

Abstract
In response to a defined panel of stimuli, immature macrophages can be classified into two major phenotypes: proinflammatory (M1) and anti-inflammatory (M2). Although both phenotypes have been implicated in several chronic inflammatory diseases, their direct role in bone resorption remains unclear. The present study investigated the possible effects of M1 and M2 macrophages on RANKL-induced osteoclastogenesis. In osteoclastogenesis assays using RAW264.7 cells or bone marrow cells as osteoclast precursors, addition of M1 macrophages significantly suppressed RANKL-induced osteoclastogenesis compared to nonstimulated conditions (M0), addition of M2 macrophages, or no macrophage addition (P < 0.05), suggesting that M1 macrophages can downregulate osteoclastogenesis. This effect was maintained when direct contact between M1 and osteoclast precursors was interrupted by cell culture insertion, indicating engagement of soluble factors released from M1. M1 macrophages developed from interferon gamma (IFN-γ) knockout (IFN-γ–KO) mice lost the ability to downregulate osteoclastogenesis. Antibody-based neutralization of interleukin-12 (IL-12), but not IL-10, produced by M1 macrophages also abrogated M1-mediated downregulation of osteoclastogenesis. Real-time PCR analyses showed that IFN-γ suppressed gene expression of NFATc1, a master regulator of osteoclastogenesis, whereas IL-12 increased the apoptosis of osteoclasts, suggesting molecular mechanisms underlying the possible roles of IFN-γ or IL-12 in M1-mediated inhibition of osteoclastogenesis. These findings were confirmed in an in vivo ligature-induced mouse periodontitis model in which adoptive transfer of M1 macrophages showed a significantly lower level of bone loss and less tartrate-resistant acid phosphatase (TRAP)-positive cell induction than M0 or M2 macrophage transfer. In conclusion, by its secretion of IFN-γ and IL-12, M1, but not M0 or M2, was demonstrated to inhibit osteoclastogenesis.
INTRODUCTION
Macrophages which originate from monocytes not only are the key effector cells in innate immunity but also play a pivotal role in the initiation of adaptive immunity (1). It is well documented that polarized macrophages can be classified mainly into two different phenotypes: proinflammatory (M1) and anti-inflammatory (M2). The production of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), by M1 macrophages promotes inflammation in the context of innate immune response, whereas the production of anti-inflammatory cytokines and arginase by M2 macrophages leads to the resolution of inflammation (2). On the other hand, it is also true that osteoclasts that are engaged in bone resorption also belong to monocyte-lineage cells. Although macrophages and osteoclasts share the same precursor, macrophage colony-stimulating factor (M-CSF)-stimulated monocytes, the possible influence of macrophages, and especially the difference between M1 and M2, on osteoclastogenesis is largely unknown.
Bone is a unique mineralized tissue which constantly undergoes a physiological remodeling process, and its homeostasis is achieved by the tuned balance between osteoclasts and bone-forming cells (osteoblasts). As such, aberrantly promoted osteoclastogenesis is attributed to the bone destruction found in bone lytic diseases such as periodontitis and rheumatoid arthritis, which affects more than 50 million people in the United States alone (3). Of importance to this study, recent research has revealed that osteoclastogenesis is regulated by the immune system. For instance, a recent study (4) revealed that γδ-T cells inhibit osteoclastogenesis by their production of interferon gamma (IFN-γ), whereas B and T cells can produce RANKL under inflammatory conditions, thus working toward the promotion of osteoclastogenesis (5). However, in the context of bone lytic diseases involving chronic inflammation, such as periodontitis and rheumatoid arthritis, infiltrations of not only B and T cells but also of macrophages are observed (6, 7). It is true that macrophages are the most abundant immune cells found in the synovial membrane in osteoarthritis (8) and in synovial fluid in rheumatoid arthritis (9), outnumbering T and B cells. Therefore, it is plausible that osteoclast differentiation is affected by local factors produced by infiltrating lymphocytes, especially macrophages. However, as noted above, it has not been established that macrophages have any regulatory effect on osteoclastogenesis. In this paper, we analyzed the possible regulatory effects of macrophages on in vitro RANKL-induced osteoclastogenesis by comparing two major polarized macrophages, M1 and M2.
MATERIALS AND METHODS
Animals.
IFN-γ knockout (KO) mice (B6: 129S7-Ifngtm1Ts/J; Jackson Laboratory, Bar Harbor, ME, USA), as well as the wild type (WT), C57BL/6j mice, were kept in the Forsyth Animal Facility. The experimental protocols used in this study were approved by the Forsyth IACUC.
Cell culture.
All cell types used in this study were cultured with Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 unit/ml penicillin, 100 μg/ml streptomycin, and 0.3 mg/ml glutamine. To induce osteoclastogenesis, mouse bone marrow (BM) cells were seeded at 105 cells/well in a 96-well plate and preincubated with M-CSF (BioLegend) (30 ng/ml) alone for 6 days. Subsequently, preincubated BM cells were stimulated with M-CSF (30 ng/ml) and RANKL (100 ng/ml, BioLegend) for an additional 7 days. In order to generate M1 and M2 macrophages, M-CSF-pretreated BM cells were stimulated with IFN-γ (BioLegend) (10 ng/ml) in the presence of lipopolysaccharide (LPS) (Sigma-Aldrich) (10 ng/ml LPS from Escherichia coli O26:B6) for M1 or with IL-4 alone (Biolegend) (20 ng/ml) for M2 for 24 h. Where stated, Porphyromonas gingivalis LPS (Invitrogen) was also used with IFN-γ (10 ng/ml) to induce M1 macrophages. To evaluate the effect of macrophages on osteoclastogenesis, M1 or M2 macrophages were harvested from the plate, washed and, applied at 6,000 cells/well to the osteoclastogenesis assay at various times after stimulation with RANKL and M-CSF. As a control, nonpolarized (M0) macrophages developed by M-CSF stimulation alone were used. In some experiments, M0, M1, or M2 macrophages housed in a cell culture insertion (96-well Transwell) (Corning Costar 96-well cell culture plates; Corning Incorporated Life Sciences) (pore size, 0.4 μm) were applied on top of the osteoclastogenesis assay prepared in the wells of a 96-well plate.
Neutralizing antibodies (Ab) for IL-10 (BioLegend) and IL-12 (Abcam) were added at a final concentration of 1 μg/ml.
TRAP staining.
After 7 days of culture with RANKL, cells were fixed by 4% paraformaldehyde, washed with phosphate-buffered saline (PBS), and stained for tartrate-resistant acid phosphatase (TRAP) using a leukocyte acid phosphatase kit (Sigma-Aldrich) as previously described (10). TRAP-positive multinucleated (>3 nuclei) cells were counted as osteoclasts.
Pit formation assay.
BM cells were preincubated with M-CSF (30 ng/ml) alone for 6 days, followed by stimulation with M-CSF (30 ng/ml) and RANKL (100 ng/ml) in the presence or absence of conditioned media collected from M0 and M1 macrophages in a 96-well Corning Osteo Assay surface plate (Corning Inc.). Seven days after RANKL addition, the plates were washed with sodium hypochlorite and air dried. Wells were imaged with a 4× objective using an EVOS cell imaging system. Image analysis was carried out with NIH ImageJ software.
Conditioned media harvested from cultured macrophages.
Conditioned media from M0, M1, and M2 macrophages were prepared as follows. After pretreatment of BM cells with M-CSF (30 ng/ml) for 6 days, the cells were stimulated with LPS (10 ng/ml), IFN-γ (10 ng/ml), or IL-4 (20 ng/ml) for 24 h or left unstimulated. In order to remove LPS, IFN-γ, or IL-4 that had been used for polarization of M0, M1, and M2 macrophages, the cultures of macrophages were washed twice with PBS, which was then replaced with fresh DMEM–10% FBS. The supernatants harvested from the subsequent incubation in fresh DMEM–10% FBS for 24 h were used as the conditioned media.
Real-time PCR.
RNA was isolated from cells using E.Z.N.A. Total RNA kit I (Omega Biotek), following the manufacturer's instructions, and was subjected to reverse transcription with a Verso cDNA synthesis kit (Thermo Scientific) in the presence of random primers and oligo(dT). Gene expression was quantified using LightCycler 480 SYBR green I Master (Roche Diagnostics), and the primer sequences used in this study are listed in Table 1. Amplification of the beta-actin gene was used as an internal control.
TABLE 1.
Sequences of the primers used in this study
| Gene product | Primer sequence |
|
|---|---|---|
| Forward | Reverse | |
| Cathepsin K | 5′-AATGCTGGACACCCAGTGGGA-3′ | 5′-GAGGCCTCCAGGTTATGGGC-3′ |
| RANK | 5′-GCTTGCATAAAGTCTGTGA-3′ | 5′-AGTCTGAGTTCCAGTGGTAGCC-3′ |
| iNOS | 5′-CACCTCACTGTGGCCGTGGT-3′ | 5′-GGAAGCCACTGACACTTCGCAC-3′ |
| CD206 | 5′-ACGTTTCGGTGGACTGTGGA-3′ | 5′-GGCAACACATCCCGCCTTTC-3′ |
| IFN-γ | 5′-ACGGCACAGTCATTGAAAGCCTAGA-3′ | 5′-TGTCACCATCCTTTTGCCAGTTCC-3′ |
| IL-10 | 5′-AATTCCCTGGGTGAGAAGCTGA-3′ | 5′-CTCTTCACCTGCTCCACTGCC-3′ |
| IL-12α | 5′-TCACACGGGACCAAACCAGC-3′ | 5′-GCAGGCAGCTCCCTCTTGTT-3′ |
| NFATc1 | 5′-CCTCGAACCCTATCGAGTGT-3′ | 5′-GCCAGACAGCACCATCTTC-3′ |
| Caspase-3 | 5′-ACATGGGAGCAAGTCAGTGGAC-3′ | 5′-GTCCACATCCGTACCAGAGCG-3′ |
| Beta-actin | 5′-TTGTTACCAACTGGGACGAC-3′ | 5′-GCTGGGGTGTTGAAGGTC-3′ |
ELISA.
Conditioned media from M0/M1 or M2 macrophages were subjected to enzyme-linked immunosorbent assay (ELISA) (Duo-set ELISA kits; R&D Systems) for measurement of IFN-γ, IL-12 p70, TNF-α, IL-10, and TGF-β levels following the instructions of the manufacturer.
Annexin V apoptosis assay.
Following 6 days of pretreatment with M-CSF alone, BM cells were cultured with M-CSF and RANKL (100 ng/ml) in the presence or absence of conditioned media of M0 or M1 macrophages with or without recombinant IL-12 (50 ng/ml) and/or IL-12 neutralizing antibody (1 μg/ml). At 96 h after the addition of RANKL with or without the other reagents noted above, apoptotic annexin V-positive cells were stained using an apoptotic/necrotic cell detection kit (Promokine, Heidelberg, Germany), following the manufacturer's protocol.
Adoptive transfer of macrophages to a mouse model of ligature-induced periodontitis.
A silk suture (Ethicon; size 5-0) was placed on the left side maxillary second molar of C57/BL6j mice (8-week-old males). The corresponding right side molar served as the nonligature control side. One day after ligature placement, M1, M2, or M0 macrophages were transferred in PBS via intraperitoneal (i.p.) injection at 107 cells/mouse. Each subset of the macrophages that were used for transfer was established ex vivo following the protocol described in the “Cell culture” section above. Four days after the macrophage transfer, mice were sacrificed. The jaw bones were defleshed by a method using Dermestid beetles (11). Alveolar bone loss was quantified by measuring the distance between the cementoenamel junction (CEJ) and the alveolar bone crest (AB) as reported previously (12), with some modifications. Briefly, the images of defleshed jaw bone were captured using a stereomicroscope on a custom-made stage holder with a reticule eyepiece at a magnification of ×25 to facilitate visualization of the cement-enamel junction and the alveolar bone ledge. A total of three measurements of the CEJ-AB distance were made for each hemimaxilla on the palatal side using Image-J (NIH), including the long axis of the distal root of the first molar and two cusps of the second molar (see Fig. 10A). The difference in the measured CEJ-AB distances between the left ligatured side and the right control side represented the alveolar bone loss of each mouse.
FIG 10.

Suppressive effects of M1 macrophage on in vivo bone resorption induced in a mouse model of periodontitis. Alveolar bone loss was induced by the placement of a silk ligature around the left maxillary second molar, while the right maxillary second molar, which did not receive the ligature, served as a control. One day after the ligature attachment, M1, M2, or M0 macrophages were adoptively transferred via systemic intraperitoneal (i.p.) injection. Four days after macrophage transfer, all groups of mice were sacrificed and alveolar bone loss (A and B) and emergence of TRAP+ osteoclasts in the periodontal tissue section (C and D) were monitored. (A) Representative photographic images of alveolar bone. The measurement line (yellow) and CEJ line (black) are shown. The bars at the top right corner indicate 1 mm. (B) The quantitative data of bone loss are shown in the histogram. Column and bar data indicate means ± SD (n = 7/group). The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by Tukey's test. (C) Representative histological images of TRAP+ osteoclasts that emerged in the alveolar bone of mice that received ligature and macrophage (M1, M2, or M0) transfer are shown. TRAP-positive cells in palatal alveolar bone are indicated by the arrows. Scale bars, 100 μm. (D) Numbers of osteoclasts per area (200 μm by 200 μm) on the palatal alveolar bone surface were quantified. Column and bar data indicate means ± SD (n = 7). The asterisks indicate significant differences (P < 0.05) by Tukey's test.
Histochemical analyses.
Maxillary jaws dissected from sacrificed mice were fixed in 4% paraformaldehyde and decalcified in 10% EDTA solution. These maxillary jaws were embedded in Tissue-Tek OTC compound (Sakura Finetek) and sectioned (7 μm in thickness) in the buccal-lingual plane using a cryostat. The resulting sections mounted on the slide glass were stained for TRAP (Sigma-Aldrich), followed by counterstaining of nuclei with hematoxylin.
Statistical analysis.
All quantitative data are shown as means ± standard deviations. Differences between the groups were analyzed using one-way analysis of variance (ANOVA) with Tukey's post hoc multiple-comparison test unless stated otherwise, and P values of <0.05 were considered to represent significant differences.
RESULTS
M1 macrophages, but not M0 or M2 macrophages, inhibit RANKL-induced osteoclastogenesis.
When M1 macrophages derived from RAW 264.7 cells (500 cells/well) were added to RAW 264.7 cells stimulated with RANKL at day 3 or day 6, M1 macrophages significantly inhibited osteoclastogenesis compared to the control group without any cell addition or the groups that received addition of M0 or M2 macrophages (Fig. 1). When macrophages were added at a lower concentration (100 cells/well), the inhibitory effect by M1 was abolished, while M0 or M2 still showed no effect (not shown), suggesting dose-dependent inhibitory effects by M1 macrophages. Importantly, inhibition of osteoclastogenesis by M1, but not M0 or M2, was also observed when BM cells were used as osteoclast precursors, as well as when they were used as a source of macrophages (Fig. 2A and B). When RNA was collected from the RANKL-stimulated, BM-derived osteoclasts 3 days after macrophage addition, the group that received M1 showed significantly diminished mRNA expression for cathepsin K, a key enzyme required for bone resorption, compared to the control group with no cell addition or the group that received either M0 or M2, indicating that M1 also suppresses the functional activity of osteoclasts (Fig. 2C).
FIG 1.
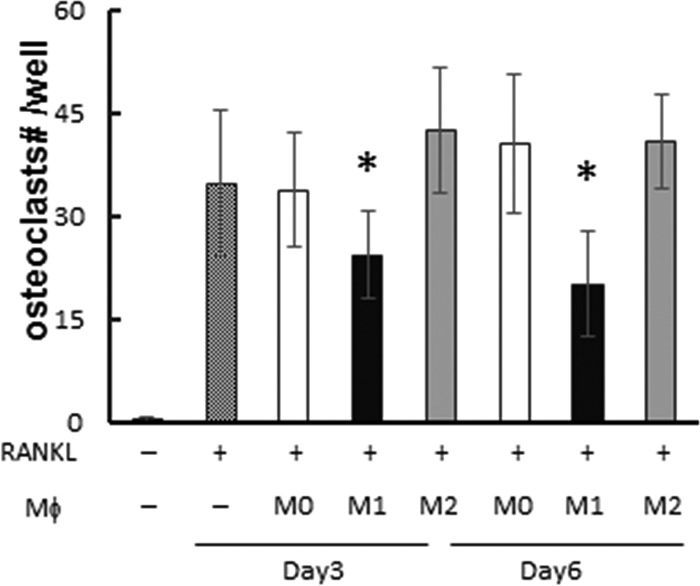
Inhibition of osteoclastogenesis by M1 macrophages in RAW264.7 cells. RAW264.7 cells were differentiated to osteoclasts by stimulation with RANKL. On either day 3 or day 6 after RANKL addition, M0, M1, or M2 derived from RAW264.7 cells were added to the well (500 cells/well). On day 7, TRAP staining was performed and multinuclear (MN) TRAP+ cells in each well were counted as mature osteoclasts. Column and bar data indicate means and standard deviations (SD), respectively (n = 5/group). *, significantly different by Dunnett's test versus no-macrophage-addition group (crosshatched column) (P < 0.05). (−), no addition of macrophages. No osteoclasts were observed without RANKL addition.
FIG 2.

Effects of M1 macrophages on osteoclastogenesis induced in BM macrophages. After BM cells were cultured with M-CSF for 6 days, RANKL was applied to the BM culture (day 0). M0, M1, or M2 macrophages were generated ex vivo by incubating BM cells with M-CSF for 6 days, which was followed by incubation using M-CSF with or without LPS, IFN-γ, or IL-4 for an additional 24 h. The harvested M1, M2, and M0 macrophages from ex vivo culture were applied to osteoclasts on day 3 or day 6 (6,000 macrophages/well). On day 7, TRAP staining was performed. (A) Images of TRAP staining for RANKL-stimulated BM cells cocultured with or without polarized macrophages are shown. Bars indicate 100 μm. (B) The number of TRAP+ multinuclear osteoclasts was calculated. Data are means ± SD (n = 5). *, significant difference by Tukey's test (P < 0.05). (C) BM cells were cultured for 6 days with M-CSF only, and on day 6, media were changed to M-CSF and RANKL. Three days after RANKL addition, M0, M1, or M2 macrophages were applied to BM culture. After coculture of BM cells and macrophages for an additional 4 days, RNA was extracted and real-time PCR performed. The readout of each sample was converted to a relative expression level (REL) value using beta-actin as the internal control. Data in the graph represent means of REL values ± SD (n = 3). *, significant difference by one-way ANOVA with post hoc Tukey's test (P < 0.05).
The soluble factor(s) produced by M1 is responsible for the inhibition of osteoclastogenesis.
In order to elucidate whether soluble factors, or membrane-bound factors, produced by M1 are responsible for the inhibition of osteoclastogenesis caused by M1 macrophages, macrophages were separated by culture insertion (pore size, 0.4 μm) from BM-derived osteoclast precursors in the coculture system. Even in the absence of cell-cell contact between macrophages and osteoclast precursors, significant inhibition of osteoclastogenesis by M1, but not by M0 or M2, was observed, suggesting that a soluble factor(s) produced by M1 is responsible for the inhibition of osteoclastogenesis (Fig. 3A). Furthermore, incubation with conditioned medium containing soluble factors only and collected from M1 macrophages but not M0 macrophages suppressed the pit formation induced by mature osteoclasts derived from bone marrow (Fig. 3B and C), suggesting that M1 can suppress the osteoclast's function to resorb bone. These results further suggested that soluble factors produced by M1 macrophages have an inhibitory effect on RANKL-mediated osteoclastogenesis.
FIG 3.

Effects of soluble factors produced by macrophages on RANKL-induced osteoclastogenesis. (A) BM cells were differentiated to osteoclasts by stimulation with RANKL on the bottom well of a culture insertion system. Three days after RANKL addition, M0, M1, or M2 macrophages were added to the upper chamber of the culture insertion. Seven days after the addition of RANKL, TRAP staining was performed on the bottom well. Data are means and SD (n = 6). The asterisks indicate significant differences by Tukey's test (P < 0.05). (B and C) BM cells stimulated with M-CSF and RANKL were incubated in the presence of conditioned media collected from M0 and M1 differentiated macrophages in a Corning Osteo Assay Surface plate for 7 days. The control group received no conditioned media. (B) Representative images of pit formation. (C) Data in the pit area in each well were quantified by the use of ImageJ. Data are means ± SD (n = 5). Asterisks show significant differences by Tukey's test (*, P < 0.05; **, P < 0.01).
Osteoclastogenesis-regulatory cytokines produced by M1 macrophages were detected.
To elucidate the mechanisms underlying the inhibition of osteoclastogenesis caused by M1 macrophages, we analyzed the expression of cytokines reported to have regulatory effects on osteoclastogenesis (13–16). At 24 h after M1 or M2 induction performed by addition of LPS, IFN-γ, or IL-4 to BM cells pretreated with M-CSF, cells were harvested for RNA extraction and analyzed by real-time PCR (Fig. 4). Furthermore, to confirm the cytokine expression at the protein level, after 24 h of stimulation with LPS, IFN-γ, or IL-4, macrophages were removed from the stimulants and recultured in fresh medium. The culture supernatants harvested from the latter reculture were subjected to cytokine ELISA (Table 2). First, the polarization of M1 and M2 from BM cells was confirmed by the expression of their signature genes, encoding inducible nitric oxide synthase (iNOS) and CD206, respectively. In addition to the polarization by E. coli LPS, M1 polarization by P. gingivalis LPS was also confirmed. M1 macrophages produced significantly higher levels of IFN-γ, IL-10, IL-12, and TNF-α than M0 or M2 macrophages. On the other hand, M2 macrophages produced significantly higher levels of TGF-β. These results indicated that at least one of the antiosteoclastogenesis factors produced by M1, e.g., IFN-γ, IL-10, or IL-12, might be engaged in the suppression of osteoclastogenesis.
FIG 4.

Gene expression levels in M0/M1/M2 macrophages. BM cells were preincubated for 6 days with M-CSF alone. At day 6, culture media were replaced with fresh medium containing the M1 or M2 differentiation factors, while M-CSF-supplemented fresh medium was applied for M0 induction. Twenty-four hours after stimulation, RNA was extracted and subject to real-time PCR. The relative expression level (REL) of each mRNA was calculated (see the Fig. 2 legend). Columns labeled with “M1” indicate results from the group stimulated with E. coli LPS at 10 ng/ml (Ec10); columns labeled with “Pg250,” “Pg50,” and“Pg10” show results from the groups stimulated with P. gingivalis LPS at concentrations of 250, 50, and 10 ng/ml, respectively. Data are means of REL and SD (n = 3). *, P < 0.05.
TABLE 2.
Cytokines produced by M0, M1, and M2 macrophagesa
| Cytokine | Macrophage level (pg/ml) ± SD |
||
|---|---|---|---|
| M0 | M1 | M2 | |
| IFN-γ | 1.8 ± 1.0 | 45.3 ± 10.4* | 3.1 ± 2.9 |
| IL-12 p70 | 7.7 ± 5.3 | 29.2 ± 3.7* | 8.3 ± 2.5 |
| TNF-α | 6.1 ± 4.7 | 765.4 ± 35.8* | 13.6 ± 5.2 |
| IL-10 | 30.0 ± 30.4 | 111.3 ± 35.9* | 27.5 ± 15.5 |
| TGF-β | 67.1 ± 25.1 | 55.1 ± 44.7 | 227.5 ± 14.7* |
After stimulation of BM cells with LPS/IFN-γ or IL-4 for 24 h, stimulants were washed out of the macrophage culture, which then received fresh medium without stimulants. The supernatants harvested during the additional culture for 24 h were subjected to cytokine ELISA. *, P < 0.05.
Among the cytokines produced by M1 macrophages, IFN-γ and IL-12, but not IL-10, have inhibitory effects on osteoclastogenesis.
The possible involvement of IL-10 in the inhibition of osteoclastogenesis by M1 macrophages was first tested using IL-10 neutralizing antibody (Fig. 5). Even with the addition of IL-10 neutralizing antibody, M1 macrophages retained their ability to inhibit osteoclastogenesis, suggesting that IL-10 was not associated with the inhibitory effect of M1 macrophages on osteoclastogenesis.
FIG 5.
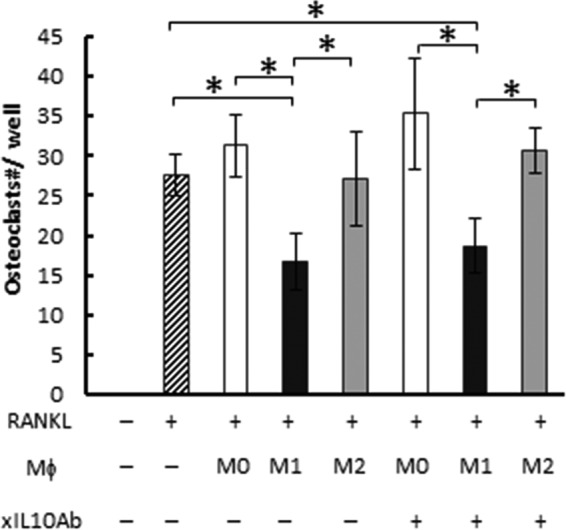
The effect of IL-10 on osteoclastogenesis. BM cells were differentiated into osteoclasts by stimulation with RANKL for 3 days. Then, the culture medium was replaced with fresh medium containing RANKL and M0, M1, or M2 macrophages derived from bone marrow cells in the presence or absence of anti-IL-10 neutralizing antibody (xIL10Ab) (1 μg/ml). After a total of 7 days of stimulation with RANKL, TRAP staining was performed. Data are means and SD (n = 6). The asterisk indicates significant difference (P < 0.05) by 2-way ANOVA post hoc Tukey's test.
Since neutralization of IL-10 present in the culture supernatant of M1 macrophages did not affect the osteoclastogenesis (Fig. 5), whether IFN-γ produced by M1 is associated with M1-mediated suppression of osteoclastogenesis was further examined. The addition of recombinant IFN-γ to BM-derived osteoclast precursors significantly suppressed RANKL-induced osteoclastogenesis, indicating that the osteoclast precursors used in this assay were susceptible to osteoclastogenesis inhibition by IFN-γ (Fig. 6A). In order to evaluate the possible involvement of IFN-γ in M1-mediated suppression of osteoclastogenesis, M1, M2, and M0 macrophages established ex vivo from BM cells of IFN-γ–KO mice were compared to those from wild-type mouse BM cells with respect to their ability to suppress RANKL-induced osteoclastogenesis (Fig. 6B). Very interestingly, M1 macrophages developed from IFN-γ–KO mice still suppressed the osteoclastogenesis compared to M0 or M2 developed from IFN-γ–KO mice (Fig. 6B), while there was no statistically significant difference in the TRAP-positive (TRAP+) osteoclast numbers between the groups that received M1 and M0/M2, unlike the results seen with the M1 macrophages derived from wild-type (WT) mice, which suppressed osteoclastogenesis significantly (Fig. 2B). On the other hand, M0 or M2 macrophages developed from IFN-γ–KO mice showed no regulatory effects on RANKL-induced osteoclastogenesis, in agreement with the results noted for wild-type mice.
FIG 6.

The effect of IFN-γ on osteoclastogenesis. (A) BM cells were differentiated to osteoclasts with RANKL only or RANKL and IFN-γ. (B) BM cells were differentiated to osteoclasts, and 3 days after RANKL addition, M0/M1/M2 macrophages derived from IFN-γ knockout (IFN-γKO) mice were added. Seven days after RANKL addition, TRAP staining was performed. Data are means and SD (n = 6). ** shows P < 0.01 by Student's t test for panel A, and n.s. indicates no significant difference by Tukey's test for panel B.
Finally, the involvement of IL-12 in the osteoclastogenesis-inhibitory effect of M1 macrophages was examined. The addition of recombinant IL-12 significantly suppressed RANKL-induced osteoclastogenesis from BM-derived osteoclast precursors (Fig. 7A), suggesting the susceptibility of osteoclast precursors to IL-12-induced inhibition of osteoclastogenesis. Antibody-mediated neutralization of IL-12 reduced the inhibitory effect on osteoclastogenesis of M1 macrophages (Fig. 7B). Very intriguingly, under IL-12-neutralized conditions, too, the addition of M1 macrophages developed from IFN-γ–KO mice showed a significantly higher level of osteoclastogenesis than that seen after the addition of M1 macrophages from wild-type mice, suggesting potential additive effects of IFN-γ.
FIG 7.

The effect of IL-12 and IFN-γ on osteoclastogenesis. (A) BM cells were differentiated to osteoclasts with RANKL (100 ng/ml) in the presence or absence of IL-12 (50 ng/ml). (B) BM cells were differentiated to osteoclasts, and 3 days after RANKL addition, M1 macrophages derived from WT or IFN-γ knockout (IFNKO) mice were added with or without IL-12 neutralizing antibody (filled columns). As a control, IL-12 neutralization antibody alone was also applied to the BM cells (3 days after RANKL addition) without addition of macrophage (slashed columns). Seven days after RANKL addition, TRAP staining was performed. Data are means and SD. No osteoclasts were observed without RANKL addition (data not shown) (n = 6). The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by Student's t test for panel A and by two-way ANOVA Tukey's test for panel B.
IFN-γ inhibits osteoclastogenesis by suppressing NFATc1 expression.
To understand the molecular mechanism underlying the inhibition of osteoclastogenesis mediated through the production of IFN-γ by M1, we analyzed the effect of IFN-γ on NFATc1 expression. NFATc1 is a master transcriptional regulator for osteoclastogenesis, and its expression is triggered by RANKL (17, 18). The induction of functionally pivotal molecules of mature osteoclasts, such as TRAP and cathepsin K, is controlled by NFATc1. Real-time PCR analysis revealed that NFATc1 expression induced by RANKL in BM-derived osteoclast precursors was remarkably suppressed by the presence of recombinant IFN-γ (Fig. 8). However, the addition of IL-12 did not show such an inhibitory effect on NFATc1 expression of RANKL-stimulated osteoclast precursors (data not shown).
FIG 8.
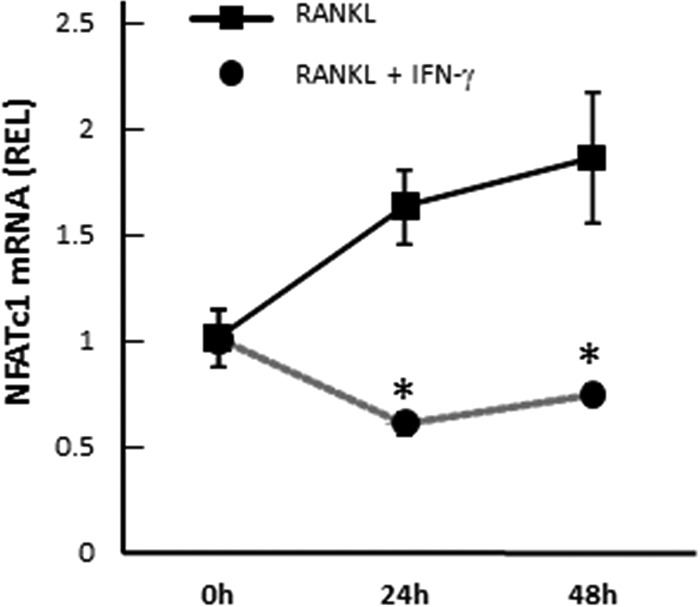
Downregulation of NFATc1 in RANKL-stimulated BM cells by IFN-γ. Following 6 days of preincubation of BM cells with M-CSF alone, NFATc1 expression was analyzed by real-time PCR in BM cells cultured with RANKL in the presence or absence of recombinant IFN-γ (10 ng/ml) for 48 h. Asterisks indicate significant differences (P < 0.05) by t test between 2 groups at the same time point. Data are means of REL and SD (n = 3).
IL-12 inhibits osteoclastogenesis by inducing apoptosis in preosteoclasts.
Since IL-12 did not suppress NFATc1 expression in RANKL-stimulated osteoclast precursors (data not shown), it was hypothesized that IL-12 might promote the apoptosis of mature osteoclasts, which, in turn, would result in a diminished number of mature osteoclasts under the influence of IL-12. As we expected, using real-time PCR, it was demonstrated that the osteoclast precursors treated with both RANKL and IL-12 showed significantly higher mRNA expression of caspase-3 than was seen with cells treated with RANKL alone, suggesting that IL-12 induced apoptosis in mature osteoclasts (Fig. 9A). In the cultures of M-CSF-treated osteoclast precursors, addition of RANKL and IL-12 increased the number of annexin V-positive cells compared to the numbers seen other groups that received (i) a control treatment (no RANKL), (ii) RANKL alone, or (iii) a combination of RANKL, IL-12, and anti-IL-12 neutralizing antibody (Fig. 9B and C), suggesting that IL-12 can directly act on osteoclasts and induce their apoptosis.
FIG 9.

Induction of caspase-3 in RANKL-stimulated BM cells by recombinant IL-12. Following 6 days of pretreatment of BM cells with M-CSF alone, the resulting BM cells were stimulated with RANKL in the presence or absence of recombinant IL-12 (50 ng/ml) for 48, 72, or 96 h and harvested for the following experiments. (A) Caspase-3 mRNA expression in the harvested cells was analyzed by real-time PCR. Asterisks indicate significant differences (P < 0.05) by t test between two groups at the same time point. Data are means of REL and SD (n = 3). (B and C) Representative images of annexin V staining (B) and percentages of annexin V-positive apoptotic cells (C) at 96 h after the stimulation with RANKL with or without IL-12 in the presence or absence of anti-IL-12 neutralizing Ab are shown. For determinations of the percentages of apoptotic cells, the number of apoptotic cells among all cells in the microscopic field (area, 100 μm by 100 μm) was counted and converted to a percentage by the following formula: [(number of apoptotic cells)/(number of all the cells) × 100]. The asterisks show significant differences (P < 0.05) by Tukey's test. Data are means of REL and SD (n = 3). The scale bar indicates 10 μm. (D and E) The effects of conditioned medium isolated from M1 and M2 of IFN-γ KO mice on the induction of annexin V expression by RANKL-stimulated BM cells were examined. Annexin V expression (D) and percentages of annexin V-positive apoptotic cells (E) are shown. After prestimulation with RANKL alone for 3 days, BM-derived macrophages were further incubated with the culture supernatant of macrophage (M0 or M1) derived from IFN-γ KO mice and RANKL in the presence or absence of anti-IL-12 neutralizing Ab for 96 h and subjected to annexin V staining. Measurement of percentages of annexin V+ apoptotic cells was carried out following the protocol shown in panel C. The asterisks indicate significant differences (P < 0.05) by Tukey's test. Data are means of REL and SD (n = 3). Scale bar, 10 μm.
M1 macrophage inhibits osteoclastogenesis in vivo.
In order to examine whether M1 macrophages can suppress osteoclastogenesis in the physiological context, a mouse model of ligature-induced periodontitis was employed so that the impact of adoptively transferred macrophages on the osteoclastogenesis induced in the periodontitis lesion could be monitored. It was previously demonstrated that periodontal bone loss is induced in mice by the ligature attachment in a RANKL-dependent manner (11). In order to confirm the migration of transferred cells into the mouse gingival tissue, M1, M2, or M0 macrophages expressing enhanced green fluorescent protein (eGFP) were adoptively transferred to ligatured mice and their migration pattern in the gingival tissue was monitored using flow cytometry analysis (see Fig. S1 in the supplemental material). As shown in Fig. 10A and B, the group of ligatured mice that received a transfer of M1 macrophage showed significantly less alveolar bone loss than those that received M0 or M2 macrophage transfer, indicating that M1 macrophages may be able to suppress periodontal bone loss. Histological analysis also demonstrated the presence of fewer TRAP-positive cells on the alveolar bone surface of the group of mice that received M1 macrophage compared to those that received M0 or M2 transfer (Fig. 10C and D). These results strongly supported the idea that M1 macrophage can inhibit osteoclastogenesis in vivo.
DISCUSSION
Among the three major macrophage subsets, M0, M1, and M2, inhibitory effects on RANKL-induced osteoclastogenesis were seen only with M1 macrophages, while M0 and M2 macrophages showed no regulatory effects on osteoclastogenesis. Further analysis found that the inhibition of osteoclastogenesis caused by M1 appeared to be mediated by the production of IFN-γ and IL-12, which downregulated the induction of NFATc1 and promoted apoptosis, respectively, thus explaining the putative mechanisms underlying the M1-mediated inhibition of osteoclastogenesis. Osteoimmunological studies have generally supported the idea of the effects of immune responses on bone metabolism (19–21). However, the possible impact of macrophage subsets on RANKL-induced osteoclastogenesis had remained unclear until the present study, which, for the first time, found that M1 macrophages, but not M0 or M2 macrophages, can suppress osteoclastogenesis in an in vitro coculture system.
Macrophages are key innate immune cells that reside in most organs of the body (22). Upon inflammatory insult, such as infection, M1 macrophages migrate to the site of infection to produce proinflammatory cytokines and remove pathogens through phagocytosis. After pathogen removal, M2 macrophages produce proresolving cytokines such as IL-4 and TGF-β in the inflammatory lesion (23). Impairment of macrophage phenotype balance is involved in many inflammatory diseases. For example, M1 macrophages are reported to accumulate in adipose tissue in diabetes and obesity, causing inflammation in obese adipose tissue and promoting insulin resistance in diabetes (24). On the other hand, diseases that involve M2 macrophage accumulation include atherosclerosis and cancer (25, 26). In atherosclerosis, the presence of M2 macrophages accounts for about 20% of the macrophages in plaque; in tumors, tumor-associated macrophages (TAM) adopt an M2-like profile, leading to immunosuppression of tumor cells (25).
Originally, it was hypothesized that M1 (inflammatory) macrophages promote osteoclastogenesis through their production of the proinflammatory cytokine TNF-α (Table 2) (27–29). However, the results from the present study have, in contrast, found that M1 macrophages have a suppressive effect on osteoclastogenesis, even while producing TNF-α. It was confirmed that the inhibitory effects mediated by IFN-γ and IL-12 appear to override the effects of TNF-α. Although additional investigation is required to explain this phenomenon, it is plausible that IFN-γ-derived cell signaling acts upstream of TNF-α's target, such as at key signaling molecule TRAF6 (30), thus nullifying its signal.
It was conceivable that IFN-γ and IL-12 downregulate the RANK expression on osteoclast precursors, in turn reducing the effect of RANKL signaling. However, in the present study, no differences in the effects of RANK expression resulting from stimulation with IFN-γ or IL-12 on osteoclast precursor cells were observed (data not shown). Instead, IFN-γ stimulation downregulated the expression of NFATc1, a master transcription factor for RANKL-induced osteoclastogenesis (17). Although we saw no effects of IL-12 on the expression of NFATc1 in RANKL-stimulated osteoclasts, the addition of IL-12 induced upregulation of mRNA for caspase-3, an important protease that mediates apoptosis, and increased the number of apoptotic cells in the RANKL-stimulated osteoclasts (Fig. 9), suggesting that IL-12 downregulated the generation of mature osteoclasts by promoting apoptosis in differentiated osteoclasts (31, 32).
To our best knowledge, no published information is available regarding the effect of human or mouse macrophages on RANKL-mediated osteoclastogenesis. Our data suggested that IFN-γ and IL-12 are responsible for the osteoclastogenesis suppression caused by M1 macrophages. More specifically, IFN-γ released by M1 suppressed gene expression of NFATc1, a master regulator of osteoclastogenesis (Fig. 7 and 8), whereas IL-12 produced from M1 increased the apoptosis of osteoclasts (Fig. 9). It is conceivable that the expression patterns of cytokines in human M1 macrophages are different from those in mouse M1 macrophages. Indeed, human M1 macrophages are reported to secrete IL-12 (33), whereas their propensity to produce IFN-γ is unknown. For these reasons, we believe that our finding, i.e., that of osteoclastogenesis inhibition by M1 macrophages, applies to humans as well based on their production of IL-12.
Although the regulation of osteoclastogenesis by macrophages has not previously been reported, macrophages and (pre)osteoclasts can colocalize in the context of inflammatory bone lesion, such as that seen in periodontal disease and arthritis (6, 7). While identification of the major macrophage phenotype in human periodontal disease remains elusive (34), we showed that LPS from P. gingivalis, a keystone pathogen in periodontal tissue, can induce M1 macrophages, which suggested that polarization toward the M1 phenotype may occur in periodontal tissue. Furthermore, it was reported that infiltration of both M1 and M2 macrophages increased in the P. gingivalis-induced mouse periodontitis lesion and that depletion of macrophages using clodronate liposomes prevented the bone resorption induced in the P. gingivalis-infected mice (7). However, since it must be noted that clodronate liposomes can also suppress osteoclastogenesis (35), further investigation may be required to establish the direct impact of either M1 or M2 macrophages on in vivo osteoclastogenesis induced in mice by P. gingivalis infection. In arthritis, it is also reported that the inflamed synovium shows a mixed phenotype of M1 and M2 accompanied by the locally produced proinflammatory cytokines TNF-α, IL-1, and IL-23, as well as by the anti-inflammatory cytokines IL-10 and TGF-β (36, 37).
Bone resorption occurring in inflammatory bone lytic diseases is irreversible. Therefore, prevention of bone resorption depends on elucidating the molecular and cellular mechanisms underlying inflammatory bone lytic diseases such as periodontitis and rheumatoid arthritis. Interestingly, our data suggest that M1 is the major phenotype among infiltrating macrophages in the periodontally diseased gingival tissue of experiment mice and that they suppress bone resorption by inhibiting osteoclastogenesis through their production of IL-12 and IFN-γ. Gonzalez et al. showed that mixed populations of M1 and M2 are present in the periodontally diseased gingival tissue of nonhuman primates and also that the proportions of polarized macrophages change with aging (38). It was also found that gene expression patterns for M1 macrophages were significantly elevated in periodontally healthy primates compared to those seen with primates with periodontitis (38). However, at this time, determination of the macrophage phenotype in periodontitis and rheumatoid arthritis in human and mouse model remains elusive. In this study, we showed, for the first time, that M1 macrophages but not M0 or M2 macrophages can suppress osteoclastogenesis in an in vitro coculture system and that adaptive transfer of M1 macrophages appeared to inhibit bone resorption in vivo. Therefore, we believe that this finding tends to shed light on bone lytic diseases. In particular, macrophages are reported to be the major cell population in the arthritis synovium, which is characterized by the presence of both osteoclast precursors and mature osteoclasts. Accordingly, M1 macrophages may also downmodulate the differentiation of osteoclast precursors, which leads to bone resorption not only in periodontitis but also in arthritis.
Since macrophage immunophenotypes are interchangeable (39), the results of this study also suggest the possibility that macrophage phenotype regulation, as a therapeutic target, may lead to the treatment of both soft tissue inflammation and associated bone resorption. A recent study demonstrated that phenotypes of macrophages and osteoclasts are also interchangeable and that (at least) osteoclast precursors can be converted to macrophage-like cells that can phagocytize and kill bacteria (40). According to the study by Jeganathan et al., differentiation of multinucleated giant cells (MGC) was induced by the incubation of RANKL-primed bone marrow-derived cells with IFN-γ plus LPS for 72 to 96 h, suggesting that preosteoclasts may differentiate into Langhans-type giant cells (41). It would intriguing to determine whether M1 cells that migrate into the inflamed gingival tissue with bone resorption lesion can act on osteoclast precursors and redirect their differentiation toward Langhans-type giant cells. To fully determine the pathophysiological role of macrophages in osteoclastogenesis and bone resorption, further experimental steps are necessary, but these steps should bring us closer to the identification and validation of therapeutic targets and new therapeutic regimens. Most certainly, we will have gained a deeper insight into the relationships among osteoclastogenesis, macrophages, and macrophage phenotype in the context of chronic inflammation as seen in periodontitis and arthritis.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by NIH grants DE-018499, DE-019917, and T32 DE 7327-12 from NIDCR.
We have no conflicts of interest regarding the content of this study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00461-16.
REFERENCES
- 1.Murray PJ, Wynn TA. 2011. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benoit M, Desnues B, Mege JL. 2008. Macrophage polarization in bacterial infections. J Immunol 181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 3.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ; on behalf of the participating members of the CDC Periodontal Disease Surveillance workgroup: James Beck (University of North Carolina, Chapel Hill, USA), Gordon Douglass (Past President, American Academy of Periodontology), Roy Page (University of Washington, Seattle, USA), Gary Slade (University of North Carolina, Chapel Hill, USA), George W. Taylor (University of Michigan, Ann Arbor, USA), Wenche Borgnakke (University of Michigan, Ann Arbor, USA), and representatives of the American Academy of Periodontology. 2012. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 91:914–920. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 4.Pappalardo A, Thompson K. 2013. Activated γδ T cells inhibit osteoclast differentiation and resorptive activity in vitro. Clin Exp Immunol 174:281–291. doi: 10.1111/cei.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP, Miranda LA, Ernst CW, Izumi Y, Taubman MA. 2006. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol 169:987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy A, Fearon U, Veale DJ, Godson C. 2011. Macrophages in synovial inflammation. Front Immunol 2:52. doi: 10.3389/fimmu.2011.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lam RS, O'Brien-Simpson NM, Lenzo JC, Holden JA, Brammar GC, Walsh KA, McNaughtan JE, Rowler DK, Van Rooijen N, Reynolds EC. 2014. Macrophage depletion abates Porphyromonas gingivalis-induced alveolar bone resorption in mice. J Immunol 193:2349–2362. doi: 10.4049/jimmunol.1400853. [DOI] [PubMed] [Google Scholar]
- 8.Moradi B, Rosshirt N, Tripel E, Kirsch J, Barié A, Zeifang F, Gotterbarm T, Hagmann S. 2015. Unicompartmental and bicompartmental knee osteoarthritis show different patterns of mononuclear cell infiltration and cytokine release in the affected joints. Clin Exp Immunol 180:143–154. doi: 10.1111/cei.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koo J, Kim S, Jung WJ, Lee YE, Song GG, Kim KS, Kim MY. 2013. Increased lymphocyte infiltration in rheumatoid arthritis is correlated with an increase in LTi-like cells in synovial fluid. Immune Netw 13:240–248. doi: 10.4110/in.2013.13.6.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han X, Kawai T, Eastcott JW, Taubman MA. 2006. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J Immunol 176:625–631. doi: 10.4049/jimmunol.176.1.625. [DOI] [PubMed] [Google Scholar]
- 11.Russel WC. 1947. Biology of the dermestid beetle with reference to skull cleaning. J Mammal 28:284–287. doi: 10.2307/1375178. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Bi L, Yu X, Kawai T, Taubman MA, Shen B, Han X. 2014. Porphyromonas gingivalis exacerbates ligature-induced, RANKL-dependent alveolar bone resorption via differential regulation of Toll-like receptor 2 (TLR2) and TLR4. Infect Immun 82:4127–4134. doi: 10.1128/IAI.02084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takayanagi H, Sato K, Takaoka A, Taniguchi T. 2005. Interplay between interferon and other cytokine systems in bone metabolism. Immunol Rev 208:181–193. doi: 10.1111/j.0105-2896.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 14.Evans KE, Fox SW. 2007. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol 8:4. doi: 10.1186/1471-2121-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohamed SG, Sugiyama E, Shinoda K, Taki H, Hounoki H, Abdel-Aziz HO, Maruyama M, Kobayashi M, Ogawa H, Miyahara T. 2007. Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone 41:592–602. doi: 10.1016/j.bone.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Kitaura H, Fujimura Y, Yoshimatsu M, Kohara H, Morita Y, Aonuma T, Fukumoto E, Masuyama R, Yoshida N, Takano-Yamamoto T. 2011. IL-12- and IL-18-mediated, nitric oxide-induced apoptosis in TNF-α-mediated osteoclastogenesis of bone marrow cells. Calcif Tissue Int 89:65–73. doi: 10.1007/s00223-011-9494-0. [DOI] [PubMed] [Google Scholar]
- 17.Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J. 2005. RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J 24:790–799. doi: 10.1038/sj.emboj.7600564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mellis DJ, Itzstein C, Helfrich MH, Crockett JC. 2011. The skeleton: a multi-functional complex organ: the role of key signalling pathways in osteoclast differentiation and in bone resorption. J Endocrinol 211:131–143. doi: 10.1530/JOE-11-0212. [DOI] [PubMed] [Google Scholar]
- 19.Guerrini MM, Takayanagi H. 2014. The immune system, bone and RANKL. Arch Biochem Biophys 561:118–123. doi: 10.1016/j.abb.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Danks L, Takayanagi H. 2013. Immunology and bone. J Biochem 154:29–39. doi: 10.1093/jb/mvt049. [DOI] [PubMed] [Google Scholar]
- 21.Del Fattore A, Teti A. 2012. The tight relationship between osteoclasts and the immune system. Inflamm Allergy Drug Targets 11:181–187. doi: 10.2174/187152812800392733. [DOI] [PubMed] [Google Scholar]
- 22.Murphy K, Travers P, Walport M, Janeway C. 2012. Janeway's immunobiology. Garland Science, London, United Kingdom. [Google Scholar]
- 23.Ariel A, Serhan CN. 2012. New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front Immunol 3:4. doi: 10.3389/fimmu.2012.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olefsky JM, Glass CK. 2010. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 25.Colin S, Chinetti-Gbaguidi G, Staels B. 2014. Macrophage phenotypes in atherosclerosis. Immunol Rev 262:153–166. doi: 10.1111/imr.12218. [DOI] [PubMed] [Google Scholar]
- 26.Cassetta L, Cassol E, Poli G. 2011. Macrophage polarization in health and disease. ScientificWorldJournal 11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locati M, Mantovani A, Sica A. 2013. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol 120:163–184. doi: 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- 28.Liu G, Yang H. 2013. Modulation of macrophage activation and programming in immunity. J Cell Physiol 228:502–512. doi: 10.1002/jcp.24157. [DOI] [PubMed] [Google Scholar]
- 29.Martinez FO, Sica A, Mantovani A, Locati M. 2008. Macrophage activation and polarization. Front Biosci 13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 30.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 31.Samejima K, Svingen PA, Basi GS, Kottke T, Mesner PW, Stewart L, Durrieu F, Poirier GG, Alnemri ES, Champoux JJ, Kaufmann SH, Earnshaw WC. 1999. Caspase-mediated cleavage of DNA topoisomerase I at unconventional sites during apoptosis. J Biol Chem 274:4335–4340. doi: 10.1074/jbc.274.7.4335. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson DW. 1999. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 33.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 34.Yu T, Zhao L, Huang X, Ma C, Wang Y, Zhang J, Xuan D. 2016. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol 2016:1–21. [DOI] [PubMed] [Google Scholar]
- 35.Frith JC, Mönkkönen J, Auriola S, Mönkkönen H, Rogers MJ. 2001. The molecular mechanism of action of the antiresorptive and antiinflammatory drug clodronate: evidence for the formation in vivo of a metabolite that inhibits bone resorption and causes osteoclast and macrophage apoptosis. Arthritis Rheum 44:2201–2210. doi:. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Hsu HC, Mountz JD. 2012. Managing macrophages in rheumatoid arthritis by reform or removal. Curr Rheumatol Rep 14:445–454. doi: 10.1007/s11926-012-0272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambarus CA, Noordenbos T, de Hair MJ, Tak PP, Baeten DL. 2012. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res Ther 14:R74. doi: 10.1186/ar3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez OA, Novak MJ, Kirakodu S, Stromberg A, Nagarajan R, Huang CB, Chen KC, Orraca L, Martinez-Gonzalez J, Ebersole JL. 2015. Differential gene expression profiles reflecting macrophage polarization in aging and periodontitis gingival tissues. Immunol Invest 44:643–664. doi: 10.3109/08820139.2015.1070269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wijesundera KK, Izawa T, Murakami H, Tennakoon AH, Golbar HM, Kato-Ichikawa C, Tanaka M, Kuwamura M, Yamate J. 2014. M1- and M2-macrophage polarization in thioacetamide (TAA)-induced rat liver lesions; a possible analysis for hepato-pathology. Histol Histopathol 29:497–511. doi: 10.14670/HH-29.10.497. [DOI] [PubMed] [Google Scholar]
- 40.Nishimura K, Shindo S, Movila A, Kayal R, Abdullah A, Savitri IJ, Ikeda A, Yamaguchi T, Howait M, Al-Dharrab A, Mira A, Han X, Kawai T. 22 June 2016. TRAP-positive osteoclast precursors mediate ROS/NO-dependent bactericidal activity via TLR4. Free Radic Biol Med doi: 10.1016/j.freeradbiomed.2016.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeganathan S, Fiorino C, Naik U, Sun HS, Harrison RE. 2014. Modulation of osteoclastogenesis with macrophage M1- and M2-inducing stimuli. PLoS One 9:e104498. doi: 10.1371/journal.pone.0104498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.