

Abstract
Chlamydia trachomatis can enter a viable but nonculturable state in vitro termed persistence. A common feature of C. trachomatis persistence models is that reticulate bodies fail to divide and make few infectious progeny until the persistence-inducing stressor is removed. One model of persistence that has relevance to human disease involves tryptophan limitation mediated by the host enzyme indoleamine 2,3-dioxygenase, which converts l-tryptophan to N-formylkynurenine. Genital C. trachomatis strains can counter tryptophan limitation because they encode a tryptophan-synthesizing enzyme. Tryptophan synthase is the only enzyme that has been confirmed to play a role in interferon gamma (IFN-γ)-induced persistence, although profound changes in chlamydial physiology and gene expression occur in the presence of persistence-inducing stressors. Thus, we screened a population of mutagenized C. trachomatis strains for mutants that failed to reactivate from IFN-γ-induced persistence. Six mutants were identified, and the mutations linked to the persistence phenotype in three of these were successfully mapped. One mutant had a missense mutation in tryptophan synthase; however, this mutant behaved differently from previously described synthase null mutants. Two hypothetical genes of unknown function, ctl0225 and ctl0694, were also identified and may be involved in amino acid transport and DNA damage repair, respectively. Our results indicate that C. trachomatis utilizes functionally diverse genes to mediate survival during and reactivation from persistence in HeLa cells.
INTRODUCTION
Chlamydia trachomatis has a characteristic biphasic developmental cycle in cell culture (1). The two dominant chlamydial forms are elementary bodies (EBs) and reticulate bodies (RBs) (1). EBs represent the infectious, nonreplicative form capable of invading host cells (1). Following entry, EBs differentiate into RBs that replicate inside parasitophorous vacuoles termed inclusions (1). Maturing RBs then redifferentiate back to EBs and exit the host cell via lysis or extrusion (2). Morphologically aberrant RBs have been observed in clinical specimens (3). This abnormal developmental cycle can be recapitulated in a laboratory setting by exposing C. trachomatis to various stressors such as host cytokines (4–6), excess amino acids (7), iron deficiency (8, 9), virus coinfections (10), and antibiotics (11, 12) (reviewed in reference 13). Under these conditions, normal C. trachomatis RBs can transition into persistent forms that do not divide, are enlarged, and have aberrant morphology (13). Upon removal of the persistence-inducing stressor, RBs transition back into normal RBs and continue normal development (13). Aberrant RB morphology is a shared feature of multiple C. trachomatis persistence models and may reflect activation of a common stress response program (13). Thus, characterizing how C. trachomatis enters, survives, and reactivates from persistence could reveal new insights into chlamydial pathogenesis.
The Th1 cytokine interferon gamma (IFN-γ) can induce differential expression of hundreds of interferon-stimulated (host) genes (ISG), and the products of many of these genes can inhibit intracellular pathogens (14, 15). Byrne and colleagues showed that IFN-γ triggers C. psittaci and C. trachomatis persistence in T24 and HeLa 229 (HeLa) cells, respectively (6, 16). In these cells, IFN-γ induces expression of indoleamine 2,3-dioxygenase (IDO1), which converts the essential amino acid tryptophan to N-formylkynurenine. It is hypothesized that this process starves chlamydia of tryptophan because C. trachomatis RBs can be reactivated when IFN-γ is removed or tryptophan is added to the cell culture medium (6, 17).
Comparison of the C. trachomatis transcriptomes during IFN-γ-induced persistence and reactivation in the presence of exogenous tryptophan or indole revealed insights into the nature of C. trachomatis persistence (18). Analysis of differentially expressed genes suggested that metabolism in persistent RBs shifts toward utilization of alternate nutrient sources and prevention of cellular damage at the expense of RB proliferation and EB production (18). For example, genes for tryptophan utilization and DNA repair and the late gene repressor euo were upregulated in persistent RBs (18). In contrast, genes encoding chlamydial histones, genes corresponding to EB outer membrane complex formation, and some cell division genes were downregulated (18). These data are consistent with the suggestion that persistence is a stress response program that is triggered in response to amino acid starvation (13).
Multiple lines of evidence suggest that IFN-γ-induced persistence and reactivation may be relevant to C. trachomatis genital infections in humans. All reference C. trachomatis strains are inhibited by IFN-γ in human epithelial cells (19), but only the genital strains encode functional tryptophan synthase enzymes that can synthesize tryptophan from serine and indole (20–24). Neither C. trachomatis bacteria nor humans produce indole, but various microorganisms that colonize the genital tract do (25–27). Interestingly, the incidence of C. trachomatis infection is also elevated in women with bacterial vaginosis (BV) (28), and the microbiomes of these women are often enriched with bacterial taxa that contain indole producers, including Prevotella spp. (25, 28–30). Moreover, indole can be detected in vaginal secretions from women with BV and aberrant RBs were observed in cervical scrapings from one patient with C. trachomatis (3). This specimen produced few infectious EBs but contained a high Chlamydia genome load, suggesting that most of the genomes corresponded to persistent organisms (3). The observations described above suggest that C. trachomatis genital strains may rely on microbiome-derived indole to circumvent IDO1-mediated tryptophan depletion (31, 32).
Tryptophan and indole similarly rescue C. trachomatis development in some IFN-γ persistence models (20, 21, 33). In addition, IFN-γ is unable to trigger C. trachomatis persistence in IDO1-deficient cells (34), confirming that the IDO1 gene is a key antichlamydial ISG. However, there is substantial variation in the IFN-γ sensitivity of isolates within subgroups of tryptophan synthase-positive genital strains (19) and tryptophan synthase-negative trachoma strains (35). It has been reported that interferon-stimulated p65 guanylate binding proteins (GBP) may contribute to IFN-γ-mediated control of C. trachomatis in HeLa cells (36) and macrophages (37), although more recent results indicate that human GBP1 (hGBP1) does not localize to C. trachomatis vacuoles and is dispensable for cell-autonomous control of this pathogen in both unprimed and IFN-γ-primed human cells (38). Thus, there is evidence that both the antichlamydial effects of IFN-γ and the C. trachomatis response could involve more than host-pathogen tryptophan competition.
In this study, we sought to expand upon the classical view of chlamydial persistence by identifying additional C. trachomatis genes outside the tryptophan synthase operon that mediate entry into, survival during, and reactivation from IFN-γ-induced persistence. Thus, a loss-of-function screen was used to identify C. trachomatis mutants that have decreased survival following indole-mediated reactivation of IFN-γ-induced persistence in HeLa cells. Our results suggest that C. trachomatis utilizes multiple strategies to allow it to survive during and reactivate from persistence.
MATERIALS AND METHODS
Cell culture and Chlamydia propagation.
McCoy and HeLa cells were obtained from the American Type Culture Collection and were cultivated in Dulbecco's modified Eagle's medium (DMEM)–high-glucose medium supplemented with 4 mM l-glutamine (HyClone), 10% fetal bovine serum (FBS; Atlanta Biologicals), sodium pyruvate (HyClone), HEPES (HyClone), and nonessential amino acids (Gibco) (DMEM-10). The cells were grown in 5% CO2 humidified incubators at 37°C, as described previously (39). C. trachomatis L2-GFP (L2-green fluorescent protein) was created by transforming C. trachomatis L2 434/Bu (C. trachomatis) with pGFP::SW2 (a kind gift from Ian Clarke, University of Southampton, United Kingdom) (40). Mutant screens were performed using a library of plaque-cloned ethane methylsulfonate (EMS)-mutagenized L2-GFP isolates (41).
Screen for Sip mutants.
The strategy for the Sip (sensitive to IFN-γ-induced persistence) mutant screen is depicted in Fig. 1A. HeLa cells were seeded in two replicate 96-well plates in DMEM-10. The following day, the medium was replaced with fresh DMEM-10 or DMEM-10 supplemented with 10 ng/ml IFN-γ (Thermo Fisher) (DMEM-10G) and the monolayers were incubated for 24 h. The untreated and IFN-γ-treated monolayers were then infected with equal volumes of mutant library isolates in sucrose-phosphate glutamic acid buffer (0.25 M sucrose, 10 mM phosphate, 5 mM l-glutamic acid, pH 7.2 [SPG]) by centrifugation at 1,600 × g for 1 h followed by rocking for 30 min at 37°C (centrifugation and rocking). The SPG was aspirated, and DMEM-10 or DMEM-10G was added. Untreated monolayers were fixed at 24 h postinfection (hpi) in 3.7% formaldehyde. IFN-γ-treated monolayers were rinsed with phosphate-buffered saline (PBS) at 24 hpi and then incubated for an additional 24 h in tryptophan-free DMEM-10 (UCSF Cell Culture Facility)–10% dialyzed FBS (DMEM-10TF)–10 μM indole. The experimental infections were fixed in 3.7% formaldehyde at 24 h postreactivation. Fluorescent inclusions were imaged using an automatic EVOS inverted fluorescence microscope at 4× magnification using a GFP filter set. Inclusions were counted using a custom program developed in FIJI (42).
FIG 1.

IFN-γ persistence screen. (A) Untreated and IFN-γ-treated HeLa cell monolayers were infected with equal volumes of isolates from an EMS-mutagenized L2-GFP library. Untreated monolayers were fixed at 24 hpi. Indole was added to the IFN-γ-treated monolayers at 24 hpi to reactivate persistent C. trachomatis. Monolayers were fixed 24 h later. (B) Secondary screening of each persistence mutant. The inclusions that each purified isolate formed under the untreated and IFN-γ-treated conditions were counted and used to calculate a ratio. This ratio was compared to the value determined with L2-GFP. Sip mutants had significantly increased untreated/IFN-γ-treated inclusion ratios compared to L2-GFP. The graph depicts the mean of the results of three experiments performed in triplicate. Error bars indicate standard deviations (SD). ***, P < 0.001; ****, P < 0.0001.
One-step growth curve analysis.
HeLa cell monolayers were infected at a multiplicity of infection (MOI) of 0.1 and then were incubated in DMEM-10 until the indicated time points. Monolayers were harvested in SPG by freeze-thaw and bead agitation (43). Serial dilutions of these lysates were used to infect fresh HeLa cells by centrifugation and rocking, and the cells were then methanol fixed at 36 hpi. Inclusions were labeled with an antichlamydial lipopolysaccharide (LPS) antibody (EVIH1), a kind gift from Harlan Caldwell (National Institutes of Health, Bethesda, MD), and with a secondary Dylight 488-conjugated anti-mouse IgG. Inclusions were imaged at 4× magnification as described above.
Indole and tryptophan reactivation assays.
HeLa cells were seeded and treated with IFN-γ as described for the Sip mutant screen. Monolayers were infected with an MOI of 0.1 for each strain. The infections were reactivated by adding fresh DMEM-TF, supplemented with either 10 μM indole or 128 mg/liter tryptophan, and then incubated for 24 h. For inclusion-forming-unit (IFU) assays, the infected monolayers were fixed in 3.7% formaldehyde at 24 hpi for the untreated plate or 24 h postreactivation for the IFN-γ-treated plate. When inclusion cross-sectional areas were determined, infected monolayers were first fixed with 3.7% formaldehyde and then permeabilized with 100% methanol and labeled with EVIH1 as described above. For infectious-progeny assays, EBs were released from monolayers by freeze-thaw in SPG and bead agitation. Serial dilutions of these lysates were used to infect fresh HeLa cells by centrifugation and rocking, and these infection reaction mixtures were fixed with methanol at 36 hpi. Inclusions were labeled with EVIH1 and Dylight 488-conjugated anti-mouse IgG. Inclusions were imaged at 20× (cross-sectional-area determinations) or 4× (infectious progeny assays) magnification as described above.
Recombination and mutation mapping.
HeLa cell monolayers in 12-well plates were infected with pairs of mutants, each at an MOI of 2, or with individual mutants at an MOI of 4 by centrifugation and rocking and were incubated in DMEM-10. The infected monolayers were lysed at 24 hpi in SPG by bead agitation. The lysates were used to infect HeLa cell monolayers that had been preincubated in DMEM-10G for 24 h by centrifugation and rocking. The infections were then incubated in DMEM-10G for 24 h, and then the medium was replaced with DMEM-10TF supplemented with 10 μM indole, and the infections were incubated for an additional 24 h. Lysates were prepared in SPG at 48 hpi by bead agitation. This process was repeated until populations of IFN-γ-resistant recombinant strains expanded in individual wells or was halted after 5 rounds if no inclusions were observed. Populations of isolates from the wells that contained IFN-γ-resistant populations were subsequently plaque cloned (44) and expanded in HeLa cells, and mutations were confirmed by DNA sequencing.
DNA sequencing.
HeLa cells infected with various mutants were harvested by bead agitation in SPG, and the lysate was clarified by centrifugation at 500 × g for 20 min. Host cell genomes were depleted by DNase treatment (45), and the remaining DNA was amplified with REPLI-g (Qiagen) according to the manufacturer's instructions. Whole-genome sequencing (WGS) libraries were prepared using a Nextera XT DNA library preparation kit according to the manufacturer's instructions. Single-end 100-bp sequencing was performed on an Illumina HiSeq 2000 sequencing system at the Center for Genomic Research and Biocomputing Core, Oregon State University, Corvallis, OR. Genome sequences were assembled and analyzed as described previously (45). Mutations were confirmed by PCR amplification of the corresponding genomic regions and Sanger sequencing.
IFN-γ sensitivity assays.
HeLa cell monolayers were incubated in DMEM-10G for 24 h, infected with the indicated strains at an MOI of 0.1, and then incubated in DMEM-10TF supplemented with IFN-γ and 128 mg/liter tryptophan. The monolayers were fixed at 24 hpi with 3.7% formaldehyde and imaged to count inclusions. The monolayers were then permeabilized with 100% methanol. Inclusions were labeled with antibodies, and the cross-sectional areas were determined as described above.
Transmission electron microscopy.
HeLa cells were infected at an MOI of 0.1 with various strains as described for the mutant screen. The monolayers were fixed 24 h postinfection or 24 h postreactivation with indole using 2.5% glutaraldehyde and 4% formaldehyde. Samples were stained with 1% osmium tetroxide and 1% tannic acid, dehydrated, and embedded in epoxy resin. Ultrathin sections were generated using an ultracut UCT ultramicrotome (Leica) and stained with uranyl acetate and lead citrate. Sections were imaged using a JEOL JEM 1010 microscope with a Gatan 890 4,000-by-4,000-pixel digital camera at the Indiana University Electron Microscopy Center in Bloomington, IN.
Quantitative real-time PCR.
A gMAX minikit (IBI Scientific) was used according to the manufacturer's instructions to purify DNA from infected cells pre- and post-indole or -tryptophan reactivation at various times. The DNA was mixed with FastStart TaqMan Probe Master mix (Roche) and 9 pmol of primers 5′-GTAGCGGTGAAATGCGTAGA-3′ and 5′-CGCCTTAGCGTCAGGTATAAA-3′ and a 5′-6-carboxyfluorescein (FAM)-ATGTGGAAG/ZEN quencher/AACACCAGT-3′ probe targeting the C. trachomatis 16S rRNA gene. Quantitative PCR was performed on an Eppendorf Realplex4 device. Standard curves were generated using a cloned copy of the C. trachomatis 16S rRNA gene. Cycling conditions were as follows: 10 min at 95°C, followed by 40 cycles of 95°C for 20 s, 60°C for 1 min, and 68°C for 20 s.
Tryptophan-free reactivation assays.
HeLa cells were seeded in DMEM-10. The following day, monolayers were rinsed with PBS and the medium was replaced with fresh DMEM-10 or DMEM-10TF. Twenty-four hours later, the monolayers were infected at an MOI of 0.1 as described above. The infections performed in DMEM-10 were fixed at 24 hpi. In contrast, infections incubated in DMEM-10TF were rinsed with PBS for 24 hpi and then incubated for an additional 24 h in either DMEM-10TF–10 μM indole or DMEM-10 and fixed at 48 hpi. Inclusions were measured and ratios were calculated and compared to L2-GFP results as described above.
Isoleucine sensitivity assay.
HeLa cells were seeded in DMEM-10 and were infected at an MOI of 0.1 as described above. The infections were then incubated in DMEM-10 supplemented with a range of concentrations of isoleucine. The infected monolayers were fixed at 44 hpi and permeabilized with methanol. The cross-sectional areas of inclusions were determined as described above.
Statistics.
Data were analyzed using GraphPad Prism version 6.0 for Windows. Statistics were calculated using one-way analysis of variance (ANOVA) and Dunnett's post hoc test for multiple comparisons.
RESULTS
Screen for IFN-γ persistence and reactivation mutants.
We screened a nonsaturating library of 2,016 chemically mutagenized L2-GFP isolates for mutants that had a reduced ability to be indole reactivated from IFN-γ-induced persistence in HeLa cells (Fig. 1A). First, the ratio of inclusions each library isolate formed in untreated HeLa cells at 24 hpi and in IFN-γ-plus-indole-treated HeLa cells at 48 hpi was calculated. In order to normalize for the differing MOIs of the infections with the different library isolates, this ratio was compared to the ratio of inclusions that the parent L2-GFP strain formed under the same sets of two conditions. Similar to previous studies, L2-GFP produced approximately 50% as many inclusions in the IFN-γ-plus-indole-treated HeLa cells as it did in the untreated cells (data not shown) (18, 20). In contrast, some library isolates formed dramatically fewer inclusions in the IFN-γ-plus-indole-treated cells than in the untreated cells, and the phenotypes of six of these mutant isolates were verified in a secondary screen (Fig. 1B). One-step growth curve analysis was used to compare the developmental kinetics of L2-GFP and these six mutant isolates (Fig. 2). The number of EBs produced by each of the mutants in cells not treated with IFN-γ was indistinguishable from the number produced by L2-GFP in untreated HeLa cells. These results indicated that the phenotypes of the six mutant strains were not measurably compromised in fitness and that the mutant phenotypes are related to their inability to enter into, survive during, and/or be reactivated from IFN-γ persistence. We named these isolates Sip mutants on the basis of the observation that they were sensitive to IFN-γ-induced persistence.
FIG 2.

Sip mutants exhibit normal growth kinetics. HeLa cell monolayers were infected with L2-GFP or Sip mutants. Monolayers were harvested at various times postinfection, and these lysates were used to infect fresh HeLa cells. The number of inclusions each isolate formed was then normalized to the inoculum. The graph depicts the mean of the results of three experiments performed in triplicate. Error bars indicate SD.
Allele mapping uncovers novel persistence genes.
A whole-genome sequencing approach was used to compare the genome sequences of the Sip mutants and L2-GFP. Each Sip mutant had between 2 to 8 mutations and contained at least two nonsynonymous substitutions in predicted coding sequences (Table 1). None of the polymorphisms in the different mutants were located in the same genes or operons. This prevented us from linking specific mutations to the phenotypes of the mutants. However, Sip1 had a missense mutation in trpB, which encodes the beta subunit of tryptophan synthase. A mutation in this gene was expected and is a key datum that validates the efficacy of the screen we developed.
TABLE 1.
Mutations in Sip mutantsa
| Mutant strain | Genome coordinate | Ref. | Mut. | Codon | Gene | CTL locus | CT locus |
|---|---|---|---|---|---|---|---|
| Sip1 (TrpBP221S) | 43026 | R | R | 818 | CTL0033 | CT664 | |
| 166676 | Y | Y | 341 | groEL | CTL0124 | CT755 | |
| 177906 | A | T | 241 | CTL0133 | CT764 | ||
| 264602 | D | N | 62 | CTL0209 | CT837 | ||
| 512700 | P | S | 221 | trpB | CTL0423b | CT170 | |
| Sip2 (CTL0225G77E) | 173557 | G | R | 298 | ftsW | CTL0129 | CT760 |
| 281464 | G | E | 77 | CTL0225b | CT852 | ||
| 290406 | P | P | 258 | cpa | CTL0233 | CT858 | |
| 324317 | V | V | 93 | gatC | CTL0257 | CT002 | |
| 404913 | S | F | 365 | CTL0325 | CT069 | ||
| 505526 | Intergenic | ||||||
| 538379 | G | E | 299 | CTL0447 | CT195 | ||
| 856877 | L | L | 64 | ispD | CTL0722 | CT463 | |
| Sip3 | 225552 | S | N | 246 | plsB | CTL0176 | CT807 |
| 248355 | Intergenic | ||||||
| 363762 | L | L | 245 | CTL0291 | CT036 | ||
| 669764 | P | L | 1128 | rpoC | CTL0566 | CT314 | |
| Sip4 | 438375 | G | R | 37 | trxB | CTL0354 | CT99 |
| 450703 | G | R | 14 | groES | CTL0366 | CT111 | |
| 528731 | G | E | 39 | tmk | CTL0440 | CT188 | |
| Sip5 | 222745 | P | S | 831 | ptr | CTL0175 | CT806 |
| 757859 | H | Y | 37 | CTL0641 | CT385 | ||
| Sip6 (CTL0694P105L) | 202648 | S | F | 71 | CTL0157 | CT789 | |
| 311659 | G | G | 39 | pmpG | CTL0250 | CT871 | |
| 387134 | P | S | 225 | CTL0312 | CT56 | ||
| 435055 | L | L | 381 | nusA | CTL0352 | CT97 | |
| 490946 | D | N | 945 | CTL0402 | CT147 | ||
| 778955 | R | R | 238 | gltT | CTL0658 | CT401 | |
| 823442 | P | L | 105 | CTL0694b | CT435 | ||
| 870246 | G | G | 296 | CTL0737 | CT476 |
Ref., reference amino acid; Mut., mutant amino acid; CTL, C. trachomatis 434/Bu; CT, C. trachomatis D/UW-3/CX.
Mapped persistence allele.
We attempted to identify the causative mutations in the Sip mutants using a counterselection lateral gene transfer (LGT) approach which takes advantage of detrimental alleles and uses them for selection. This technique produces markerless recombinants that have swapped the detrimental mutant allele for a wild-type copy (41). HeLa cells were coinfected with different pairs of the Sip mutants, and then coinfections were repeatedly passaged in HeLa cells cultivated under the persistence screen conditions. Phenotypic revertants arose from three of the coinfection experiments (Sip1 × Sip2, Sip1 × Sip6, and Sip2 × Sip6). In contrast, no revertants arose in control infections in which twice-as-large inoculums of individual Sip mutants were passaged (data not shown). This indicated that the IFN-γ-resistant isolates resulted from recombination and loss of the detrimental causative allele and were not genetic revertants or suppressor mutants. L2-GFP and recombinant Sip (rSip) strains produced similar numbers of inclusions following indole reactivation (Fig. 3). Sanger sequencing revealed that each of the rSip isolates differed from one of its two mutant parents by one nonsynonymous mutation. This analysis linked nonsynonymous mutations in genes encoding the beta subunit of tryptophan synthase (TrpB), a putative integral membrane protein (CTL0225), and a putative oxidoreductase (CTL0694) to the phenotypes of Sip1 (TrpBP221S), Sip2 (CTL0225G77E), and Sip6 (CTL0694P105L), respectively (Table 1). We focused our subsequent efforts on characterization of the phenotypes of these mapped mutants.
FIG 3.

Sip recombinants have wild-type persistence phenotypes. Recombinant Sip isolates formed numbers of inclusions similar to those formed by L2-GFP isolates in persistence reactivation experiments. Inclusion counts from corresponding control and reactivation experiments were used to calculate ratios for each isolate, and these ratios were compared to L2-GFP data. The graph depicts the mean of the results of three experiments performed in triplicate. Error bars indicate SD. n.s., nonsignificant.
Sip mutants can enter into a persistent state.
Results from our primary screen could not be used to determine whether Sip mutants were compromised in their abilities to enter, survive during, or reactivate from IFN-γ persistence. To address this issue, we used established methods to examine each mutant and determine at which step the mutation caused a defect in the development of its persistence program. Light microscopy and transmission electron microscopy (TEM) were used to examine the inclusions of the Sip mutants and the L2-GFP strain in untreated and IFN-γ-plus-indole-treated HeLa cells to determine if the Sip mutants had a defect in entering persistence. Inclusions of the Sip mutants were significantly (P < 0.0001) smaller (but were not fragmented in the manner observed in a prior study of severely tryptophan-starved C. trachomatis [46]) than those of L2-GFP following reactivation, despite being similar or larger in size under normal growth conditions (Fig. 4A and B). No obvious differences in EB or RB morphology were detected in the untreated HeLa cells, consistent with the results of the one-step growth curve experiments (Fig. 2 and 5A to D). However, in IFN-γ-plus-indole-treated HeLa cells, only the inclusions of the Sip mutants still contained aberrant RBs (Fig. 5F to H) following reactivation, indicating that they maintained the ability to enter persistence. In contrast, dividing RBs were frequently observed in HeLa cells infected with L2-GFP (Fig. 5E). As expected, dividing RBs were also observed in HeLa cells infected with rSip strains (see Fig. S1 in the supplemental material). These results suggested that the Sip mutants could enter persistence but were impaired in their ability to survive during or to be reactivated from persistence.
FIG 4.

Sip mutants make smaller inclusions following reactivation. (A) L2-GFP and mapped Sip mutant inclusion morphology was examined by fluorescence microscopy. Micrographs depict representative fields of GFP-positive (GFP+) inclusions in untreated and in IFN-γ-plus-indole-treated HeLa cells. (B and C) The control (B) and reactivation (C) infections whose results are presented in panel A were fixed, and the inclusions were labeled with antibodies and imaged at 20× to calculate their cross-sectional areas. At least 1,000 inclusions were measured for each strain and condition in each of three independent experiments. Cross-sectional areas were compared to L2-GFP data, and the error bars indicate SD. ****, P < 0.0001; n.s, nonsignificant.
FIG 5.

Sip mutant inclusions contain aberrant RBs. Transmission electron microscopy was used to compare RB and EB morphologies in untreated (A to D) and in IFN-γ-plus-indole-treated (E to H) HeLa cells at 24 hpi and 24 h postreactivation, respectively. Arrowheads indicate aberrant RBs.
Sip mutants are not reactivated by tryptophan or indole.
Since the Sip mutants produced fewer inclusions, we also tested if the remaining inclusions produced infectious EBs. We infected untreated HeLa cells with lysates from IFN-γ-plus-indole-treated infections and counted inclusions at 34 hpi. All three of the Sip mutant strains produced significantly (P < 0.0001) fewer infectious EBs than L2-GFP (Fig. 6A). This result confirmed that the Sip mutant strains not only formed fewer inclusions but also were impaired in their ability to produce infectious progeny.
FIG 6.
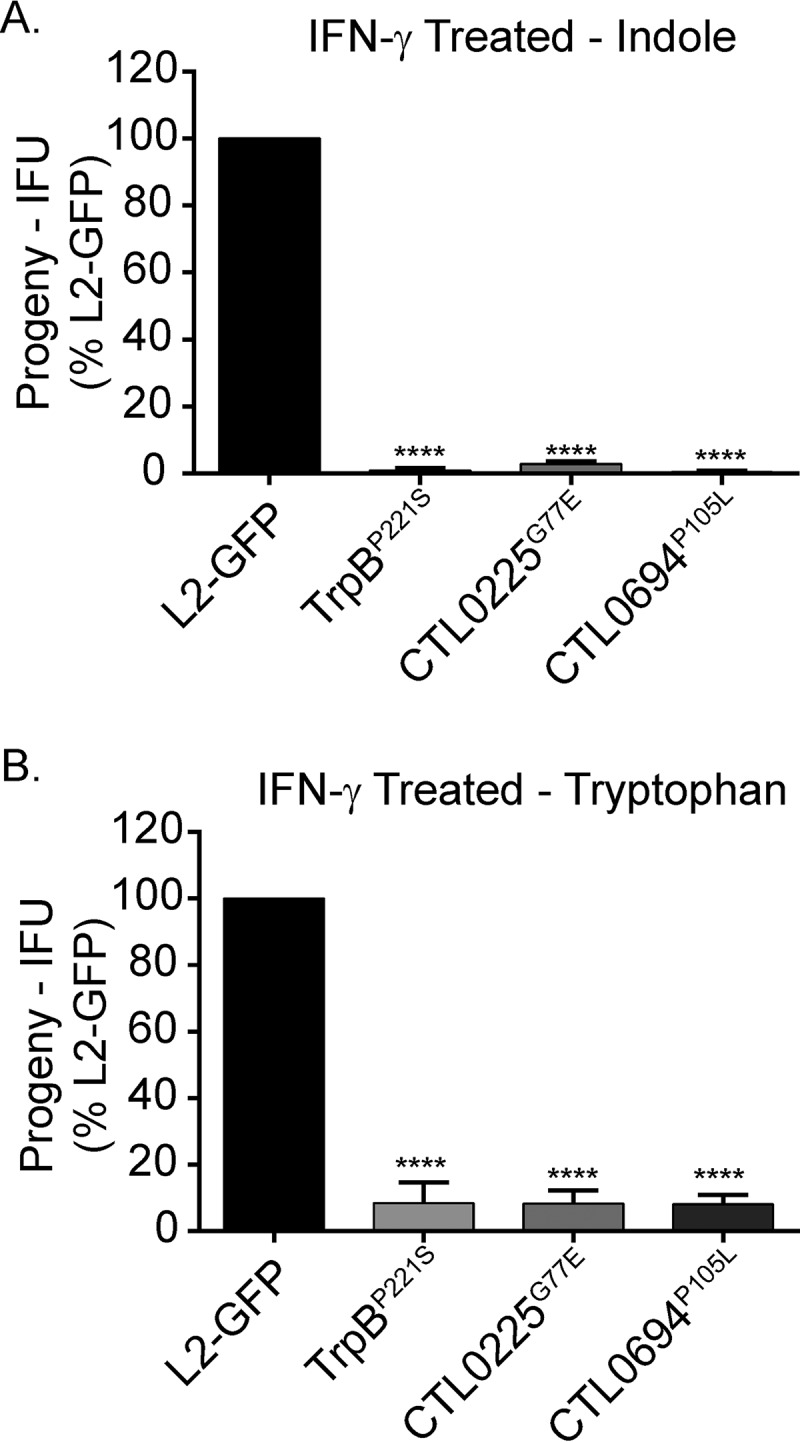
Sip mutant EB production cannot be rescued by tryptophan or indole treatment. IFN-γ-treated HeLa cells were infected with L2-GFP or Sip mutants, and then the infections were reactivated with (A) indole or (B) tryptophan. The infections were harvested 24 h postreactivation and the EBs in the lysates enumerated in untreated HeLa cells. The number of inclusions that each strain formed was normalized to the inoculum and was then compared to L2-GFP data. The graph depicts the mean of the results of three experiments performed in triplicate. The error bars indicate SD. ****, P < 0.0001.
Next, we tested if the Sip mutants produced infectious EBs following tryptophan reactivation to determine if the mutants had defects in acquiring indole or converting indole to tryptophan. A C. trachomatis trpB null mutant can be reactivated only with tryptophan, whereas strains that encode a functional tryptophan synthase can be reactivated by tryptophan or indole (47). Tryptophan and indole similarly rescued L2-GFP EB production (Fig. 6), consistent with the results of previous studies (20, 21, 33, 47). In contrast, tryptophan had little effect on the EB production seen with the Sip mutants (Fig. 6B). Surprisingly, even the TrpBP221S mutant was not rescued by treatment with tryptophan. This suggested that the defects of the Sip mutants were unrelated to tryptophan metabolism and that the mutant trpB allele in the TrpBP221S strain might produce a protein with an altered function.
Sip mutants are differentially sensitive to IFN-γ.
Since IFN-γ upregulates the expression of diverse ISG, we tested if the Sip mutants could form inclusions in IFN-γ-treated cells under tryptophan-sufficient conditions. Untreated and IFN-γ-treated HeLa cells were independently infected with L2-GFP or the Sip mutants. IFN-γ plus excess tryptophan were immediately added following infection to prevent entry into persistence. The numbers of inclusions formed (Fig. 7A) and their relative sizes (Fig. 7B) were assessed 24 hpi. The TrpBP221S and CTL0225G77E mutants produced slightly more inclusions than L2-GFP under the tryptophan-sufficient conditions. Tryptophan represses the expression of trpAB, and tryptophan treatment restored TrpBP221S strain inclusion formation; thus, this result suggested that the mutant trpB allele in the TrpBP221S strain was detrimental to chlamydial survival only during persistence. The CTL0225G77E inclusions were significantly larger (P < 0.0001) than those seen with L2-GFP in IFN-γ-plus-tryptophan-treated cells. This is consistent with the CTL0225G77E inclusions being significantly (P < 0.0001) larger than L2-GFP inclusions under normal growth conditions (Fig. 4B). In contrast, the numbers of inclusions seen with the CTL0694P105L mutant were similar to the numbers of inclusions seen with L2-GFP under these conditions but the inclusions were significantly smaller (P < 0.0001), indicating that a tryptophan-independent effect of IFN-γ may partially affect the development and subsequence size of CTL0694P105L inclusions during persistence.
FIG 7.
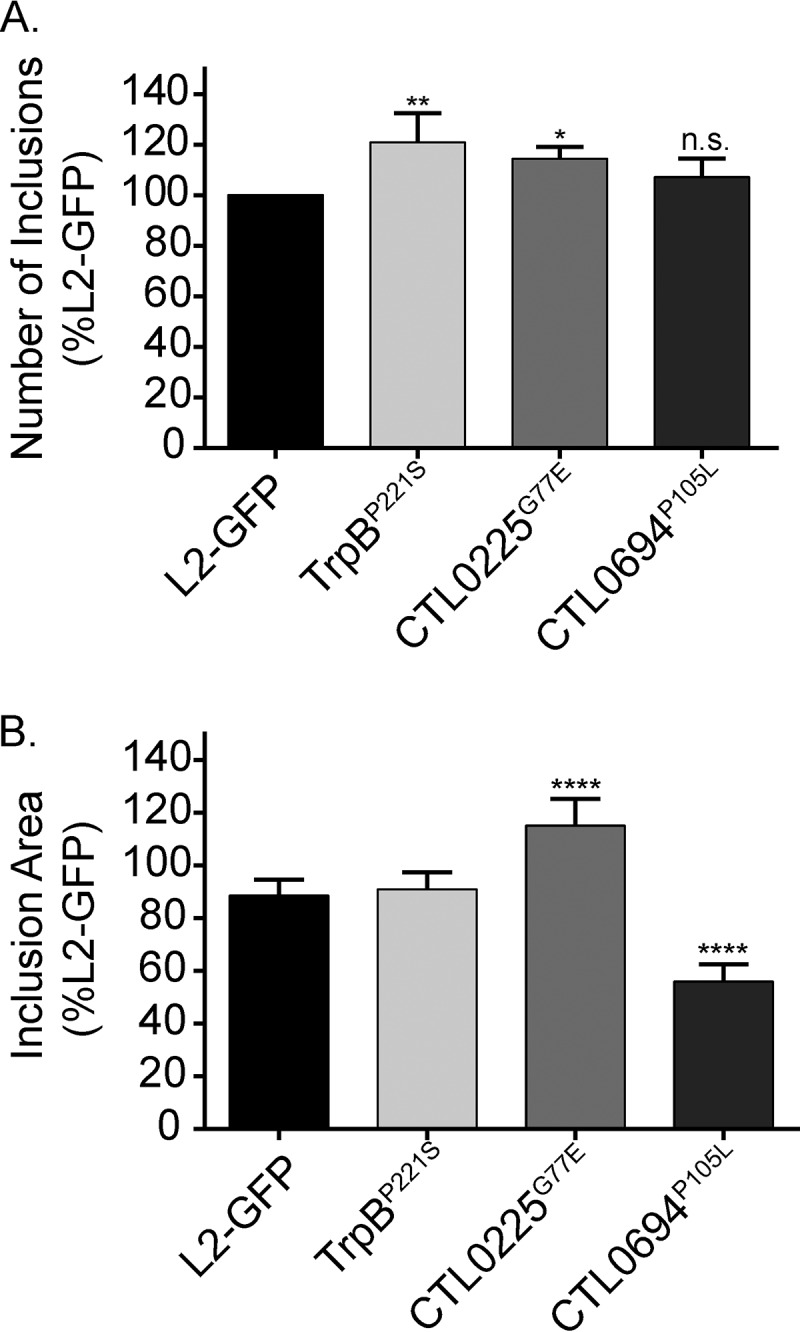
The IFN-γ sensitivity phenotypes of Sip mutants differ. Untreated and IFN-γ-treated HeLa cells were infected with L2-GFP or Sip mutants. Medium supplemented with tryptophan and IFN-γ was added immediately after infection. (A) The number of inclusions that the Sip mutants formed in untreated versus IFN-γ-treated HeLa cells was converted to a ratio, and then the ratio was compared to L2-GFP data. The graph depicts the mean of the results of three experiments performed in triplicate. Error bars depict SD. *, P < 0.05; **, P < 0.01; n.s., nonsignificant. (B) The cross-sectional areas of inclusions in IFN-γ-treated cells at 24 hpi. At least 1,000 inclusions were measured for each strain in 2 experiments with 6 replicates, and the graph depicts the mean of the results of each of the experiments. Error bars depict SD. ****, P < 0.0001.
Genome replication during persistence.
We next used quantitative PCR to measure and compare the rates of L2-GFP and Sip mutant genome accumulation during IFN-γ treatment and reactivation. Consistent with previous reports that C. trachomatis genome replication continues during persistence, albeit more slowly (48), L2-GFP and Sip mutant genome copies slowly accumulated from 2 to 24 h postinfection in IFN-γ-treated HeLa cells. The patterns of TrpBP221S genome accumulation diverged during indole and tryptophan reactivation. The rate of accumulation of genome copies of the TrpBP221S strain slightly increased 36 h after indole addition (Fig. 8A) but increased approximately 5-fold during the same interval after tryptophan addition (Fig. 8B). This result suggested that tryptophan, but not indole, was able to partially mitigate the detrimental effects of TrpBP221S on genome accumulation. Genome accumulation of CTL0225G77E was decreased and delayed compared to the L2-GFP results during indole and tryptophan reactivation, suggesting that this mutant might be unable to exit persistence and resume rapid genome replication. Finally, CTL0694P105L genome accumulation lagged only slightly behind L2-GFP genome accumulation during reactivation with indole and tryptophan. Considering the results of the indole and tryptophan reactivation experiments, where the CTL0694P105L strain formed aberrant RBs (Fig. 5H) and few infectious EBs (Fig. 6), these results suggest that genome replication is uncoupled from EB production during CTL0694P105L reactivation.
FIG 8.
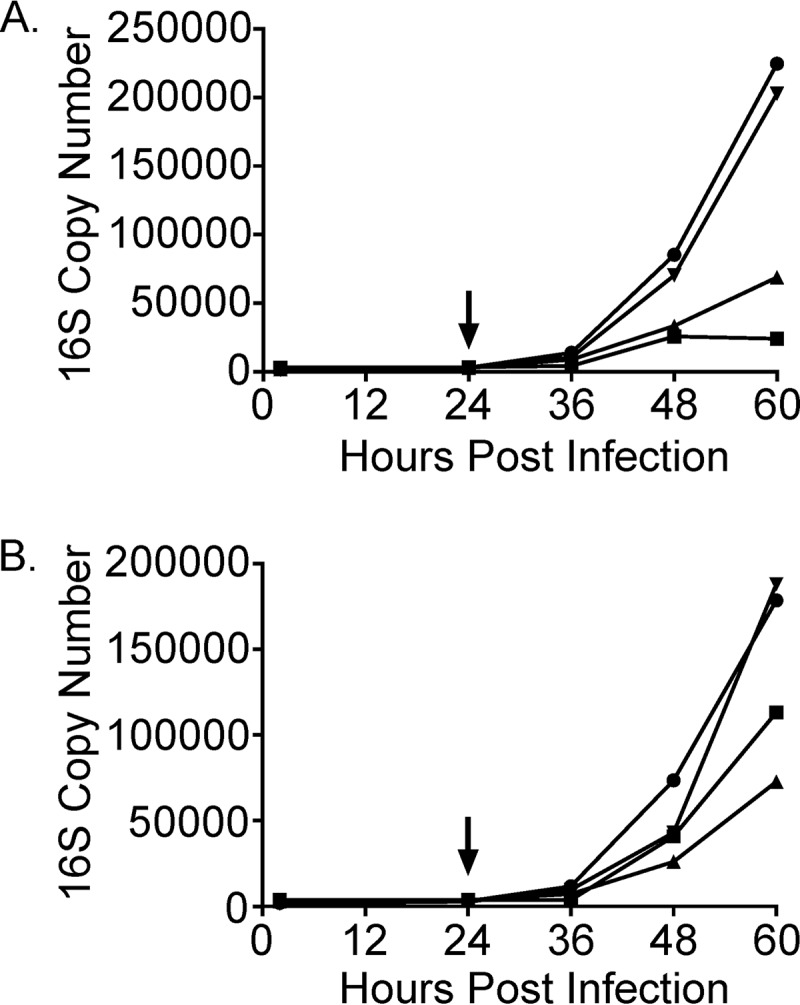
Genome replication is altered during Sip mutant reactivation. Genome copy numbers of L2-GFP and the Sip mutants were characterized pre- and post-indole (A) or -tryptophan (B) reactivation using primers and a probe set targeting the C. trachomatis 16S rRNA gene. The arrows indicate the times of indole or tryptophan addition. Data points represent the mean numbers of genome copies from two experiments performed in duplicate. Circles, L2-GFP; squares, TrpBP221S; triangles, CTL0225G77E; inverted triangles, CTL0694P105L.
Sip mutants have similar phenotypes in low-tryptophan and IFN-γ-induced persistence models.
It was unclear if the defects in the Sip mutants in the IFN-γ persistence model were primarily due to tryptophan depletion or were due to other effects of IFN-γ. C. trachomatis also enters persistence in HeLa cells in the absence of IFN-γ when tryptophan is limiting and can be reactivated later by the addition of tryptophan or indole to the cell culture medium (46). We compared the number of inclusions seen with L2-GFP, the Sip mutants, and the Sip revertants formed under tryptophan-replete conditions at 24 hpi to the number seen under tryptophan-free conditions when we added tryptophan or indole 24 h postinfection and measured inclusions at 48 hpi. Inclusion formation seen with all three of the mutants, but not that seen with L2-GFP or the Sip revertants, was impaired to a degree similar to that observed in the IFN-γ reactivation experiments under the tryptophan-free reactivation conditions (see Fig. S2 in the supplemental material). These results suggested that tryptophan limitation was the main factor that was responsible for the phenotypes of the Sip mutants in the IFN-γ persistence model.
CTL0225 may be an amino acid transporter.
Addition of high levels of some amino acids to the infection medium can cause C. trachomatis to enter an alternate persistence-like state (7). In explanation of this phenomenon, Braun et al. proposed that isoleucine-mediated inhibition of C. trachomatis growth was mediated by competition for the branched-chain amino acid transporter BrnQ (49). In their system, excess isoleucine caused C. trachomatis to form smaller inclusions that contained aberrant RBs. To further differentiate the persistence phenotypes of the Sip mutants, we compared the levels of sensitivity of the mutants to isoleucine using a range of isoleucine concentrations. Addition of isoleucine similarly reduced the sizes of the L2-GFP, TrpBP221S, CTL0694P105L, and rSip2 strains in a dose-dependent manner. In contrast, CTL0225G77E formed significantly (P < 0.0001) smaller inclusions at the same isoleucine concentrations (see Fig. S3 in the supplemental material). This suggests that CTL0225 might function similarly to BrnQ.
DISCUSSION
Our results indicate that IFN-γ elicits multiple stresses that impact C. trachomatis persistence. Since none of the mutants that we identified had mutations in the same genes or operons, this indicates that a limitation of our approach was that our screen did not reach saturation. As expected, we were able to isolate a Sip mutant that contained a mutation in tryptophan synthase. Additionally, five other mutants were isolated that contained no mutations with obvious links to tryptophan metabolism. Among these five, we were able to map the persistence gene in two Sip mutants, the CTL0225G77E and CTL0694P105L mutants, using a counterselection LGT technique. We also attempted to map the Sip mutants using an antibiotic-driven, positive-selection LGT approach (50). However, the rifampin- and ofloxacin-resistant parents that we isolated for these crosses exhibited altered persistence phenotypes, consistent with previous reports that endogenous C. trachomatis antibiotic resistance alleles can have phenotypic costs (51–54). Our experience suggests that counterselection LGT performs optimally when mutants have strong phenotypes, and further refinement of the counterselection approach may allow us to map the remaining mutants in the future (41).
Kari et al. reported that trachoma isolates that lack tryptophan synthase and a C. trachomatis serovar D trpB null mutant can be fully reactivated from IFN-γ-induced persistence with tryptophan (47). In contrast, the IFN-γ sensitivity of our TrpBP221S mutant was reversed when tryptophan was added to the cell culture medium prior to, but not after, the time point at which IFN-γ persistence was induced (Fig. 6B). This could indicate that aspects of the persistence models used in our study and the study by Kari et al. differed or that the TrpBP221S mutation causes production of a TrpB protein with an altered function. We were unable to compare the phenotypes of the TrpBP221S mutant, the serovar D TrpB null mutant studied by Kari et al. (47), and a C. trachomatis L2 trpA allelic exchange mutant recently described by the Fields group (55) directly because the growth rates of these strains differ substantially (data not shown). However, C. trachomatis trpBA expression is normally tightly repressed by TrpR in the presence of tryptophan (20, 47, 56, 57). Since we observed that more TrpBP221S genomes accumulated in the tryptophan reactivation experiments than in the indole reactivation experiments (Fig. 8), this supports the hypothesis that TrpBP221S might be toxic. It has been previously shown that Salmonella enterica serovar Typhimurium tryptophan synthase can produce ammonia and pyruvate from serine by a β-elimination reaction in the absence of indole (31). We speculate that TrpBP221S may not be able to bind indole but may still be able to produce ammonia, based on a hypothesis that Aiyar and colleagues proposed to explain the inactivation of tryptophan synthase in independent trachoma lineages (31). Future studies will determine if the mutant TrpB is associated with altered catalysis of the serine-indole condensation and β-elimination reactions.
In addition to the predicted trpB mutant, we identified genes not previously understood to be involved in IFN-γ persistence. CTL0225 and a potential paralog, CTL0226, are predicted to be members of the small neutral amino acid transporter (SNAT) family (58, 59). Highly conserved CTL0225 orthologs are present in all sequenced Chlamydia spp. (≥89% identity), and the closest homologs outside the Chlamydia genus are >30%-identical hypothetical proteins in Buchnera and Coxiella spp. (60). SNAT proteins are predicted to be dimer-forming inner membrane proteins that usually contain 6 transmembrane domains (59, 61). TMHMM Server 2.0 analysis suggests that CTL0225G77E would have altered topology and lack a transmembrane domain present in native CTL0225 (see Fig. S4 in the supplemental material) (62). SnatA, the only characterized SNAT protein, was identified in a search for amino acid transporters of Thermococcus sp. strain KS-1, a thermophilic archaeon that is auxotrophic for several amino acids (59). Recombinant Thermococcus sp. strain KS-1 SnatA permitted growth of an Escherichia coli glycine/d-alanine transporter (cycA) mutant on minimal alanine medium (59). SnatA also mediated high-affinity energy- and proton-dependent glycine transport that was inhibited by a variety of neutral amino acids, including tryptophan (59). Chlamydia spp. are auxotrophic for numerous amino acids and respond to amino acid starvation by forming aberrant RBs (7). Paradoxically, high concentrations of leucine, isoleucine, methionine, and phenylalanine can also trigger formation of aberrant RBs that can be reactivated by valine addition (49). In this case, valine starvation appears to be mediated by amino acid antagonism for the chlamydial branched-chain amino acid transporter BrnQ (49). Interestingly, we observed that CTL0225G77E, but not the other Sip mutants, was also sensitive to excess isoleucine (see Fig. S4) and our preliminary results suggest that this phenotype can be reversed by valine addition (data not shown). Thus, we hypothesize that CTL0225 is an amino acid transporter that is dispensable under normal growth conditions but not during persistence under conditions in which amino acids other than tryptophan may be limiting. The identification of additional CTL0225 substrates could determine if this protein plays a narrow accessory role in tryptophan metabolism or a broader role in acquisition of amino acids whose availability is restricted by the presence of IFN-γ.
CTL0694 is a hypothetical oxidoreductase that shares protein homology (28%) with E. coli CysJ (60). The canonical function of E. coli CysJ is in the sulfite reduction pathway (63), but C. trachomatis appears to lack all of the other enzymes in this pathway except for CysQ (60). E. coli CysJ can also interact with YcbX (64) to reduce 6-N-hydroxylaminopurine, a powerful mutagen, to adenine (65). In this complex, CysJ provides the necessary reducing equivalents to the molybdenum cofactor in YcbX (65), but CysJ-like redox proteins have also been hypothesized to provide reducing equivalents to multiple acceptor proteins (66). Similar N-hydroxylated-compound-detoxification systems have been identified in organisms as diverse as humans (67) and green algae (68). Oxidative radicals promote generation of hydroxylated nucleobases (69). Knockdown of mitochondrial amidoxime reducing component 2 sensitizes HeLa cells to aminopurine (70), indicating that HeLa cells constitutively express machinery to cope with hydroxylated aminopurines. IFN-γ treatment also elevates generation of oxidative radicals (71), suggesting that this could lead to the generation of hydroxylated nucleobases in IFN-γ-treated HeLa cells. Since Chlamydia spp. scavenge ribonucleotide triphosphates from their hosts and cannot synthesize purines and pyrimidines (72), they may need to cope with endogenously generated as well as imported hydroxylated nucleobases. We hypothesize that the reduced size of CTL0694P105L inclusions under IFN-γ-plus-tryptophan conditions could reflect DNA damage and altered protein production. Additionally, the disparity between CTL0694P105L genome accumulation and EB production in reactivation experiments could indicate that genome replication continues but produces damaged chromosomes leading to fewer infectious EBs and that this is may also be independent of inclusion size. The function of CTL0694 and its potential interaction partner(s) in chlamydia will need to be determined in future studies.
In summary, C. trachomatis survival and reactivation during IFN-γ persistence are not solely mediated by tryptophan synthase. The genes identified in the screen could open new lines of investigation that alter the classical view of chlamydial persistence. Extending our approach by generating a larger mutant library could help to uncover additional genes that play roles in persistence. Testing if these genes contribute to C. trachomatis pathogenesis would be ideal, but this not feasible with all of the genes due to the lack of a relevant animal model. However, since CTL0225 and CTL0694 are highly conserved in other Chlamydia spp., including C. muridarum, a future direction would be to test if these genes contribute to chlamydial survival and pathogenesis in mice.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeffrey Chang, Stan Spinola, Richard Morrison, Raphael Valdivia, and Byron Batteiger and members of the Nelson laboratory for their help in reviewing drafts of the manuscript and experimental advice.
Work described in this article was supported by NIH grant AI099278 to D.E.N. The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00356-16.
REFERENCES
- 1.Moulder JW. 1985. Comparative biology of intracellular parasitism. Microbiol Rev 49:298–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis ME, Belland RJ, AbdelRahman YM, Beatty WL, Aiyar AA, Zea AH, Greene SJ, Marrero L, Buckner LR, Tate DJ, McGowin CL, Kozlowski PA, O'Brien M, Lillis RA, Martin DH, Quayle AJ. 2014. Morphologic and molecular evaluation of Chlamydia trachomatis growth in human endocervix reveals distinct growth patterns. Front Cell Infect Microbiol 4:71. doi: 10.3389/fcimb.2014.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanna L, Merigan TC, Jawetz E. 1966. Inhibition of TRIC agents by virus-induced interferon. Proc Soc Exp Biol Med 122:417–421. doi: 10.3181/00379727-122-31150. [DOI] [PubMed] [Google Scholar]
- 5.Shemer-Avni Y, Wallach D, Sarov I. 1988. Inhibition of Chlamydia trachomatis growth by recombinant tumor necrosis factor. Infect Immun 56:2503–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrne GI, Lehmann LK, Landry GJ. 1986. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun 53:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coles AM, Reynolds DJ, Harper A, Devitt A, Pearce JH. 1993. Low-nutrient induction of abnormal chlamydial development: a novel component of chlamydial pathogenesis? FEMS Microbiol Lett 106:193–200. doi: 10.1111/j.1574-6968.1993.tb05958.x. [DOI] [PubMed] [Google Scholar]
- 8.Raulston JE. 1997. Response of Chlamydia trachomatis serovar E to iron restriction in vitro and evidence for iron-regulated chlamydial proteins. Infect Immun 65:4539–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson CC, Carabeo RA. 2011. An optimal method of iron starvation of the obligate intracellular pathogen, Chlamydia trachomatis. Front Microbiol 2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deka S, Vanover J, Dessus-Babus S, Whittimore J, Howett MK, Wyrick PB, Schoborg RV. 2006. Chlamydia trachomatis enters a viable but non-cultivable (persistent) state within herpes simplex virus type 2 (HSV-2) co-infected host cells. Cell Microbiol 8:149–162. doi: 10.1111/j.1462-5822.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto A, Manire GP. 1970. Electron microscopic observations on the fine structure of cell walls of Chlamydia psittaci. J Bacteriol 104:1332–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kramer MJ, Gordon FB. 1971. Ultrastructural analysis of the effects of penicillin and chlortetracycline on the development of a genital tract Chlamydia. Infect Immun 3:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wyrick PB. 2010. Chlamydia trachomatis persistence in vitro: an overview. J Infect Dis 201(Suppl 2):S88–S95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meunier E, Broz P. 2016. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol 18:168–180. doi: 10.1111/cmi.12546. [DOI] [PubMed] [Google Scholar]
- 16.Beatty WL, Byrne GI, Morrison RP. 1993. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc Natl Acad Sci U S A 90:3998–4002. doi: 10.1073/pnas.90.9.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasui H, Takai K, Yoshida R, Hayaishi O. 1986. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurrence in cancer patients. Proc Natl Acad Sci U S A 83:6622–6626. doi: 10.1073/pnas.83.17.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belland RJ, Nelson DE, Virok D, Crane DD, Hogan D, Sturdevant D, Beatty WL, Caldwell HD. 2003. Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc Natl Acad Sci U S A 100:15971–15976. doi: 10.1073/pnas.2535394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison RP. 2000. Differential sensitivities of Chlamydia trachomatis strains to inhibitory effects of gamma interferon. Infect Immun 68:6038–6040. doi: 10.1128/IAI.68.10.6038-6040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, Maclean I, Mohammed Z, Peeling R, Roshick C, Schachter J, Solomon AW, Stamm WE, Suchland RJ, Taylor L, West SK, Quinn TC, Belland RJ, McClarty G. 2003. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest 111:1757–1769. doi: 10.1172/JCI17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fehlner-Gardiner C, Roshick C, Carlson JH, Hughes S, Belland RJ, Caldwell HD, McClarty G. 2002. Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J Biol Chem 277:26893–26903. [DOI] [PubMed] [Google Scholar]
- 22.Carlson JH, Porcella SF, McClarty G, Caldwell HD. 2005. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect Immun 73:6407–6418. doi: 10.1128/IAI.73.10.6407-6418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris SR, Clarke IN, Seth-Smith HM, Solomon AW, Cutcliffe LT, Marsh P, Skilton RJ, Holland MJ, Mabey D, Peeling RW, Lewis DA, Spratt BG, Unemo M, Persson K, Bjartling C, Brunham R, de Vries HJ, Morre SA, Speksnijder A, Bebear CM, Clerc M, de Barbeyrac B, Parkhill J, Thomson NR. 2012. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet 44:413–419. doi: 10.1038/ng.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 25.Sasaki-Imamura T, Yoshida Y, Suwabe K, Yoshimura F, Kato H. 2011. Molecular basis of indole production catalyzed by tryptophanase in the genus Prevotella. FEMS Microbiol Lett 322:51–59. doi: 10.1111/j.1574-6968.2011.02329.x. [DOI] [PubMed] [Google Scholar]
- 26.Romanik M, Martirosian G, Wojciechowska-Wieja A, Cieslik K, Kazmierczak W. 2007. Co-occurence of indol-producing bacterial strains in the vagina of women infected with Chlamydia trachomatis. Ginekol Pol 78:611–615. (In Polish.) [PubMed] [Google Scholar]
- 27.Lloyd D, Lauritsen FR, Degn H. 1991. The parasitic flagellates Trichomonas vaginalis and Tritrichomonas foetus produce indole and dimethyl disulphide: direct characterization by membrane inlet tandem mass spectrometry. J Gen Microbiol 137:1743–1747. doi: 10.1099/00221287-137-7-1743. [DOI] [PubMed] [Google Scholar]
- 28.Brotman RM, Klebanoff MA, Nansel TR, Yu KF, Andrews WW, Zhang J, Schwebke JR. 2010. Bacterial vaginosis assessed by Gram stain and diminished colonization resistance to incident gonococcal, chlamydial, and trichomonal genital infection. J Infect Dis 202:1907–1915. doi: 10.1086/657320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fredricks DN, Fiedler TL, Marrazzo JM. 2005. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med 353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- 30.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ. 2011. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A 108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aiyar A, Quayle AJ, Buckner LR, Sherchand SP, Chang TL, Zea AH, Martin DH, Belland RJ. 2014. Influence of the tryptophan-indole-IFNgamma axis on human genital Chlamydia trachomatis infection: role of vaginal co-infections. Front Cell Infect Microbiol 4:72. doi: 10.3389/fcimb.2014.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McClarty G, Caldwell HD, Nelson DE. 2007. Chlamydial interferon gamma immune evasion influences infection tropism. Curr Opin Microbiol 10:47–51. doi: 10.1016/j.mib.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Nelson DE, Virok DP, Wood H, Roshick C, Johnson RM, Whitmire WM, Crane DD, Steele-Mortimer O, Kari L, McClarty G, Caldwell HD. 2005. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A 102:10658–10663. doi: 10.1073/pnas.0504198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas SM, Garrity LF, Brandt CR, Schobert CS, Feng GS, Taylor MW, Carlin JM, Byrne GI. 1993. IFN-gamma-mediated antimicrobial response. Indoleamine 2,3-dioxygenase-deficient mutant host cells no longer inhibit intracellular Chlamydia spp. or Toxoplasma growth. J Immunol 150:5529–5534. [PubMed] [Google Scholar]
- 35.Kari L, Whitmire WM, Carlson JH, Crane DD, Reveneau N, Nelson DE, Mabey DC, Bailey RL, Holland MJ, McClarty G, Caldwell HD. 2008. Pathogenic diversity among Chlamydia trachomatis ocular strains in nonhuman primates is affected by subtle genomic variations. J Infect Dis 197:449–456. doi: 10.1086/525285. [DOI] [PubMed] [Google Scholar]
- 36.Tietzel I, El-Haibi C, Carabeo RA. 2009. Human guanylate binding proteins potentiate the anti-chlamydia effects of interferon-gamma. PLoS One 4:e6499. doi: 10.1371/journal.pone.0006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Zeer MA, Al-Younes HM, Lauster D, Abu Lubad M, Meyer TF. 2013. Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG-inducible guanylate binding proteins. Autophagy 9:50–62. doi: 10.4161/auto.22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston AC, Piro A, Clough B, Siew M, Virreira Winter S, Coers J, Frickel EM. 10 March 2016. Human GBP1 does not localize to pathogen vacuoles but restricts Toxoplasma gondii. Cell Microbiol doi: 10.1111/cmi.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajaram K, Nelson DE. 2015. Chlamydia muridarum infection of macrophages elicits bactericidal nitric oxide production via reactive oxygen species and cathepsin B. Infect Immun 83:3164–3175. doi: 10.1128/IAI.00382-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brothwell JA, Muramatsu MK, Toh E, Rockey DD, Putman TE, Barta ML, Hefty PS, Suchland RJ, Nelson DE. 13 July 2016. Interrogating genes that mediate Chlamydia trachomatis survival in cell culture using conditional mutants and recombination. J Bacteriol doi: 10.1128/JB.00161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajaram K, Giebel AM, Toh E, Hu S, Newman JH, Morrison SG, Kari L, Morrison RP, Nelson DE. 2015. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun 83:2870–2881. doi: 10.1128/IAI.00106-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto A, Izutsu H, Miyashita N, Ohuchi M. 1998. Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J Clin Microbiol 36:3013–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Putman TE, Suchland RJ, Ivanovitch JD, Rockey DD. 2013. Culture-independent sequence analysis of Chlamydia trachomatis in urogenital specimens identifies regions of recombination and in-patient sequence mutations. Microbiology 159:2109–2117. doi: 10.1099/mic.0.070029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonhardt RM, Lee SJ, Kavathas PB, Cresswell P. 2007. Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infect Immun 75:5105–5117. doi: 10.1128/IAI.00668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A 108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gérard HC, Krausse-Opatz B, Wang Z, Rudy D, Rao JP, Zeidler H, Schumacher HR, Whittum-Hudson JA, Köhler L, Hudson AP. 2001. Expression of Chlamydia trachomatis genes encoding products required for DNA synthesis and cell division during active versus persistent infection. Mol Microbiol 41:731–741. doi: 10.1046/j.1365-2958.2001.02550.x. [DOI] [PubMed] [Google Scholar]
- 49.Braun PR, Al-Younes H, Gussmann J, Klein J, Schneider E, Meyer TF. 2008. Competitive inhibition of amino acid uptake suppresses chlamydial growth: involvement of the chlamydial amino acid transporter BrnQ. J Bacteriol 190:1822–1830. doi: 10.1128/JB.01240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suchland RJ, Sandoz KM, Jeffrey BM, Stamm WE, Rockey DD. 2009. Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother 53:4604–4611. doi: 10.1128/AAC.00477-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Binet R, Maurelli AT. 2009. The chlamydial functional homolog of KsgA confers kasugamycin sensitivity to Chlamydia trachomatis and impacts bacterial fitness. BMC Microbiol 9:279. doi: 10.1186/1471-2180-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Binet R, Maurelli AT. 2005. Fitness cost due to mutations in the 16S rRNA associated with spectinomycin resistance in Chlamydia psittaci 6BC. Antimicrob Agents Chemother 49:4455–4464. doi: 10.1128/AAC.49.11.4455-4464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binet R, Maurelli AT. 2007. Frequency of development and associated physiological cost of azithromycin resistance in Chlamydia psittaci 6BC and C. trachomatis L2. Antimicrob Agents Chemother 51:4267–4275. doi: 10.1128/AAC.00962-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Binet R, Bowlin AK, Maurelli AT, Rank RG. 2010. Impact of azithromycin resistance mutations on the virulence and fitness of Chlamydia caviae in guinea pigs. Antimicrob Agents Chemother 54:1094–1101. doi: 10.1128/AAC.01321-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mueller KE, Wolf K, Fields KA. 2016. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. mBio 7(1):e01817-15. doi: 10.1128/mBio.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carlson JH, Wood H, Roshick C, Caldwell HD, McClarty G. 2006. In vivo and in vitro studies of Chlamydia trachomatis TrpR:DNA interactions. Mol Microbiol 59:1678–1691. doi: 10.1111/j.1365-2958.2006.05045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akers JC, Tan M. 2006. Molecular mechanism of tryptophan-dependent transcriptional regulation in Chlamydia trachomatis. J Bacteriol 188:4236–4243. doi: 10.1128/JB.01660-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akahane S, Kamata H, Yagisawa H, Hirata H. 2003. A novel neutral amino acid transporter from the hyperthermophilic archaeon Thermococcus sp. KS-1. J Biochem 133:173–180. [DOI] [PubMed] [Google Scholar]
- 60.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsu BV, Saier MH Jr. 2015. The LysE superfamily of transport proteins involved in cell physiology and pathogenesis. PLoS One 10:e0137184. doi: 10.1371/journal.pone.0137184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 63.Eschenbrenner M, Coves J, Fontecave M. 1995. The flavin reductase activity of the flavoprotein component of sulfite reductase from Escherichia coli. A new model for the protein structure. J Biol Chem 270:20550–20555. [DOI] [PubMed] [Google Scholar]
- 64.Li C, Peck HD Jr, Przybyla AE. 1987. Cloning of the 3′-phosphoadenylyl sulfate reductase and sulfite reductase genes from Escherichia coli K-12. Gene 53:227–234. doi: 10.1016/0378-1119(87)90011-4. [DOI] [PubMed] [Google Scholar]
- 65.Kozmin SG, Wang J, Schaaper RM. 2010. Role for CysJ flavin reductase in molybdenum cofactor-dependent resistance of Escherichia coli to 6-N-hydroxylaminopurine. J Bacteriol 192:2026–2033. doi: 10.1128/JB.01438-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White RH. 2010. The twists and turns of enzyme function. J Bacteriol 192:2023–2025. doi: 10.1128/JB.00087-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Havemeyer A, Grunewald S, Wahl B, Bittner F, Mendel R, Erdelyi P, Fischer J, Clement B. 2010. Reduction of N-hydroxy-sulfonamides, including N-hydroxy-valdecoxib, by the molybdenum-containing enzyme mARC. Drug Metab Dispos 38:1917–1921. doi: 10.1124/dmd.110.032813. [DOI] [PubMed] [Google Scholar]
- 68.Chamizo-Ampudia A, Galvan A, Fernandez E, Llamas A. 2011. The Chlamydomonas reinhardtii molybdenum cofactor enzyme crARC has a Zn-dependent activity and protein partners similar to those of its human homologue. Eukaryot Cell 10:1270–1282. doi: 10.1128/EC.05096-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simandan T, Sun J, Dix TA. 1998. Oxidation of DNA bases, deoxyribonucleosides and homopolymers by peroxyl radicals. Biochem J 335(Pt 2):233–240. doi: 10.1042/bj3350233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plitzko B, Havemeyer A, Kunze T, Clement B. 2015. The pivotal role of the mitochondrial amidoxime reducing component 2 in protecting human cells against apoptotic effects of the base analog N6-hydroxylaminopurine. J Biol Chem 290:10126–10135. doi: 10.1074/jbc.M115.640052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aune TM, Pogue SL. 1989. Inhibition of tumor cell growth by interferon-gamma is mediated by two distinct mechanisms dependent upon oxygen tension: induction of tryptophan degradation and depletion of intracellular nicotinamide adenine dinucleotide. J Clin Invest 84:863–875. doi: 10.1172/JCI114247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tipples G, McClarty G. 1993. The obligate intracellular bacterium Chlamydia trachomatis is auxotrophic for three of the four ribonucleoside triphosphates. Mol Microbiol 8:1105–1114. doi: 10.1111/j.1365-2958.1993.tb01655.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.