleotide per generation in mutation accumulation strains [10]), it is unlikely that the approximately 107 cells spread on each plate included cells with preexisting point mutations. The frequency of gene duplication has not been measured in E. coli, but at most sites in Salmonella enterica, it is in the range of 10−5 to 10−4 (11). Assuming a similar range of duplication frequencies in E. coli, 100 to 1,000 of the 107 cells might have had a preexisting duplication of the region surrounding the proBA* operon.
Screening of 77 large colonies using the Melt-MAMA SNP detection assay revealed that most contained one of the three point mutations shown in Fig. 2 (Table 3). The large colonies that lacked one of these point mutations contained an amplification of a large region surrounding the proBA* operon. It may seem surprising that we observed more large colonies with point mutations than with segmental amplifications given that the frequency of gene duplication is much higher than the frequency of point mutations. It is important to recognize that the frequency of large colonies bearing a particular genetic change is not equivalent to the frequency at which the genetic change occurs. Detection of a genetic change depends upon the frequency at which that change occurs, the time at which it occurs, and its effect on growth rate. Segmental duplication, the prerequisite for segmental amplification, would be expected to result in less than a 2-fold increase in growth rate due to the burden of maintaining the extra genetic material as well as producing elevated levels of proteins from the duplicated genes. In contrast, point mutations M1, M2, and M3 increase the growth rate by 3.2- to 5.1-fold. Thus, the preponderance of the point mutations observed in our experiments is likely due to their large effects on growth rates, which result in visibly larger colonies.
TABLE 3.
Frequencies of large colonies containing previously identified point mutations or segmental amplifications containing the proBA* operon obtained after growth of the ΔargC proA* strain on M9/glucose plates
| No. of colonies screened (×105) | No. of mutants found | Colonies with: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 |
M2 |
M3 |
Amplification |
||||||
| No. | Frequency (×10−5) | No. | Frequency (×10−5) | No. | Frequency (×10−5) | No. | Frequency (×10−5) | ||
| 1.4 ± 0.4 | 25 | 3 | 2.2 | 19 | 13.7 | 1 | 0.7 | 2 | 1.4 |
| 2.1 ± 0.7 | 23 | 2 | 1.0 | 18 | 8.6 | 3 | 1.4 | 0 | 0 |
| 1.1 ± 0.5 | 22 | 3 | 2.8 | 13 | 11.9 | 2 | 1.8 | 4 | 3.7 |
| Avg | 2.0 ± 0.9 | 11.4 ± 2.6 | 1.3 ± 0.6 | 1.7 ± 1.9 | |||||
We identified the extent of the amplified region in four strains: JK293b A1 to A4 (Fig. 4). The amplified regions of A1, A2, and A3 all end in an IS5A site, but they start at variable positions, resulting in a range of sizes from 15 to 35 kb. The boundaries of the amplified region in A4 are approximate, as PCRs using primers that span the expected junction region are inefficient and clean sequence reads cannot be obtained, suggesting that rearrangements within the tandem array have led to heterogeneity in the junctions between segments. Table S6 in the supplemental material shows the precise junction regions for A1, A2, and A3. Table S7 in the supplemental material lists the genes and operons included in the four characterized segmental amplifications.
We examined the effect of copy number on growth rate using strains containing the four segmental amplifications diagrammed in Fig. 4. Each strain was streaked onto M9/glucose plates and grown for 5 days at 37°C. Individual colonies were used to inoculate 5-ml M9/glucose cultures. At mid-log phase, each strain was diluted into fresh M9/glucose to give an OD600 of 0.0005. Five serial transfers at mid-log phase were performed (∼35 generations of growth). Aliquots of each culture were frozen at intervals to capture intermediate stages in the amplification process.
Aliquots of each frozen culture (i.e., multiple time points for each of the four amplification strains) were spread onto plates containing M9/glucose. Three colonies from each plate were inoculated into M9/glucose. When the cultures reached mid-log phase, the cells were harvested and washed. One portion of the cells was inoculated into M9/glucose medium for analysis of growth rate, and the other was used to determine copy number of the proBA* operon using qPCR. Figure 5 shows that growth rate increases with the copy number of the proBA* operon in each case and levels off at high copy numbers. None of the strains achieved a growth rate equivalent to that of the wild-type strain under the same conditions.
FIG 5.
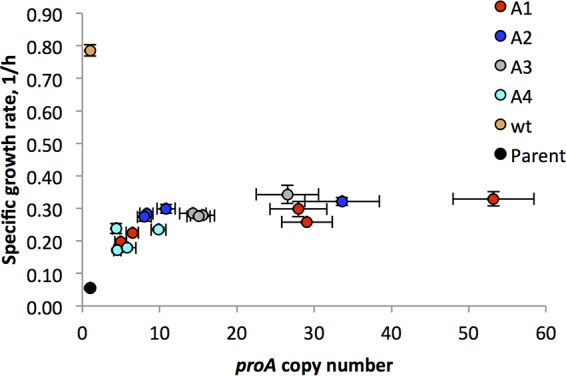
Specific growth rate for cells harboring segmental amplifications encompassing the proBA* operon as a function of copy number. Designations of amplification strains correspond to those shown in Fig. 4. Error bars represent standard deviations from 3 replicates.
Effects of the point mutations M1, M2, and M3 and gene amplification on expression of proB and proA*.
We assessed the level of expression of proB and proA* in the wild-type BW25113 strain, the parental ΔargC proA* strain, and strains containing M1, M2, and M3 using RT-qPCR (Fig. 6). Expression of proB and proA* in the parental strain was elevated relative to the wild-type strain by 1.5- and 2.0-fold, respectively. M1 is the most effective promoter mutation, increasing expression of proB and proA* by more than by 8-fold relative to the parental strain. M2 increases expression of proB and proA* by more than 4-fold. In contrast, M3 increases expression of proA* by 9.5-fold relative to the parental strain, but expression of proB is actually slightly decreased.
FIG 6.
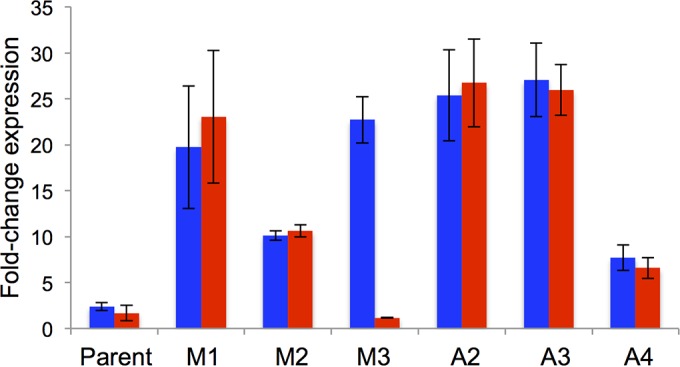
Levels of expression of proA (blue) and proB (red) in the parental ΔargC proA* strain and in strains containing M1, M2, M3 or segmental amplifications (A2, 12.8-fold; A3, 22-fold; A4, 5.2-fold) relative to their expression in wild-type E. coli BW25113. Error bars represent standard deviations from three biological replicates. (cysG and idnT were used as internal references.) Error bars represent 95% confidence intervals from 3 replicates.
We also analyzed the expression and copy number of proB and proA* in three strains harboring segmental amplifications. Clones of strains A2 and A3 that had 13 and 22 copies of the proBA* operon, respectively, showed comparable elevations of about 13-fold in expression of proB and proA* relative to the parental strain. A clone of strain A4 that had 5 copies of the proBA* operon showed an elevation in expression of proB and proA* of about 3.5-fold relative to the parental strain.
Synonymous mutation M3 strengthens an inefficient promoter in proB.
The qRT-PCR results shown in Fig. 6 suggest that M3 creates a new transcription start site in proB. The location of this transcription start site was mapped by 5′-RACE (Fig. 2C). Because the method uses a dA-tailing step at the 5′ end of the cDNA generated by reverse transcription of the mRNA of interest, a poly(T) sequence ultimately precedes the coding region. Consequently, it is not possible to tell whether transcription starts at the T or the G indicated in Fig. 2C. Interestingly, transcripts with the same transcription start site as well as transcripts that began 3 to 8 bases upstream of M3 were identified in the parental ΔargC proA* strain.
Synonymous mutation M3 increases production of ProA* by 2-fold.
Given the large discrepancy between levels of expression of proB and proA* in the strain containing M3, it was of interest to determine the effect of M3 on the levels of the proteins themselves. We examined the effect of M3 on the levels of ProA and ProB in the ΔargC proA* strain by label-free mass spectrometry analysis. We detected a total of 31 unique peptides in tryptic digests of cell extracts that could be unambiguously assigned to ProA* (69.8% coverage) and 6 peptides that could be unambiguously assigned to ProB (21.5% coverage) in three replicate runs. Figure 7 compares the counts of peptides assigned to ProB and ProA* in tryptic digests in extracts of cells grown in M9/glucose. The level of ProB was not affected by M3, but the level of ProA* was increased by 2-fold. (Note that this type of analysis allows us to compare the levels of individual proteins in two different samples but does not indicate anything about the absolute levels of ProA and ProB, as the peptide counts depend upon the sequence of the peptide as well as its abundance.)
FIG 7.
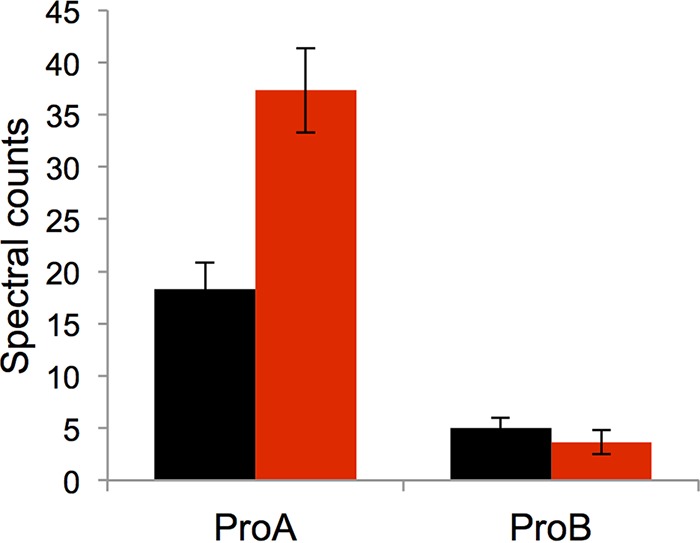
Spectral counts from ProA* and ProB peptides in the ΔargC proA* strain (black) and in JK313, a ΔargC proA* strain in which M3 had been introduced (red). Data were obtained from 3 biological replicates. The error bars represent standard deviations of the means.
Mutations that increase expression of proA* do not preclude segmental amplification.
Because neither M1, M2, nor M3 increased the growth rate of the ΔargC proA* strain to wild-type levels, we continued to grow each strain in M9/glucose to determine whether segmental duplication/amplification would occur and, if so, to what level. By 200 generations, segmental amplification occurred in the populations carrying M2 and M3, which accumulated 3.8 and 14.4 copies of proA*, respectively. No amplification was observed in the strain carrying M1 after 200 generations. However, by 1,300 generations, the copy number of proA* in the population had increased to 4.
DISCUSSION
ΔargC proA* E. coli has a severe growth defect relative to wild-type E. coli due to a poor ability to synthesize both proline and arginine. Thus, there is strong selective pressure for evolution of strategies to ameliorate the growth restriction caused by the inefficiency of the bifunctional ProA*. In theory, growth might be improved by a mutation that increases the efficiency of one or both activities or increases expression of the inefficient weak-link enzyme. It appears that the latter strategy is most accessible; we identified four genetic changes that increase expression of proA* by three different mechanisms.
Segmental amplifications that include the proBA* operon occur readily in the genome of the ΔargC proA* strain when the cells are grown either on M9/glucose plates or in liquid M9/glucose. After approximately 23 generations of growth on plates, colonies with up to 11 copies of the proBA* operon were identified. Further growth in liquid M9/glucose cultures resulted in amplification up to 53 copies. The leveling off of the growth rate observed with high numbers of copies of proA* indicates that the benefit of extra copies eventually diminishes due to the costs of maintaining a large number of copies of the region surrounding the proBA* operon and of overexpressing genes present in multiple copies.
We identified the boundaries of the amplified segment in four strains. In each case, the amplified segment ended at an IS5A element, suggesting that this site is a hot spot for the recombination event that generates the initial duplication. The observation that gene amplification increases the growth rate of the ΔargC proA*strain is not surprising. Gene amplifications are found in many circumstances when an increase in gene dosage improves growth rate. Gene amplifications contribute to loss of growth control and to resistance to therapeutic agents in tumors (12–15), to the emergence of antibiotic resistance in pathogens (16, 17), and to improved growth of S. enterica in which a single inefficient enzyme serves functions in both histidine and tryptophan biosynthesis (18). Duplications that include genes encoding permeases increase the growth rate of Salmonella enterica serovar Typhimurium under carbon-limiting conditions (19), and amplification of a region containing the clc cluster of genes that enable degradation of chlorocatechol allows a strain of Pseudomonas putida to grow on chlorobenzene as the sole carbon source (20).
Although an increase in the number of copies of the proBA* operon led to an increase in growth rate, neither expression of the proBA* operon nor growth rate was directly proportional to copy number. Expression was elevated by 3.5-fold when 5 copies were present but only 13-fold when 22 copies were present. The diminishing return in expression gained by additional copies may be due to a shortage of RNA polymerase molecules to transcribe the additional copies of the proBA* operon. In E. coli, the number of free RNA polymerase molecules in a cell is less than the number of promoters (21). Consequently, the promoter of the proBA* operon must compete with hundreds of other promoters for a relatively small pool of RNA polymerase molecules. Thus, an increase in the number of promoters present in the genome would be expected to lead to a concomitant decrease in the availability of RNA polymerase molecules and thus a decrease in the level of transcription of each individual gene.
The growth rate of the ΔargC proA* strain was also improved substantially by mutations in the promoter of the proBA* operon. M1 changes the −35 region of the promoter to the consensus sequence for E. coli and increases expression of the proBA* operon by more than 8-fold. M2 affects the −10 region of the proBA promoter and increases expression of the proBA* operon by more than 4-fold.
M3 is the most intriguing of the point mutations we detected; this synonymous mutation in proB increases expression of proA* by 9.5-fold without increasing expression of proB. Figure 2 shows that M3 creates a region that differs from the E. coli −10 consensus region at only one position and is actually a better match than the −10 region of the wild-type proBA promoter. The region upstream of this new −10 region bears no resemblance to the consensus −35 region for E. coli promoters. However, the −35 region is not strictly required for transcription initiation; two promoters of the gal operon in E. coli lack the −35 element entirely, and the λ Pre promoter has a nearly unrecognizable −35 element with only one match to the six positions of the consensus −35 element (22, 23). Furthermore, the wild-type sequence has the ex-10 TG element upstream of the putative −10 region that can compensate for a poor or missing −35 element. Analysis of mRNA isolated from the ΔargC proA* strain containing M3 by 5′-RACE (rapid amplification of cDNA ends) indicates that there is indeed a transcript initiated 12 or 13 bp downstream of M3. We detected a similar transcript in the parental ΔargC proA* strain, as well. Thomason et al. also detected a transcript initiating at this point in E. coli MG1655 (24). Transcription from this cryptic promoter is evidently very weak, as the levels of expression of proB and proA* are indistinguishable in the ΔargC proA* strain and all other improved strains, except the one containing M3. Generation of a promoter by a synonymous mutation in an upstream gene is not unprecedented. A synonymous mutation in mabA, the first gene in the mabA-inhA operon in Mycobacterium tuberculosis, generates an alternative promoter for inhA. The consequent overexpression of inhA, which encodes the target for the clinically important drug isoniazid, leads to isoniazid resistance (25).
M3 selectively increases expression of proA* and is thus the most “precise” of the genetic changes we detected in that it accomplishes an increase in the level of the weak-link enzyme, ProA*, without a collateral increase in either the amount of DNA the cells must carry or the expression of other proteins. The increase in growth rate due to M3 is less than that due to massive amplifications of the region surrounding the proBA* operon. M3 increases the growth rate by 5.1-fold; amplification of the proBA* operon in strain A3 to 27 copies increased the growth rate by 6.2-fold, and amplification in strain A1 to 53 copies increased growth rate by 6-fold. These data suggest that additional expression of proA* might be achieved by segmental amplification in strains carrying M3 as well as in strains carrying mutations in the canonical promoter. Indeed, multiple copies of the proA* allele were detected after continued adaptation, although the extent of amplification was lower than that seen in the absence of M1, M2, and M3. Thus, occurrence of these mutations would slow, but not prevent, divergence of the gene encoding proA* to two genes encoding specialist enzymes for reduction of γ-glutamyl phosphate and N-acetyl glutamyl phosphate.
Because mRNA levels often do not correlate well with protein levels (26–28), we investigated the effect of M3 on the levels of ProA* and ProB using label-free quantitative proteomic analysis. Quantitation by this method is based upon the number of times peptides that can be assigned to an individual protein are detected in a tryptic digest of total proteins. The number of times a given peptide is detected is correlated with protein abundance. Further, more peptides are generally detectable from more abundant proteins. Because the number of spectral counts correlates with the abundance of the protein, the method can be used to compare protein abundances between biological samples that are prepared and processed in parallel (29–31). This method is widely accepted in the proteomics community and has been used to identify differences in protein levels in Shewanella oneidensis grown under oxic and anoxic conditions (32), the relative levels of 3,274 mouse proteins in the brain, heart, kidney, liver, lung, and placenta (28), and changes in protein levels in Mycobacterium tuberculosis during reactivation from a dormant state (33). We found that M3 results in a 2-fold increase in the level of ProA*, whereas the level of ProB is unaffected. Another example of adaptive mutations that adjust the relative levels of two enzymes in a metabolic pathway was reported by Chou et al. (34) using a system in which the endogenous genes for formaldehyde oxidation in Methylobacterium extorquens were removed and two genes encoding enzymes for a different formaldehyde oxidation pathway were expressed from a plasmid. In this case, the initial expression levels appeared to be too high, as adaptive mutations decreased expression of both enzymes. Notably, fitness was highest when single mutations or combinations of mutations decreased expression of the second enzyme in the pathway more than expression of the first enzyme in the pathway.
The discrepancy between the increase in proA* mRNA (9.5-fold) and the increase in ProA* (2-fold) caused by M3 is particularly interesting. There are two possible explanations for this phenomenon. First, translation of an upstream coding sequence is believed to increase the availability of ribosomes in the vicinity of the Shine-Dalgarno sequence of the downstream coding sequence (35, 36). Loss of the upstream coding sequence for ProB in the transcript that begins at the new transcriptional start site created by M3 will abrogate the possibility of translational coupling. Second, the Shine-Dalgarno sequence upstream of the start codon of proA* is in a different structural context in the proA* transcript initiated downstream of M3 and may not be as accessible for ribosome binding.
The 5-fold increase in growth rate when the level of ProA* is only doubled by M3 indicates that the growth rate of the ΔargC proA* strain is ultrasensitive to the concentration of ProA*. Ultrasensitivity is defined as a fractional change in a response, Y (growth rate in our case), that is larger than the fractional change in an input, X (the level of ProA* in our case), as shown by the following equation (4):
Ultrasensitivity can be caused by a number of mechanisms (4), including allosteric interactions in multimeric proteins, homo-multimerization of proteins, signal amplification cascades, positive-feedback loops, covalent modifications by antagonistic enzymes (e.g., kinases and phosphatases) operating near the zero-order regime, and cooperativity due to multiple covalent modifications. Most relevant to the effect of M3 on growth rate is ultrasensitivity caused by competition between a high-affinity ligand and a low-affinity ligand for binding to a macromolecule. In such cases, the low-affinity ligand has no opportunity to bind until the concentration of the macromolecule exceeds that of the high-affinity ligand (the “equivalence” point). Once that threshold is reached, an ultrasensitive response may occur if the concentration of the low-affinity ligand is higher than that of the macromolecule. The Hill coefficient of the response, which can be used as an approximate measure of the level of ultrasensitivity, varies, depending upon a number of physical parameters. Theoretical as well as experimental analyses suggest that high Hill coefficients are observed when (i) the low-affinity ligand is present at a concentration much greater than its KD (equilibrium dissociation constant), (ii) the affinities of the two ligands are very different, and (iii) the high-affinity ligand is more abundant than the low-affinity ligand. Ultrasensitive responses due to this molecular titration mechanism have been observed for phosphorylation of Wee1 by Cdk1 in the presence of other protein substrates (38), translation of mRNAs in the presence of microRNAs (39), the output of genetic circuits in which a transcriptional regulator is sequestered by another protein (40), and binding of transcription factors to promoters in the presence of decoy sites in the genome (41).
The ultrasensitivity revealed by the 5-fold increase in growth rate of the ΔargC proA* strain due to a 2-fold increase in the level of ProA* is likely due to a similar molecular titration mechanism in which two substrates with different affinities compete for the active site of ProA*. γ-Glutamyl phosphate is believed to be channeled from the active site of ProB (glutamyl kinase) to the active site of ProA (42–44) to prevent loss due to cyclization (45). Thus, the ProB–γ-glutamyl phosphate complex competes with N-acetyl glutamyl phosphate for the active site of ProA*. The ProB–γ-glutamyl phosphate complex is likely to have a higher affinity for ProA* than N-acetyl glutamyl phosphate. This expectation is supported by the data shown in Fig. 3. Strains containing M1 or M2, both of which overexpress the entire proBA* operon, seem to be primarily handicapped by a poor ability to make arginine; addition of arginine to the medium (blue bars) improves growth more than addition of proline (red bars). This difference is much less pronounced in the strain carrying M3, suggesting that M3 has a disproportionate effect on the synthesis of arginine. M3 increases the concentration of ProA* but not ProB. The resulting increase in the ratio of ProA* to ProB should improve the access of N-acetyl glutamyl phosphate to the active site of ProA*. If the concentration of N-acetyl glutamyl phosphate exceeds its KD for ProA* (a reasonable possibility since it would be expected to accumulate in the absence of ArgC), then reduction of N-acetyl glutamyl phosphate, and consequently growth rate, might well show ultrasensitivity with respect to the concentration of ProA*.
M3 is also interesting because it causes a polar effect on the expression of a downstream gene. Such effects have been recognized since the 1960s, but in most reported cases, polar effect mutations are detrimental, interfering in some way with proper transcription or translation of a downstream gene (46–50). The predominantly adverse effects of reported polar mutations may be due to previous selection for bases or codons that function well under typical growth conditions. The strong beneficial effect of M3 in this case occurs when the cells are forced to overcome a challenge posed by the need to recruit a promiscuous enzyme to serve a new function. This is a situation that must have arisen on many occasions during the diversification of life as microbes adapted to new environments. Polar-effect mutations such as M3 may also have played a role in the recruitment of proteins to play new regulatory or structural roles by increasing the level of a protein encoded by a downstream gene in an operon and thereby allowing it to interact with new binding partners. Thus, polar effect mutations may have played an unappreciated role in the evolution of microbial genomes.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00262-16.
REFERENCES
- 1.Copley SD. 2003. Enzymes with extra talents: moonlighting functions and catalytic promiscuity. Curr Opin Chem Biol 7:265–272. doi: 10.1016/S1367-5931(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 2.Khanal A, Yu McLoughlin S, Kershner JP, Copley SD. 2015. Differential effects of a mutation on the normal and promiscuous activities of orthologs: implications for natural and directed evolution. Mol Biol Evol 32:100–108. doi: 10.1093/molbev/msu271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu McLoughlin S, Copley SD. 2008. A compromise required by gene sharing enables survival: implications for evolution of new enzyme activities. Proc Natl Acad Sci U S A 105:13497–13502. doi: 10.1073/pnas.0804804105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q, Bhattacharya S, Andersen ME. 2013. Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open Biol 3:130031. doi: 10.1098/rsob.130031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birdsell DN, Pearson T, Price EP, Hornstra HM, Nera RD, Stone N, Gruendike J, Kaufman EL, Pettus AH, Hurbon AN, Buchhagen JL, Harms NJ, Chanturia G, Gyuranecz M, Wagner DM, Keim PS. 2012. Melt analysis of mismatch amplification mutation assays (Melt-MAMA): a functional study of a cost-effective SNP genotyping assay in bacterial models. PLoS One 7:e32866. doi: 10.1371/journal.pone.0032866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zwietering MH, Jongenburger I, Rombouts FM, van't Riet K. 1990. Modeling of the bacterial growth curve. Appl Environ Microbiol 56:1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Webb AM, Kershner JP, Blaskowski S, Copley SD. 2014. A versatile and highly efficient method for scarless genome editing in Escherichia coli and Salmonella enterica. BMC Biotechnol 14:84. doi: 10.1186/1472-6750-14-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A 109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson P, Roth J. 1981. Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc Natl Acad Sci U S A 78:3113–3117. doi: 10.1073/pnas.78.5.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui A, Ihara T, Suda H, Mikami H, Semba K. 2013. Gene amplification: mechanisms and involvement in cancer. Biomol Concepts 4:567–582. doi: 10.1515/bmc-2013-0026. [DOI] [PubMed] [Google Scholar]
- 13.Dahl F, Stenberg J, Fredriksson S, Welch K, Nilsson M, Bicknell D, Bodmer WF, Davis RW, Ji H. 2007. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci U S A 104:9387–9392. doi: 10.1073/pnas.0702165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang L, Fang JY, Xu J. 2016. Gastric cancer and gene copy number variation: emerging cancer drivers for targeted therapy. Oncogene 35:1475–1482. doi: 10.1038/onc.2015.209. [DOI] [PubMed] [Google Scholar]
- 15.Despierre E, Moisse M, Yesilyurt B, Sehouli J, Braicu I, Mahner S, Castillo-Tong DC, Zeillinger R, Lambrechts S, Leunen K, Amant F, Moerman P, Lambrechts D, Vergote I. 2014. Somatic copy number alterations predict response to platinum therapy in epithelial ovarian cancer. Gynecol Oncol 135:415–422. doi: 10.1016/j.ygyno.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Sandegren L, Andersson DI. 2009. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol 7:578–588. doi: 10.1038/nrmicro2174. [DOI] [PubMed] [Google Scholar]
- 17.Laehnemann D, Pena-Miller R, Rosenstiel P, Beardmore R, Jansen G, Schulenburg H. 2014. Genomics of rapid adaptation to antibiotics: convergent evolution and scalable sequence amplification. Genome Biol Evol 6:1287–1301. doi: 10.1093/gbe/evu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasvall J, Sun L, Roth JR, Andersson DI. 2012. Real-time evolution of new genes by innovation, amplification, and divergence. Science 338:384–387. doi: 10.1126/science.1226521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonti RV, Roth JR. 1989. Role of gene duplications in the adaptation of Salmonella typhimurium to growth on limiting carbon sources. Genetics 123:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravatn R, Studer S, Springael D, Zehnder AJ, van der Meer JR. 1998. Chromosomal integration, tandem amplification, and deamplification in Pseudomonas putida F1 of a 105-kilobase genetic element containing the chlorocatechol degradative genes from Pseudomonas sp. strain B13. J Bacteriol 180:4360–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patrick M, Dennis PP, Ehrenberg M, Bremer H. 2015. Free RNA polymerase in Escherichia coli. Biochimie 119:80–91. doi: 10.1016/j.biochi.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Keilty S, Rosenberg M. 1987. Constitutive function of a positively regulated promoter reveals new sequences essential for activity. J Biol Chem 262:6389–6395. [PubMed] [Google Scholar]
- 23.Busby S, Truelle N, Spassky A, Dreyfus M, Buc H. 1984. The selection and characterisation of two novel mutations in the overlapping promoters of the Escherichia coli galactose operon. Gene 28:201–209. doi: 10.1016/0378-1119(84)90257-9. [DOI] [PubMed] [Google Scholar]
- 24.Thomason MK, Bischler T, Eisenbart SK, Forstner KU, Zhang A, Herbig A, Nieselt K, Sharma CM, Storz G. 2015. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197:18–28. doi: 10.1128/JB.02096-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ando H, Miyoshi-Akiyama T, Watanabe S, Kirikae T. 2014. A silent mutation in mabA confers isoniazid resistance on Mycobacterium tuberculosis. Mol Microbiol 91:538–547. doi: 10.1111/mmi.12476. [DOI] [PubMed] [Google Scholar]
- 26.Griffin TJ, Gygi SP, Ideker T, Rist B, Eng J, Hood L, Aebersold R. 2002. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics 1:323–333. doi: 10.1074/mcp.M200001-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Gygi SP, Rochon Y, Franza BR, Aebersold R. 1999. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19:1720–1730. doi: 10.1128/MCB.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. 2006. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 29.Wong JW, Cagney G. 2010. An overview of label-free quantitation methods in proteomics by mass spectrometry. Methods Mol Biol 604:273–283. doi: 10.1007/978-1-60761-444-9_18. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. 2003. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal Chem 75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 31.Roy SM, Becker CH. 2007. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling. Methods Mol Biol 359:87–105. doi: 10.1007/978-1-59745-255-7_6. [DOI] [PubMed] [Google Scholar]
- 32.Fang R, Elias DA, Monroe ME, Shen Y, McIntosh M, Wang P, Goddard CD, Callister SJ, Moore RJ, Gorby YA, Adkins JN, Fredrickson JK, Lipton MS, Smith RD. 2006. Differential label-free quantitative proteomic analysis of Shewanella oneidensis cultured under aerobic and suboxic conditions by accurate mass and time tag approach. Mol Cell Proteomics 5:714–725. [DOI] [PubMed] [Google Scholar]
- 33.Gopinath V, Raghunandanan S, Gomez RL, Jose L, Surendran A, Ramachandran R, Pushparajan AR, Mundayoor S, Jaleel A, Kumar RA. 2015. Profiling the proteome of Mycobacterium tuberculosis during dormancy and reactivation. Mol Cell Proteomics 14:2160–2176. doi: 10.1074/mcp.M115.051151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou HH, Delaney NF, Draghi JA, Marx CJ. 2014. Mapping the fitness landscape of gene expression uncovers the cause of antagonism and sign epistasis between adaptive mutations. PLoS Genet 10:e1004149. doi: 10.1371/journal.pgen.1004149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levin-Karp A, Barenholz U, Bareia T, Dayagi M, Zelcbuch L, Antonovsky N, Noor E, Milo R. 2013. Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. ACS Synth Biol 2:327–336. doi: 10.1021/sb400002n. [DOI] [PubMed] [Google Scholar]
- 36.Govantes F, Andujar E, Santero E. 1998. Mechanism of translational coupling in the nifLA operon of Klebsiella pneumoniae. EMBO J 17:2368–2377. doi: 10.1093/emboj/17.8.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Kim SY, Ferrell JE Jr. 2007. Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell 128:1133–1145. doi: 10.1016/j.cell.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 39.Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43:854–859. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchler NE, Cross FR. 2009. Protein sequestration generates a flexible ultrasensitive response in a genetic network. Mol Syst Biol 5:272. doi: 10.1038/msb.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee TH, Maheshri N. 2012. A regulatory role for repeated decoy transcription factor binding sites in target gene expression. Mol Syst Biol 8:576. doi: 10.1038/msb.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seddon AP, Zhao KY, Meister A. 1989. Activation of glutamate by gamma-glutamate kinase: formation of gamma-cis-cycloglutamyl phosphate, an analog of gamma-glutamyl phosphate. J Biol Chem 264:11326–11335. [PubMed] [Google Scholar]
- 43.Marco-Marin C, Gil-Ortiz F, Perez-Arellano I, Cervera J, Fita I, Rubio V. 2007. A novel two-domain architecture within the amino acid kinase enzyme family revealed by the crystal structure of Escherichia coli glutamate 5-kinase. J Mol Biol 367:1431–1446. doi: 10.1016/j.jmb.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 44.Arentson BW, Sanyal N, Becker DF. 2012. Substrate channeling in proline metabolism. Front Biosci 17:375–388. doi: 10.2741/3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishnaswamy PR, Pamiljans V, Meister A. 1962. Studies on the mechanism of glutamine synthesis: evidence for the formation of enzyme-bound activated glutamic acid. J Biol Chem 237:2932–2940. [Google Scholar]
- 46.Yanofsky C, Ito J. 1966. Nonsense codons and polarity in the tryptophan operon. J Mol Biol 21:313–334. doi: 10.1016/0022-2836(66)90102-1. [DOI] [PubMed] [Google Scholar]
- 47.Fink GR, Martin RG. 1967. Translation and polarity in the histidine operon. II. Polarity in the histidine operon. J Mol Biol 30:97–107. [DOI] [PubMed] [Google Scholar]
- 48.Newton WA, Beckwith JR, Zipser D, Brenner S. 1965. Nonsense mutants and polarity in the lac operon of Escherichia coli. J Mol Biol 14:290–296. doi: 10.1016/S0022-2836(65)80250-9. [DOI] [PubMed] [Google Scholar]
- 49.Mendez J, Fernandez L, Menendez A, Reimundo P, Perez-Pascual D, Navais R, Guijarro JA. 2009. A chromosomally located traHIJKCLMN operon encoding a putative type IV secretion system is involved in the virulence of Yersinia ruckeri. Appl Environ Microbiol 75:937–945. doi: 10.1128/AEM.01377-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alifano P, Ciampi MS, Nappo AG, Bruni CB, Carlomagno MS. 1988. In vivo analysis of the mechanisms responsible for strong transcriptional polarity in a “sense” mutant within an intercistronic region. Cell 55:351–360. doi: 10.1016/0092-8674(88)90058-X. [DOI] [PubMed] [Google Scholar]
- 51.Hawley DK, McClure WR. 1983. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res 11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. 2007. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res 35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BO. 2009. The transcription unit architecture of the Escherichia coli genome. Nat Biotechnol 27:1043–1049. doi: 10.1038/nbt.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baba T, Ara T, Hasegawa M., Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herring CD, Glasner JD, Blattner FR. 2003. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311:153–163. [DOI] [PubMed] [Google Scholar]
- 56.Herring CD, Blattner FR. 2004. Conditional lethal amber mutations in essential Escherichia coli genes. J Bacteriol 186:2673–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.