

ABSTRACT
We tested pairwise combinations of classical base analog mutagens in Escherichia coli to study possible mutagen synergies. We examined the cytidine analogs zebularine (ZEB) and 5-azacytidine (5AZ), the adenine analog 2-aminopurine (2AP), and the uridine/thymidine analog 5-bromodeoxyuridine (5BrdU). We detected a striking synergy with the 2AP plus ZEB combination, resulting in hypermutability, a 35-fold increase in mutation frequency (to 53,000 × 10−8) in the rpoB gene over that with either mutagen alone. A weak synergy was also detected with 2AP plus 5AZ and with 5BrdU plus ZEB. The pairing of 2AP and 5BrdU resulted in suppression, lowering the mutation frequency of 5BrdU alone by 6.5-fold. Sequencing the mutations from the 2AP plus ZEB combination showed the predominance of two new hot spots for A·T→G·C transitions that are not well represented in either single mutagen spectrum, and one of which is not found even in the spectrum of a mismatch repair-deficient strain. The strong synergy between 2AP and ZEB could be explained by changes in the dinucleoside triphosphate (dNTP) pools.
IMPORTANCE Although mutagens have been widely studied, the mutagenic effects of combinations of mutagens have not been fully researched. Here, we show that certain pairwise combinations of base analog mutagens display synergy or suppression. In particular, the combination of 2-aminopurine and zebularine, analogs of adenine and cytidine, respectively, shows a 35-fold increased mutation frequency compared with that of either mutagen alone. Understanding the mechanism of synergy can lead to increased understanding of mutagenic processes. As combinations of base analogs are used in certain chemotherapy regimens, including those involving ZEB and 5AZ, these results indicate that testing the mutagenicity of all drug combinations is prudent.
INTRODUCTION
Mutagen-induced mutations have been the subject of intensive investigation for decades (e.g., see references 1 to 4; see reviews in references 5 to 7). However, there are far fewer studies of combinations of mutagens. We are studying possible synergies between mutagens in Escherichia coli and initially examined a set of base analog mutagens (Fig. 1): the cytidine analogs zebularine (ZEB) and 5-azacytidine (5AZ), the adenine analog 2-aminopurine (2AP), and the uridine/thymidine analog 5-bromodeoxyuridine (5BrdU). ZEB lacks the amino group of cytidine (8) and causes C·G→T·A changes in the rpoB/rifampin resistance (Rifr) system in E. coli (9). It is used in chemotherapy as a demethylating agent (8, 10, 11) to reverse the effects of gene-silenced tumor suppressor gene (12–15) and because the hydrated form is a potent inhibitor of cytidine deaminase (16). 5AZ, another cytidine analog that is used as a demethylating agent in chemotherapy (12, 13), possesses a unique mutagenic specificity, stimulating only C·G→G·C changes (17–19). 5AZ and ZEB have been used in combination in chemotherapy (20). 2-Aminopurine results principally in G·C→A·T and A·T→G·C changes (e.g., see references 18 and 21 to 25), as does 5BrdU (24–26). 2AP has been used to enhance the oncolytic activity of E1b-deleted adenovirus in hepatocellular carcinoma cells (27), and 5BrdU has been used in human glioma cell combination treatments with 1-nitrosourea and cisplatin (28). The initial studies of 1-nitrosourea and cisplatin focused on their direct pairing effects, but already in the early 1980s, it became apparent that for 2AP and 5BrdU, other processes, such as dinucleoside triphosphate (dNTP) pool changes, might be involved (e.g., see references 23 to 25 and references therein). It is now clear that there are several routes to mutagenesis other than direct mispairing that could emanate from the presence of some of the four compounds studied here. This includes partial induction of the SOS system in E. coli and related bacteria (6), the partial or complete saturation of the mismatch repair system (7, 26, 29, 30), and potential effects on altering dNTP pools, as the integrity of these pools is crucial to replication fidelity (e.g., see references 24, 25, and 31; see Discussion). Here, we report that strong synergy between certain pairs of these mutagens results in hypermutation. In particular, the combination of 2AP and ZEB creates a dramatic increase in mutation frequency that is 35-fold higher than that of either mutagen alone. The combinations of 2AP plus 5AZ and 5BrdU plus ZEB display weaker synergistic effects, and in contradistinction, the combination of 2AP and 5BrdU shows a suppressive effect on the mutation frequency of 5BrdU alone. Sequencing results for 2AP plus ZEB reveal two mutational hot spots not well represented in the spectrum of either 2AP or ZEB alone. We discuss possible explanations for this, including changes in the dNTP pools, and the potential applications and implications of these results.
FIG 1.
The structures of the four base analogs used in this study.
MATERIALS AND METHODS
E. coli strains.
The nucleotide diphosphate kinase (NDK)-deficient E. coli strain used here is from the Keio collection, described in Baba et al. (32), made from the starting strain BW25113 (33). This starting strain (lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 mutant) is used as the wild type (WT) in the experiments reported here, unless otherwise stated. The NDK-deficient mutant carries a complete deletion of the respective gene, with a kan insert in place of the gene.
Media.
The media (34) used were LB (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl) and minimal A [10.5 g/liter K2HPO4, 4.5 g/liter KH2PO4, 1 g/liter (NH4)2SO4, 0.5 g/liter sodium citrate·2H2O], supplemented with 10 ml of 20% glucose, 1 ml of 1 M MgSO4, and 0.5 ml of 1% thiamine hydrochloride (vitamin B1) per liter.
Growth conditions.
Unless otherwise stated, all genetic methods are as described by Miller (34). Overnight cultures containing different concentrations of a given mutagen were seeded with approximately 1 × 103 cells by inoculating 2-ml cultures with 50 μl of a 10−4 dilution of an overday culture. After 18 h of incubation at 37°C on a rotor at 50 rpm, the cells were plated on specific media.
Determination of mutant frequencies.
We inoculated 100 to 1,000 cells in a series of cultures of LB that were then grown for 18 h at 37°C with aeration, prior to plating on the appropriate media (LB plates with or without 100 μg/ml rifampin). The frequencies of Rifr mutants were determined as described previously (35, 36). Briefly, mutation frequency (f) was determined as the median frequency of mutation from a set of cultures (the number of cultures varied from 12 to 65), and the mutation rate (μ) was determined by the formula of Drake (37). The 95% confidence limits were determined according to the method of Dixon and Massey (38).
Chromosomal DNA isolation and sequencing.
The chromosomal DNA isolation and sequencing steps were carried out exactly as described by Garibyan et al. (35).
Chemicals.
Rifampin (Rif), 2-aminopurine (2AP), 5-azacytidine (5AZ), and 5-bromodeoxyuridine (5BrdU) were purchased from Sigma (St. Louis, MO). Zebularine (ZEB) was a gift from Victor Marquez.
RESULTS
Mutation frequencies resulting from mutagen pairs.
We used the rpoB/Rifr system (35, 39) to examine mutation frequencies resulting from different pairwise combinations of the four base analog mutagens 2AP, ZEB, 5AZ, and 5BrdU. We detected a strong synergy between 2AP and ZEB. Figure 2 displays the dramatic 35-fold increase in mutation frequency compared with that of either base analog alone at the concentrations used. We also detected weak synergies between 2AP and 5AZ (2.5-fold higher than the sum of each mutagen alone) and between 5BrdU and ZEB (2.7-fold) (Table 1). On the other hand, the pair 2AP plus 5BrdU gave a 6-fold lower mutation frequency than that of 5BrdU alone, indicating that 2AP is significantly suppressing the mutagenic effects of 5BrdU. The pair 5AZ plus ZEB gave a weak suppression effect, and the pair 5AZ plus 5BrdU gave no significant effect (Table 1).
FIG 2.
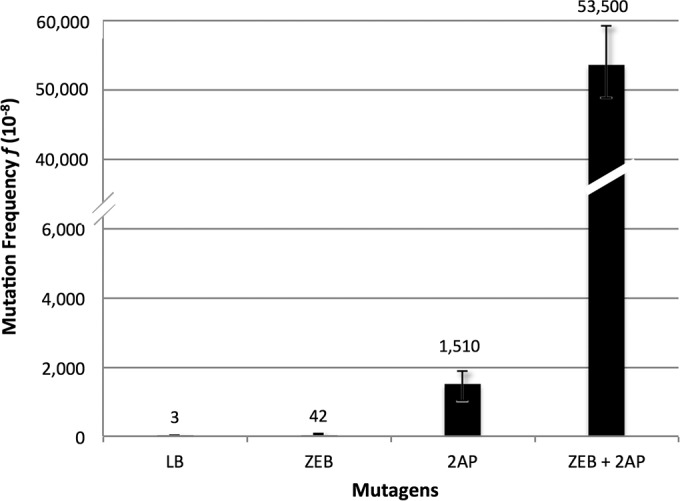
The frequencies of Rifr mutants found in cultures grown with either no mutagen (LB), 5 μg/ml ZEB, 20 μg/ml 2AP, or 5 μg/ml ZEB plus 500 μg/ml 2AP. Error bars represent 95% confidence intervals.
TABLE 1.
rpoB mutation frequencies in the BW25113 strain background with various mutagens
| Mutagen(s) | Concn (μg/ml) | No. of replicates | f (10−8) in rpoB (95% CI)a |
|---|---|---|---|
| None | 39 | 2.7 (1.8–4.2) | |
| 2AP | 500 | 62 | 1,505 (1,110–2,000) |
| ZEB | 5 | 58 | 42.1 (31–56.5) |
| 5BrdU | 5 | 42 | 12,950 (11,700–14,800) |
| 5AZ | 20 | 20 | 1,045 (735–1,470) |
| 5AZ | 50 | 26 | 982 (802–1,280) |
| 2AP + ZEB | 500, 5 | 65 | 53,500 (48,800–59,300) |
| 2AP + 5AZ | 500, 20 | 53 | 6,280 (5,210–7,140) |
| 2AP + 5BrdU | 500, 5 | 28 | 2,100 (1,860–2,580) |
| 5BrdU + ZEB | 5, 5 | 32 | 34,700 (25,800–40,600) |
| 5AZ + ZEB | 50, 5 | 17 | 544 (297–669) |
| 5AZ + 5BrdU | 50, 5 | 15 | 9,680 (6,080–12,300) |
95% CI, 95% confidence interval.
Sequence of mutational changes in rpoB for the 2AP plus ZEB pair.
There are at least 92 base substitution mutations in rpoB that can lead to the Rifr phenotype at 37°C, and each of the 6 possible base substitutions is well represented (35, 39, 40). We can detect 80 of these mutations with a single primer pair in one segment of the rpoB gene; we therefore analyzed this segment in this study. There are 11 A·T→G·C mutations, 17 G·C→A·T mutations, 9 A·T→T·A mutations, 10 A·T→CG mutations, 18 G·C→T·A mutations, and 15 G·C→C·G mutations included in this set. Because many mutagens have preferred hot spots resulting from aspects of the surrounding sequence (e.g., see references 2, 22, and 35), a system that monitors numerous sites allows one to define a mutagenic fingerprint for different mutagens and mutagenic pathways. Thus, we determined the sequences of the mutations resulting from the 2AP plus ZEB combination and compared them with those we found for ZEB alone (9) and for 2AP alone (this study). All mutations analyzed were of independent origin (see Materials and Methods for details). Figure 3 displays the results, showing only the two transition substitutions A·T→G·C and G·C→A·T, since these substitution sites comprise virtually all of the mutations found in these three spectra, with 90 of 90 detected mutations for 2AP, 147 of 155 detected mutations for ZEB, and 87 of 88 mutations for 2AP plus ZEB. The ZEB spectrum shows a major hot spot at position 1600 (G·C→A·T, 86 of 156 mutations detected [55%]) and a minor hot spot at position 1691 (G·C→A·T, 22 of 156 mutations detected [14%]). The 2AP spectrum shows a major hot spot at position 1576 (G·C→A·T, 26 of 90 mutations detected [29%]), and secondary hot spots at position 1601 (G·C→AT, 19 of 90 mutations detected [20%]) and at position 1532 (A·T→G·C, 14 of 90 mutations detected [16%]). Surprisingly, the spectrum for the combination of 2AP plus ZEB is not dominated by the 2AP or ZEB major hot spot but rather a new major hot spot at position 1598 (A·T→G·C, 38 of 88 mutations detected [43%]) and a second hot spot at position 1547 (A·T→G·C, 22 of 88 mutations detected [25%]). A minor hot spot is seen at position 1601 (G·C→AT, 12 of 88 mutations detected [14%]) that is also seen in the 2AP spectrum, although here, it is 23-fold more frequent (see below). The hot spot at position 1547 is also seen in the spectra of spontaneous mutations in wild-type mismatch repair-deficient strains, as well as for NDK- and deoxycytidine deaminase (DCD)-deficient strains (35, 36, 39, 41, 42). However, the A·T→G·C mutations at position 1598 are only rarely seen (see Discussion) and constitute only 5.5% of the mutations in the 2AP spectrum (5 of 90 mutations detected).
FIG 3.

The spectrum of mutations in rpoB caused by 2AP or ZEB (bottom) or the combination of 2AP plus ZEB (top). The transitions (G·C→A·T and A·T→G·C) are shown as a percentage of the total mutations in each sample.
We can get a better picture of the increase in mutation frequencies at certain sites by multiplying the frequencies of Rifr mutants (Fig. 2 and Table 1) by the fraction of mutants with changes at each specific site, resulting in the frequency of mutants with changes at each site. Table 2 shows these frequencies. When we discount the mutants in the 2AP plus ZEB spectrum that are represented by only one or two occurrences, we can see that although mutations at a number of sites are increased, mutations at three sites have both very high frequencies and high to very high increases in frequencies in the combination over that in the two single spectra. Mutations at the new hotspot at position 1598 (A·T→G·C) occur at a frequency of 22,900 × 10−8 (2.3 × 10−4), 270-fold higher in the 2AP plus ZEB combination than with either 2AP or ZEB alone, those at position 1547 (A·T→G·C) occur at a frequency of 13,250 × 10−8 that represents at least several thousand-fold over the background single frequencies, and mutations at position 1601 (G·C→A·T) are at a frequency of 7,230 × 10−8, 23-fold higher than that found in 2AP.
TABLE 2.
Distribution of mutation frequencies in rpoB
| Site (bp) | bp change |
f (10−8) with: |
||
|---|---|---|---|---|
| 2AP | ZEB | 2AP + ZEB | ||
| 1522 | AT→GC | 0 | 0 | 600a |
| 1532 | AT→GC | 234 | 0.54 | 1,200a |
| 1534 | AT→GC | 33 | 0.27 | 3,000 |
| 1538 | AT→GC | 17 | 0 | 1,200a |
| 1547 | AT→GC | 0 | 0.27 | 13,250 |
| 1552 | AT→GC | 0 | 0 | 1,200a |
| 1577 | AT→GC | 0 | 0 | 0 |
| 1598 | AT→GC | 84 | 0.27 | 22,900 |
| 1702 | AT→GC | 0 | 0 | 0 |
| 1703 | AT→GC | 0 | 0 | 0 |
| 1715 | AT→GC | 0 | 0 | 0 |
| 1520 | GC→AT | 0 | 0 | 0 |
| 1535 | GC→AT | 134 | 1.1 | 0 |
| 1545 | GC→AT | 17 | 0 | 0 |
| 1546 | GC→AT | 50 | 1.4 | 0 |
| 1547 | GC→AT | 17 | 0 | 0 |
| 1565 | GC→AT | 0 | 0 | 0 |
| 1576 | GC→AT | 435 | 0.27 | 1,200a |
| 1585 | GC→AT | 0 | 2.7 | 0 |
| 1586 | GC→AT | 0 | 0 | 0 |
| 1592 | GC→AT | 0 | 0 | 0 |
| 1595 | GC→AT | 0 | 0 | 0 |
| 1600 | GC→AT | 50 | 22.6 | 600a |
| 1601 | GC→AT | 318 | 0.81 | 7,230 |
| 1609 | GC→AT | 0 | 0.54 | 0 |
| 1610 | GC→AT | 0 | 0 | 0 |
| 1691 | GC→AT | 100 | 5.9 | 0 |
| 1708 | GC→AT | 0 | 1.1 | 0 |
| 1721 | GC→AT | 17 | 0.81 | 0 |
Based on only one or two occurrences.
Sequence of mutational changes in rpoB for the 2AP plus 5AZ pair.
We also sequenced the rpoB gene in mutants resulting from the combination of 2AP plus 5AZ, as well as from 2AP alone (see above) and 5AZ alone. The results are shown in Table 3, which lists all the transition sites (G·C→A·T and A·T→G·C), and the G·C→C·G sites, since these 41 sites encompass virtually all of the resulting mutations. 5AZ alone has a unique G·C→C·G specificity (17, 18), and this specificity is also seen in rpoB from our previous study (35) and in this study (Table 3). The combination of 2AP plus 5AZ shows much weaker synergy than that for the 2AP plus ZEB combination, so there is a significant contribution from the backgrounds of 2AP alone (e.g., positions 1576 and 1691, G·C→AT) and 5AZ alone (G·C→C·G) in the combined spectrum. Note that the sample size here for 2AP is larger than for either 5AZ or for the combination of 2AP plus 5AZ. The spectrum of the 5AZ plus 2AP combination does not show a dramatic difference from the combined spectra of 5AZ alone and 2AP alone.
TABLE 3.
Distribution of mutations in rpoB
| Site (bp) | bp change | No. of mutations with: |
||
|---|---|---|---|---|
| 5AZ | 2AP | 5AZ + 2AP | ||
| 1522 | AT→GC | 0 | 0 | 0 |
| 1532 | AT→GC | 0 | 14 | 2 |
| 1534 | AT→GC | 0 | 2 | 0 |
| 1538 | AT→GC | 0 | 1 | 1 |
| 1547 | AT→GC | 0 | 0 | 2 |
| 1552 | AT→GC | 0 | 0 | 0 |
| 1577 | AT→GC | 0 | 0 | 0 |
| 1598 | AT→GC | 0 | 5 | 4 |
| 1702 | AT→GC | 0 | 0 | 0 |
| 1703 | AT→GC | 0 | 0 | 0 |
| 1715 | AT→GC | 0 | 0 | 0 |
| 1520 | GC→AT | 0 | 0 | 0 |
| 1535 | GC→AT | 0 | 8 | 1 |
| 1545 | GC→AT | 0 | 1a | 0 |
| 1546 | GC→AT | 0 | 3 | 0 |
| 1547 | GC→AT | 0 | 1 | 0 |
| 1565 | GC→AT | 0 | 0 | 0 |
| 1576 | GC→AT | 0 | 26 | 5 |
| 1585 | GC→AT | 0 | 0 | 0 |
| 1586 | GC→AT | 0 | 0 | 0 |
| 1592 | GC→AT | 0 | 0 | 0 |
| 1595 | GC→AT | 0 | 0 | 0 |
| 1600 | GC→AT | 0 | 3 | 5 |
| 1601 | GC→AT | 0 | 19 | 8 |
| 1609 | GC→AT | 0 | 0 | 0 |
| 1610 | GC→AT | 0 | 0 | 0 |
| 1691 | GC→AT | 0 | 6 | 2 |
| 1708 | GC→AT | 0 | 0 | 0 |
| 1721 | GC→AT | 0 | 1 | 0 |
| 1527 | GC→CG | 1 | 0 | 2 |
| 1537 | GC→CG | 4 | 0 | 0 |
| 1574 | GC→CG | 0 | 0 | 2 |
| 1576 | GC→CG | 16 | 0 | 2 |
| 1578 | GC→CG | 4 | 0 | 0 |
| 1585 | GC→CG | 1 | 0 | 0 |
| 1592 | GC→CG | 1 | 0 | 0 |
| 1594 | GC→CG | 0 | 0 | 2 |
| 1597 | GC→CG | 0 | 0 | 0 |
| 1600 | GC→CG | 0 | 0 | 1 |
| 1601 | GC→CG | 0 | 0 | 0 |
| 1691 | GC→CG | 1 | 0 | 2 |
| 1709 | GC→CG | 0 | 0 | 0 |
| 1716 | GC→CG | 16 | 0 | 8 |
| 2059 | GC→CG | 0 | 0 | 0 |
| Total | 44 | 90 | 49 | |
Part of a two-base change with position 1576.
Thymidine deprivation by ZEB.
In mammalian cells, ZEB results in thymidine deprivation from directly inhibiting thymidylate synthase and from the inhibition of dCMP deaminase by deoxyzebularine monophosphate (dZMP) (43). This has not been determined in bacteria, so we tested the effect of adding thymidine to ZEB-treated cultures, in both minimal and rich (LB) media. As Fig. 4 shows, thymidine protects cells from killing by ZEB at a range of concentrations in both minimal and rich media, indicating that ZEB results in thymidine deprivation in E. coli.
FIG 4.

Growth potentiation by thymidine in the presence of different concentrations of ZEB in LB medium (A) and minimal medium (Min) (B). Error bars represent 95% confidence intervals.
Synergy in an NDK-deficient background.
One explanation for the synergy between 2AP and ZEB involves altered dNTP pools, such as that seen in a number of E. coli mutants, for example DCD-deficient and NDK-deficient strains (41, 42, 44). We therefore decided to examine the 2AP plus ZEB combination in an NDK-deficient strain to determine whether the increased mutation frequency might be even further enhanced in this background. Table 4 shows that the frequencies of 2AP-, ZEB-, and 5BrdU-induced mutations are increased in this background, but so are the frequencies of the mutations induced by the combination of ZEB and 2AP. In the case of the combination, the levels of Rifr mutants are 185,000 per 108 cells, or approximately 1 per 560 cells! We sequenced 17 Rifr mutants from this combination and found that all 17 have A·T→G·C changes, with 6 changes at position 1534, 5 changes at position 1547, and 6 changes at position 1598. Thus, the frequency of a specific mutation at each of these three positions is approximately 60,000 per 108 cells, or one out of every 1,670 cells. These high mutation frequencies are not restricted to the rpoB region, as we found similar high levels for mutations in the thyA gene (after selection for trimethoprim resistance; data not shown). We also tested for auxotrophy among 100 to 200 colonies in the ndk background for several cultures grown on LB plates after growth in LB broth in the presence of ZEB, 2AP, and 2AP plus ZEB. We found 39% of the colonies from 2AP plus ZEB to be auxotrophs, versus 11% for 2AP alone, 1% for ZEB alone, and 0% for LB alone.
TABLE 4.
rpoB mutation frequency in an NDK-deficient strain background with various mutagens
| Mutagen(s) | Concn (μg/ml) | No. of replicates | f (10−8)a in rpoB (95% CI)a |
|---|---|---|---|
| None | 18 | 28.2 (16–55.2) | |
| 2AP | 500 | 18 | 11,650 (9,780–15,600) |
| ZEB | 5 | 12 | 150 (85.5–535) |
| 2AP + ZEB | 500, 5 | 27 | 185,000 (144,000–250,000) |
| 5BrdU | 5 | 31 | 116,000 (96,700–134,000) |
95% CI, 95% confidence interval.
DISCUSSION
What is the effect of using more than one mutagenic treatment or mutagen at the same time? Will certain combinations of mutagens unlock unexpected pathways of mutagenesis? Many single mutagens themselves are no longer considered to have only one type of effect (e.g., see references 7 and 45). Even for base analogs, one can recognize a number of different pathways to mutagenesis that might be activated: (i) direct mispairing involving different tautomeric forms of either the analog or the conventional base (see review in reference 6), although the actual tautomers involved may be different than those originally envisioned (46, 47); (ii) up- or downregulation of different operons, such as the partial or complete induction of the SOS system (6); (iii) saturation of certain repair systems, in particular, the mismatch repair system (7, 29, 30); (iv) alteration of the dNTP pools (31); and (v) inhibition of specific enzymes (43). In this respect, there are then several possible routes to synergistic effects, including but not limited to the induction of specific pathways, dNTP pool changes, or specific mispairing between different base analogs. Here, we examined a set of four base analog mutagens, 2AP, ZEB, 5AZ, and 5BrdU, and looked for synergistic effects on mutagenesis resulting from pairwise combinations. As Table 1 and Fig. 2 show, three combinations gave higher levels of Rifr mutants than either single mutagen alone. The increase in levels with the 2AP plus ZEB combination is striking (Fig. 2). Moreover, the analysis of the mutations occurring in rpoB indicates that new mutational hot spots emerge that are not prominent in the spectra of either 2AP or ZEB alone (Fig. 3 and Table 2). One possibility is that 2AP can make a specific mispair with ZEB that leads to the increased mutation rate. However, this would have to involve two rare tautomers pairing, and it appears unlikely that this could account for the very high levels of mutagen synergy we observe. A more likely explanation involves imbalances in the dNTP pools.
There is an exquisite balance between dNTP levels and ratios and DNA replication speed (e.g., see references 48 and 49) and mutation rates. Long-standing work in both microbial and mammalian cells has shown that alterations of the dNTP pools can affect mutation rates (31, 41, 42, 44, 50–54). More recently, studies in E. coli have shown that perturbations in the absolute levels of all four dNTPs, even when the ratios are not altered, leads to changes in mutation rates (26, 55, 56). Increases in the levels raise mutation rates (55, 56), and decreases in the levels lead to lower mutation rates (26). (Raising dNTP levels leads to the suppression of 3′-exonuclease proofreading, resulting in increased misincorporation and increased mutagenesis; see below [57–59]). We can look to results from the analysis of different mutants with altered dNTP ratios for clues about the cause of the synergistic effects seen in the work reported here, since the spectrum of mutations in any given system (in this case rpoB) can be fingerprints of specific processes. Mutants defective in deoxycytidine deaminase (DCD) or nucleotide diphosphate kinase (NDK) have increased dCTP and dGTP (41, 44, 49, 50; but also see reference 60), decreased dATP (41, 44), and higher rates of certain base substitutions (36, 41, 44, 50, 51). The double mutant deficient in both DCD and NDK has a more extreme imbalance and a more extreme mutation rate increase (42). These findings are relevant, because ZEB-treated E. coli cells are likely to be phenotypically DCD deficient. This stems from the fact that deoxyzebularine monophosphate, generated in vivo, inhibits mammalian DCD (43). Moreover, zebularine itself inhibits mammalian thymidylate synthase (43). Both of these inhibitions lead to thymidine deprivation (43). Indeed, we find that E. coli cells grown in the presence of ZEB are severely thymidine limited, as seen in Fig. 4. In fact, Fig. 4 is similar to the respective figure we constructed for DCD-deficient cells (61). The spectrum of rpoB mutations in the 2AP plus ZEB combination does share one hot spot with the spectra of DCD-, NDK-, or DCD- and NDK-deficient strains, the A·T→G·C mutation at position 1547 (36, 41, 42), although this site also represents a hot spot for spontaneous mutations in either a wild-type or mismatch repair-deficient background (35, 39). What really stands out in Fig. 3 is the major hot spot for 2AP plus ZEB, the A·T→G·C change at position 1598, a site that is poorly represented in most spectra (e.g., see references 35, 36, 39, 41 and 42). However, this site is well induced in only one situation described in the literature, and this allows us to pinpoint the likely source of the increased mutation level. Ahluwalia and Schaaper have reported that specific engineered mutations in the gene encoding ribonucleotide reductase (RNR) can result in a high mutation rate and a spectrum in rpoB that has hot spots at several of four particular A·T→G·C sites in rpoB, these being positions 1532, 1538, 1547, and 1598 (62; for related work in yeast, see also reference 63). Moreover, the altered RNR generates a dNTP imbalance, with increased dCTP and dGTP and reduced dATP. This favors the misincorporation of dGTP across from template T. The four sites are favored over other A·T→G·C sites in rpoB because the next nucleotide incorporated is either dGTP (three cases) or dCTP (in the case of position 1547) (62; see also earlier work in references 57 to 59). It is likely that the simple addition of two base analogs, 2AP and ZEB, results in a related situation, although more pronounced, yielding even higher frequencies, for at least two of these sites. The sequences surrounding these two sites are as follows: position 1547, 5′-CTG-GTC-CAT-3′, and position 1598, 5′-GCA-CTC-GGC-3′. Thus, with replication proceeding on the other strand in the 5′→3′ direction, when G is misincorporated across from T (in bold), the next nucleotides are CC and G for positions 1547 and 1598, respectively. The increased replication errors resulting from the 2AP plus ZEB combination inevitably saturate the mismatch repair system, thus resulting in the observed hypermutability. Neither analog alone fully saturates mismatch repair, as one sees a marked increase in the mutation frequency in ZEB-treated mismatch repair (MMR)-deficient cells compared to wild-type-treated cells (9), and the major hot spot seen in an MMR-deficient strain (A·T→G·C, position 1547) (35) does not appear in the 2AP spectrum among 90 sequenced mutations (Table 3). This is despite the fact that the spontaneous mutation frequencies in rpoB in a MutS-deficient strain and a 2AP-treated wild-type strain are similar. 2AP does appear to partially saturate MMR, however (29, 30).
One prediction of the notion that the 2AP plus ZEB combination mimics the dNTP imbalance seen in the mutant with an altered RNR is that there would be even more potent mutagenesis in an NDK strain, as the dNTP imbalance in NDK-deficient strains (41, 44) is in the same direction as the presumed imbalance generated by 2AP plus ZEB. Table 4 shows that this is indeed the case, as the rpoB mutation frequency increases from 53,000 × 10−8 in the starting strain to 185,000 × 10−8 in the NDK-deficient strain. This is, in fact, an extraordinarily high mutation frequency, particularly when the majority of mutations are at two sites (the A·T →G·C hot spots at positions 1547 and 1598; see Results). A remaining unexplained result is that in the wild-type background, we observe a large increase in a 2AP secondary hot spot in the 2AP plus ZEB combination (relative to the 2AP-alone spectrum; 23-fold), involving a G·C→A·T change at position 1601 in rpoB (Tables 2 and 3). This indicates that there are additional complexities in these synergies. Also intriguing is the suppression of 5BrdU mutagenesis by 2AP (Table 1). One might try to explain this either by specific mispairing models involving both 2AP and 5BrdU, or by some aspect of the pool imbalances (24, 25). However, definitive elucidation of this suppression effect is a topic for subsequent experiments.
Ongoing studies are aimed at examining a larger set of potential mutagen synergies, both with the aim of uncovering additional pathways of mutagenesis, and also of examining combinations that are currently in use in chemotherapy. For example, the agents cisplatin (CPT), ZEB, 5AZ, and brostallicin (BRC) are used in different combinations in chemotherapy (64–66). Specifically, BRC plus CPT is used to treat patients with advanced solid tumors (64), ZEB plus CPT has been used in human carcinoma cell lines (65), and ZEB plus BRC has been used in prostate cancer cell lines (66). We note that the ZEB plus 5AZ combination studied in this paper (Table 1) has been used successfully in mice with L1210 leukemia (20).
ACKNOWLEDGMENT
Part of this research was funded by a faculty research grant from the University of California.
REFERENCES
- 1.Benzer S, Freese E. 1958. Induction of specific mutations with 5-bromouracil. Proc Natl Acad Sci U S A 44:112–119. doi: 10.1073/pnas.44.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benzer S. 1961. On the topography of the genetic fine structure. Proc Natl Acad Sci U S A 47:403–415. doi: 10.1073/pnas.47.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crick FH, Barnett L, Brenner S, Watts-Tobin RJ. 1961. General nature of the genetic code for proteins. Nature 192:1227–1232. doi: 10.1038/1921227a0. [DOI] [PubMed] [Google Scholar]
- 4.Freese E. 1959. The specific mutagenic effect of base analogs on phage T4. J Mol Biol 1:87–105. [Google Scholar]
- 5.Drake JW, Baltz RH. 1976. The biochemistry of mutagenesis. Annu Rev Biochem 45:11–37. doi: 10.1146/annurev.bi.45.070176.000303. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Seide W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- 7.Miller JH. 2005. Perspective on mutagenesis and repair: the standard model and alternate modes of mutagenesis. Crit Rev Biochem Mol Biol 40:155–179. doi: 10.1080/10409230590954153. [DOI] [PubMed] [Google Scholar]
- 8.Marquez VE, Kelley AJ, Agbaria R, Ben-Kasus T, Cheng JC, Yoo CB, Jones PA. 2005. Zebularine: a unique molecule for an epigenetically based strategy in cancer chemotherapy. Ann N Y Acad Sci 1058:246–254. doi: 10.1196/annals.1359.037. [DOI] [PubMed] [Google Scholar]
- 9.Lee G, Wolff E, Miller JH. 2004. Mutagenicity of the cytidine analog zebularine in Escherichia coli. DNA Repair 3:155–161. doi: 10.1016/j.dnarep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Scott SA, Lakshimikuttysamma A, Sheridan DP, Sanche SE, Geyer CR, DeCoteau JF. 2007. Zebularine inhibits human acute myeloid leukemia cell growth in vitro in association with p151NK4B demethylation and reexpression. Exp Hematol 35:263–273. doi: 10.1016/j.exphem.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Herranz M, Martin-Caballero J, Fraga MF, Ruiz-Cabello J, Flores JM, Desco M, Marquez VE, Esteller M. 2006. The novel DNA methylation inhibitor zebularine is effective against the development of T-cell lymphoma. Blood 107:1174–1177. doi: 10.1182/blood-2005-05-2033. [DOI] [PubMed] [Google Scholar]
- 12.Gowher H, Jeltsch A. 2004. Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer Biol Ther 3:10620–11068. [DOI] [PubMed] [Google Scholar]
- 13.Yoo CB, Jones PA. 2006. Epigenetic therapy of cancer: past, present, and future. Nat Rev Drug Discov 5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 14.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. 2003. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 15.Cheng JC, Weisenberger DJ, Gonzales FA, Gangning L, Zu GI, Hu YG, Marquez VE, Jones PA. 2004. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol 24:1270–1278. doi: 10.1128/MCB.24.3.1270-1278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlow D, Wolfenden R. 1998. Substrate connectivity effects in the transition state for cytidine deaminase. Biochemistry 37:11873–11878. doi: 10.1021/bi980959n. [DOI] [PubMed] [Google Scholar]
- 17.Levin DE, Ames BN. 1986. Classifying mutagens as to their specificity in causing the six possible transitions and transversions: a simple analysis using the Salmonella mutagenicity assay. Environ Mutagen 8:9–28. doi: 10.1002/em.2860080103. [DOI] [PubMed] [Google Scholar]
- 18.Cupples C, Miller JH. 1989. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A 86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. 1997. Mutagenicity of 5-aza-2′deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci U S A 94:4681–4685. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemaire M, Momparler LF, Raynal NJ-M, Bernstein ML, Momparler RI. 2009. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2′-deoxycytidine. Cancer Chemother Pharmacol 63:411–416. doi: 10.1007/s00280-008-0750-6. [DOI] [PubMed] [Google Scholar]
- 21.Yanofsky C, Ito J, Horn V. 1966. Amino acid replacements and the genetic code. Cold Spring Harbor Symp Quant Biol 31:151–162. doi: 10.1101/SQB.1966.031.01.023. [DOI] [PubMed] [Google Scholar]
- 22.Coulondre C, Miller JH. 1977. Genetic studies of the lac repressor IV. Mutagenic specificity in the lacI gene of Escherichia coli. J Mol Biol 117:577–606. [DOI] [PubMed] [Google Scholar]
- 23.Persing DH, McGinty L, Adams CW, Fowler RG. 1981. Mutational specificity of the base analogue 2-aminopurine in Escherichia coli. Mutat Res 83:25–37. doi: 10.1016/0027-5107(81)90068-3. [DOI] [PubMed] [Google Scholar]
- 24.Goodman MF, Hopkins RL, Lasken R, Mhaskar DN. 1985. The biochemical basis of 5-bromouracil- and 2-aminopurine-induced mutagenesis. Basic Life Sci 31:409–423. [DOI] [PubMed] [Google Scholar]
- 25.Hopkins RL, Goodman MF. 1980. Deoxyribonucleotide pools, base pairing, and sequence configuration affecting bromodeoxyuridine- and 2-aminopurine-induced mutagenesis. Proc Natl Acad Sci U S A 77:1801–1805. doi: 10.1073/pnas.77.4.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becket E, Tse L, Yung M, Cosico A, Miller JH. 2012. Polynucleotide phosphorylase plays an important role in the generation of spontaneous mutations in Escherichia coli. J Bacteriol 194:5613–5620. doi: 10.1128/JB.00962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharon D, Schuemann M, Sheena M, McPherson R, Chaurasiya S, Shaw A, Hitt MM. 2013. 2-Aminopurine enhances the oncolytic activity of E1b-deleted adenovirus in hepatocellular carcinoma cells. PLoS One 8:e65222. doi: 10.1371/journal.pone.0065222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mancini WR, Glaze ER, Stetson PL, Greenberg HS. 1999. Sensitization of 1,3-bis (2-chloroethyl)-1-nitrosourea, and cisplatin cytotoxicity by 5-bromo-2′-deoxyuridine in human glioma. J Phar Exp Ther 289:1404–1409. [PubMed] [Google Scholar]
- 29.Cupples CG, Cabrera M, Cruz C, Miller JH. 1990. A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics 125:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matic I, Babic A, Radman M. 2003. 2-Aminopurine allows interspecies recombination by a reversible inactivation of the Escherichia coli mismatch repair system. J Bacteriol 185:1459–1461. doi: 10.1128/JB.185.4.1459-1461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunz BA, Kohalmi SE, Kunkel TA, Mathews TA, McIntosh EM, Reidy JA. 1994. Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat Res 318:1–64. doi: 10.1016/0165-1110(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 32.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 35.Garibyan L, Huang T, Kim TM, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. 2003. Use of the rpoB gene to determine the specificity of base specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair 2:593–608. doi: 10.1016/S1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 36.Miller JH, Funchain P, Clendenin W, Huang T, Nguyen A, Wolff E, Yeung A, Chiang J, Garibyan L, Slupska MM, Yang H. 2002. Escherichia coli strains (ndk) lacking nucleoside diphosphate kinase are powerful mutators for base substitutions and frameshifts in mismatch repair deficient strains. Genetics 162:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drake JW. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A 88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dixon WJ, Massey FJ Jr. 1969. Introduction to statistical analysis. McGraw-Hill, New York, NY. [Google Scholar]
- 39.Wolff E, Kim M, Hu K, Yang H, Miller JH. 2004. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol 186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corzett CH, Goodman MF, Finkel SE. 2013. Competitive fitness during feast and famine: how SOS DNA polymerases influence physiology and evolution in Escherichia coli. Genetics 194:409–420. doi: 10.1534/genetics.113.151837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaaper R, Mathews CK. 2013. Mutational consequences of dNTP pool imbalances in E. coli. DNA Repair (Amst) 12:73–79. doi: 10.1016/j.dnarep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tse L, Kang TM, Yuan J, Mihora D, Becket E, Maslowska KH, Schaaper RM, Miller JH. 2016. Extreme dNTP pool changes and hypermutability in dcd ndk strains. Mut Res 784:16–24. doi: 10.1016/j.mrfmmm.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo CB, Valente R, Congiatu C, Gavazza F, Angel A, Siddiqui MA, Jones PA, McGuigan C, Marquez VE. 2008. Activation of p16 gene silenced by DNA methylation in cancer cells by phosphoramidate derivatives of 2′-deoxyzebularine. J Med Chem 51:7593–7601. doi: 10.1021/jm8005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Q, Zhang X, Almaula N, Mathews CK, Inouye M. 1995. The gene for nucleoside diphosphate kinase functions as a mutator gene in Escherichia coli. J Mol Biol 254:337–341. doi: 10.1006/jmbi.1995.0620. [DOI] [PubMed] [Google Scholar]
- 45.Arima Y, Nishigori C, Takeuchi T, Oka S, Morimoto K, Utani A, Miyachi Y. 2006. 4-Nitroquinoline 1-oxide forms 8-hydroxydeoxyguanosine in human fibroblasts through reactive oxygen species. Toxicol Sci 91:382–392. doi: 10.1093/toxsci/kfj161. [DOI] [PubMed] [Google Scholar]
- 46.Sowers LC, Boulard Y, Fazakerly GV. 2000. Multiple structures for the 2-aminopurine-cytosine mispair. Biochemistry 39:7613–7620. doi: 10.1021/bi992388k. [DOI] [PubMed] [Google Scholar]
- 47.Kimsey IJ, Perzold K, Sathyamoorthy B, Stein ZW, Al-Hashimi HM. 2015. Visualizing transient Watson-Crick-like mispairs in DNA and RNA duplexes. Nature 519:315–320. doi: 10.1038/nature14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gon S, Camara JE, H, Klungsøyr HK, Crooke E, Skarstad K, Beckwith J. 2006. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J 25:1137–1147. doi: 10.1038/sj.emboj.7600990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nordenskjold BA, Skoog L, Brown NC, Reichard P. 1970. Deoxyribonucleotide pools and deoxyribonucleic acid synthesis in cultured mouse embryo cells. J Biol Chem 245:5360–5368. [PubMed] [Google Scholar]
- 50.Sánchez A, Sharma S, Rozenzhak S, Roguev A, Krogan NJ, Chabes A, Russell P. 2012. Replication fork collapse and genome instability in a deoxycytidylate deaminase mutant. Mol Cell Biol 32:4445–4454. doi: 10.1128/MCB.01062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinberg GL, Ullman B, Martin DW Jr. 1981. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc Natl Acad Sci U S A 78:2447–2451. doi: 10.1073/pnas.78.4.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinberg GL, Ullman B, Wright CM, Martin DW Jr. 1985. The effects of exogenous thymidine on endogenous deoxynucleotides and mutagenesis in mammalian cells. Somat Cell Mol Genet 11:413–419. doi: 10.1007/BF01534835. [DOI] [PubMed] [Google Scholar]
- 53.Meuth M. 1989. The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp Cell Res 181:305–316. doi: 10.1016/0014-4827(89)90090-6. [DOI] [PubMed] [Google Scholar]
- 54.Meuth M, Aufreiter E, Reichard P. 1976. Deoxyribonucleotide pools in mouse-fibroblast cell lines with altered ribonucleotide reductase. Eur J Biochem 71:39–43. doi: 10.1111/j.1432-1033.1976.tb11087.x. [DOI] [PubMed] [Google Scholar]
- 55.Wheeler LJ, Rajagopal I, Mathews CK. 2005. Stimulation of mutagenesis by proportional deoxynucleoside triphosphate accumulation in Escherichia coli. DNA Repair 4:1450–1456. doi: 10.1016/j.dnarep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Gon S, Napolitano R, Rocha W, Coulon S, Fuchs RP. 2011. Increase in dNTP pool size during the DNA damage response plays a key role in spontaneous and induced-mutagenesis in Escherichia coli. Proc Natl Acad Sci U S A 108:19311–19316. doi: 10.1073/pnas.1113664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clayton LK, Goodman MF, Branscomb EW, Galas DJ. 1979. Error induction and correction by mutant and wild-type T4 DNA polymerases. Kinetic error discrimination mechanisms. J Biol Chem 254:1902–1912. [PubMed] [Google Scholar]
- 58.Fersht A. 1979. Fidelity of replication of phage ΦX174 DNA polymerase III holoenzyme: spontaneous mutation by misincorporation. Proc Natl Acad Sci U S A 76:4946–4950. doi: 10.1073/pnas.76.10.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kunkel TA, Schaaper RM, Beckman RA, Loeb LA. 1981. On the fidelity of DNA replication. Effect of the next nucleotide on proofreading. J Biol Chem 256:9883–9889. [PubMed] [Google Scholar]
- 60.Nordman J, Wright A. 2008. The relationship between dNTP pool levels and mutagenesis in an Escherichia coli DNP kinase mutant. Proc Natl Acad Sci U S A 105:10197–10202. doi: 10.1073/pnas.0802816105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang TM, Yuan J, Zhou A, Beppler C, Miller JH. 2014. Deoxycytidine deaminase-deficient Escherichia coli strains display acute sensitivity to cytidine, adenosine, and guanosine and increased sensitivity to a range of antibiotics, including vancomycin. J Bacteriol 196:1950–1957. doi: 10.1128/JB.01383-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahluwalia D, Schaaper RM. 2013. Hypermutability and error catastrophe due to defects in ribonucleotide reductase. Proc Natl Acad Sci U S A 110:18596–18601. doi: 10.1073/pnas.1310849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res 39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caponigro F, Lorusso D, Fornari G, Barone C, Meriano M, Airoldi M, Schena M, MacArthur R, Weitman S, Jannuzzo MG, Crippa S, Fiorentini F, Petroccione A, Comis S. 2010. Phase I dose-escalation study of brostallicin, a minor groove binder, in combination with cisplatin in patients with advanced solid tumors. Cancer Chemother Pharmacol 66:389–394. doi: 10.1007/s00280-009-1175-6. [DOI] [PubMed] [Google Scholar]
- 65.Suzuki M, Shinohara F, Nishimura K, Echigo S, Rikiishi H. 2007. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int J Oncol 31:1449–1456. doi: 10.3892/ijo.31.6.1449. [DOI] [PubMed] [Google Scholar]
- 66.Sabatino M, Geroni C, Ganzinelli M, Ceruti R, Broggini M. 2013. Zebularine partially reverses GST methylation in prostate cancer cells and restores sensitivity to the DNA minor groove binder brostallicin. Epigenetics 8:656–665. doi: 10.4161/epi.24916. [DOI] [PMC free article] [PubMed] [Google Scholar]