

Abstract
Chronic infection with Toxoplasma gondii becomes established in tissues of the central nervous system, where parasites may directly or indirectly modulate neuronal function. Epidemiological studies have revealed that chronic infection in humans is a risk factor for developing mental diseases. However, the mechanisms underlying parasite-induced neuronal dysfunction in the brain remain unclear. Here, we examined memory associated with conditioned fear in mice and found that T. gondii infection impairs consolidation of conditioned fear memory. To examine the brain pathology induced by T. gondii infection, we analyzed the parasite load and histopathological changes. T. gondii infects all brain areas, yet the cortex exhibits more severe tissue damage than other regions. We measured neurotransmitter levels in the cortex and amygdala because these regions are involved in fear memory expression. The levels of dopamine metabolites but not those of dopamine were increased in the cortex of infected mice compared with those in the cortex of uninfected mice. In contrast, serotonin levels were decreased in the amygdala and norepinephrine levels were decreased in the cortex and amygdala of infected mice. The levels of cortical dopamine metabolites were associated with the time spent freezing in the fear-conditioning test. These results suggest that T. gondii infection affects fear memory through dysfunction of the cortex and amygdala. Our findings provide insight into the mechanisms underlying the neurological changes seen during T. gondii infection.
INTRODUCTION
Toxoplasma gondii is one of the most successful brain parasites, infecting approximately one-third of the human population (1). T. gondii can persist in brain and muscle throughout the host's life, and chronic infection is asymptomatic in immunocompetent humans (2). However, recent studies have suggested that T. gondii infection is a risk factor for developing mental diseases, such as schizophrenia and depression, as well as human behavior and personality changes and suicide (3, 4). Interestingly, T. gondii infection increases the risk of schizophrenia roughly 2.7 times, which is higher than that for genes associated with schizophrenia (5). Several studies have also suggested that rodents infected with T. gondii exhibit decreased avoidance behavior in response to cat odors, indicating manipulation of the host's behavior by T. gondii to facilitate the parasite's transmission and complete sexual replication in the definitive host (6–11).
To date, research on the mechanism(s) underlying behavioral changes following T. gondii infection has been conducted primarily from two points of view. First, the relationship between parasite localization in the brain and behavioral changes has been investigated, with a previous study reporting that T. gondii has no obvious tropism in the brain (12–15). However, another study found that tissue cyst density in amygdalar areas (the medial and basolateral amygdala) is 2-fold higher than that in nonamygdalar areas (9), whereas the presence of tissue cysts in the forebrain contributes to the attenuation of predator odor aversion and anxiety-like behavior (16). Overall, these studies suggest that the T. gondii cyst distribution contributes to behavioral changes, but this still requires further investigation.
Second, research on the mechanisms underlying behavioral changes following T. gondii infection has examined the effect of the infection on neuronal cell biology, including neurotransmitter synthesis, signal transduction, gene expression, and epigenetic modulation (14, 17–21). One study reported that dopaminergic cells are upregulated by infection, suggesting that T. gondii affects the central nervous system to manipulate host behavior (22). In support of this finding, dopamine (DA) levels in T. gondii-infected mice are higher than those in control mice (17). Furthermore, increased DA release is observed in acutely infected male mice (18), and increased DA levels are observed in the striatum of infected mice at 6 days postinfection (dpi) (20). Moreover, treatment of T. gondii-infected rats with haloperidol, an antipsychotic that is known to affect the dopaminergic system, reverses the behavioral effect of T. gondii infection (23). In their recent study, Hari Dass et al. indicated that T. gondii infection induces hypomethylation of the arginine vasopressin promoter in the medial amygdala (21). They also showed that decreased aversion to cat odors in the T. gondii-infected rat is recovered by systemic hypermethylation (21). Despite these findings, the mechanism(s) underlying the behavioral changes induced by T. gondii infection remains unclear.
The presence of an aversive stimulus is transmitted to the amygdala via the cortex and thalamus. The activated amygdala then facilitates stimulation of the hypothalamic-pituitary-adrenal (HPA) axis (24). The HPA axis is essential for adaptation to a stressful environment (25). Activation of the HPA axis facilitates secretion of corticosterone (CORT), which plays an important role in expressing emotional behavior (24). The cortex, particularly the prefrontal cortex, is implicated in stress regulation. Lesions in the cortex decrease or increase the CORT response to stress (26). The amygdala receives dense serotonergic innervation from the dorsal raphe nucleus, and activation of the dorsal raphe nucleus increases amygdala 5-hydroxytryptamine (5-HT; serotonin) levels and CORT secretion (27). CORT modulates serotonergic activity in the amygdala (28). A previous study suggested that T. gondii infection causes dendritic retraction of basolateral amygdala neurons and decreases the amounts of CORT, both basal CORT levels and the levels of CORT induced by aversive cat odors (29). Additionally, it has been known for decades that the noradrenergic system is involved in memory consolidation (30). Noradrenergic stimulation of the amygdala enhances memory consolidation (31). Aversive stimuli enhance the secretion of norepinephrine (NE) from the locus coeruleus to the cortex and amygdala, resulting in enhanced fear memory consolidation modulated by stress hormone regulation (32).
In addition to attenuation of predator odor aversion, learning and memory deficits, as well as effects on intact memory, have been demonstrated in rodents infected with T. gondii (9, 15, 33–35). The effects of T. gondii infection on rodent behavior vary with the experimental design, including differences in rodent species, route of infection, parasite strain, dosage and stage of parasites, time postinfection, and type of behavior test (36, 37). These differences make it difficult to clarify the characteristics of the brain pathology associated with behavioral changes following T. gondii infection. Therefore, the use of one behavioral paradigm and experimental design to examine both the brain histopathological and neurological changes in infected rodents would further the understanding of the mechanisms of the behavioral changes induced by T. gondii infection. In this study, we investigated the brain parasite distribution, histopathological lesion severity, and neurotransmitter (DA, 5-HT, and NE) levels to evaluate how latent T. gondii infection affects host fear memory.
MATERIALS AND METHODS
Ethics statement.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (38). The protocol was approved by the Committee on the Ethics of Animal Experiments at the Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan (permit numbers 23-64, 24-17, 25-66, and 26-68). Mice were decapitated without anesthesia for brain sampling, and all efforts were made to minimize animal suffering.
Mice.
Mice (male C57BL/6 mice; age, 8 weeks) were obtained from CLEA Japan (Tokyo, Japan). Mice were housed (four to six mice per cage) under a 12-h light and 12-h dark cycle (light from 8:00 a.m. to 8 p.m.) in the animal facility of the National Research Center for Protozoan Diseases at the Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan. All mice were treated using the guiding principles for the care and use of research animals endorsed by the Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan. All animal experiments began after 1 week of habituation.
Parasite culture.
T. gondii (strain PLK; type II) parasites were passaged using monkey kidney adherent epithelial cells (Vero cells) in Eagle's minimum essential medium (Sigma, St. Louis, MO, USA) containing 8% fetal bovine serum. Infected cells were syringe lysed using a 27-gauge needle to release tachyzoites into RPMI 1640 medium (Sigma) and then filtered using a 5.0-μm-pore-size filter (Millipore, Bedford, MA, USA).
Parasite infection and experimental groups.
T. gondii tachyzoites (1 ′ 103) were intraperitoneally inoculated into 9-week-old mice. Body weight measurements were taken daily for 30 days after infection. This study consisted of six experiments, and the experimental trials are described in Fig. S1 in the supplemental material. All behavioral experiments were performed at 37 to 41 dpi, commencing at 7:00 to 8:30 a.m. under a light intensity of 300 lx. For high-performance liquid chromatography (HPLC) analysis, uninfected and infected mice were sacrificed at 40 and 52 dpi. Times of 40 and 52 dpi were selected to evaluate the impact of T. gondii infection because these days corresponded to those for the start and end of the fear-conditioning test, respectively. We examined the correlation coefficients between the percentage of time spent freezing in the fear-conditioning test and the levels of neurotransmitters using the samples collected at 52 dpi. Moreover, mice were sacrificed at 45 and 54 dpi for histopathological analysis and for quantitation of the parasite load using quantitative PCR, respectively. Successful establishment of latent infection was confirmed using an enzyme-linked immunosorbent assay for detecting antibodies to the T. gondii dense granule protein 7 (TgGRA7) (39). Mice with no anti-TgGRA7 antibodies were excluded from the experiments.
Fear-conditioning test.
We performed contextual and cued fear-conditioning tests to evaluate learning and memory. The fear-conditioning test is a behavioral experiment that assesses the ability of mice to learn the association between an environmental cue and an aversive stimulus. On the first day, the mice were placed in a conditioning chamber and given pairings of an auditory cue and a mild foot shock. On the following 2 days, the mice were exposed to the same conditioning chamber (context test) and a differently shaped chamber, and the auditory cue was presented (tone test). Freezing behavior during the test was measured as an index of fear memory. Therefore, if a mouse normally learned the association between the conditioned cues and the foot shocks, it spent longer in the freezing state than a mouse that had an incomplete memory. On the last day, the mice received 30 successive auditory cues without the foot shock (extinction test). The normal mouse spent increasingly less time in the freezing state during the test. However, if a mouse had a deficit in fear extinction, it showed high levels of freezing until late in the session. To measure associative-type long-term fear memory, fear-conditioning tests were performed from 37 to 41 dpi according to methods used in earlier studies (9, 40), but with some modifications (see Fig. S2 in the supplemental material). In a fear-conditioning test box (18 cm by 17 cm; Muromachi, Tokyo, Japan), freezing was recorded using a video-tracking system (Comp Act VAS, version 3.0x; Muromachi). The test consisted of four phases, conditioning, context, tone, and extinction, as described in Fig. S2 in the supplemental material. On test day 1, mice were placed in the box for habituation (120 s). An auditory tone (75 dB, 300 Hz) was then presented for 28 s, with a mild foot shock (0.5 mA) being paired with the auditory tone for 2 s. An interval of 60 s preceded a second identical trial. After the last foot shock presentation, mice were kept in the box for an additional 30 s. On test day 2, mice were placed in the same spatial and olfactory context for 5 min to measure the contextual fear-conditioned response. On test day 3, mice were placed in the box in a novel chamber and allowed to habituate for 3 min. The auditory tone was then presented for 3 min. On test day 4, to determine the extinction rate of cued fear conditioning, mice were presented with 30 successive auditory tones (75 dB and 300 Hz for 10 s with 50-s interval durations). Freezing was measured during the first tone before it was paired to a foot shock (28 s) in the unconditioning phase, during the second tone after it was paired to foot shock (28 s) in the conditioning phase, during observation (300 s) in the context test, during habituation (180 s), and during the tone (180 s) in the tone test. The freezing ratio (in percent) was calculated by dividing the time spent freezing by the total amount of time of each session. In the extinction test, the freezing ratio (in percent) was repeatedly calculated by dividing the time spent freezing by 5 min for every 5 min of the extinction test.
DNA extraction and quantitative PCR.
To measure the T. gondii burden in mouse brain at 54 dpi, one hemisphere from each mouse brain was divided into eight regions: cortex, hippocampus, caudoputamen, amygdala, thalamus, hypothalamus, midbrain, and cerebellum (see Fig. S1 in the supplemental material). The method used for dissection of these brain regions is described in detail in Fig. S3 in the supplemental material. Tissue was immediately stored at −30°C. DNA was isolated from the brain regions, and parasite counts were analyzed by real-time PCR using the B1 gene, as described previously (14). PCR was performed using an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA), and amplification was monitored using the SYBR green method (Applied Biosystems). A standard curve was constructed with 10-fold serial dilutions of T. gondii DNA extracted from 1 ′ 105 parasites. The curve ranged from 10,000 parasites to 0.01 parasite. The parasite number was calculated by plotting the threshold cycle values on the standard curve.
Histopathological analysis.
After being fixed with 4% paraformaldehyde solution, at 45 dpi brain samples were cut coronally, embedded in paraffin wax, sectioned at 4 μm, and then stained with hematoxylin and eosin (see Fig. S1 in the supplemental material). Pathological lesion severity was scored using the following scheme: 0, no lesion; 1, slight lesion; 2, mild lesion; 3, moderate lesion; and 4, severe lesion. Representative examples of the scoring are shown in Fig. S4 in the supplemental material. Pathological scores ranging from 0 to 3 were determined for two types of lesions, meningitis, including ventriculitis, and perivascular cuffs. Pathological scores ranging from 0 to 4 were also determined for inflammatory cells, which included glial cell, macrophage, and lymphocyte infiltration.
High-performance liquid chromatography.
At 40 and 52 dpi, neurotransmitter levels in the brains were measured by HPLC (see Fig. S1 in the supplemental material). The brains were divided into two regions: cortex and amygdala (regions related to emotional behavior and memory) (see Fig. S3 in the supplemental material). The collected tissue was immediately stored at −80°C. Each brain sample was homogenized using a BioMasher homogenizer (Funakoshi, Tokyo, Japan), and then 300 μl/10 mg tissue of 0.2 M perchloric acid (containing 100 μM EDTA-2Na) was added. Isoproterenol HCl (Sigma) was used as a monoamine internal standard. Homogenates were placed on ice for 30 min and then centrifuged at 20,000 ′ g for 15 min at 0°C. Supernatants were mixed with 1 M sodium acetate to adjust the pH to 3.0 and filtered using an Ultrafree MC device (Millipore). The final products were injected into an HTEC-500 HPLC system (electrochemical detector; Eicom, Kyoto, Japan) equipped with an SC-5ODS column for monoamines. Chromatograms were analyzed using PowerChrom software (version 2.5; eDAQ Pty. Ltd., Densitone East, Australia).
Correlation analysis.
The correlation coefficients for the percentage of time spent freezing in the fear-conditioning test and the levels of cortical neurotransmitters were calculated using the Pearson correlation coefficient (r). Previous studies have shown that the strength of the linear association between pairs of variables can be determined using the Pearson correlation coefficients, as follows: |r| = 0.70, strong correlation; 0.5 < |r| < 0.7, moderately strong correlation; and |r| = 0.3 to 0.5, weak to moderate correlation (41).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism (version 6.0) software (GraphPad Software, San Diego, CA, USA). Statistically significant differences between two groups were analyzed using two-tailed unpaired t tests, except for the extinction test, in which statistically significant differences were determined using repeated-measures analysis of variance (ANOVA) with the Bonferroni test as the post hoc test. With three groups or more, statistically significant differences were determined using one-way ANOVA followed by Tukey's multiple-comparison test. For the correlation analysis, significant differences were determined using the Pearson correlation coefficient. P values of <0.05 represent statistically significant differences.
RESULTS
Impaired long-term fear memory consolidation in T. gondii-infected mice.
We performed fear-conditioning tests to evaluate learning and memory. During the conditioning phase, there were no significant behavioral differences between infected and control mice (Fig. 1A). However, infected mice showed significantly reduced freezing behavior in the conditioned context (Fig. 1B) and following habituation for 3 min in a novel chamber (Fig. 1C) compared with that of control mice. The percentage of time spent in freezing behavior did not change between habituation and tone conditioning in T. gondii-infected mice, but freezing behavior increased with tone conditioning in uninfected mice (Fig. 1C). These results indicate that mice infected with T. gondii have an impaired ability to consolidate fear memory. When mice were subjected to 30 successive tones over 30 min, the percentage of time spent freezing by both control and T. gondii-infected mice gradually decreased (Fig. 1D). Compared with uninfected animals, T. gondii-infected mice exhibited significantly reduced freezing during the first 5 min. This difference might have influenced our conditioned context results.
FIG 1.

Impaired long-term fear memory consolidation in uninfected and T. gondii-infected mice. The ordinate shows the percentage of time spent freezing. (A) The unconditioned trial shows freezing during the first tone before pairing to foot shock, and the conditioned trial shows freezing during the second tone after pairing to foot shock on test day 1. (B) Contextual conditioned freezing time. (C) Tone-conditioned freezing time. (D) Extinction of tone-conditioned freezing time. Significant differences were determined by unpaired t tests (****, P < 0.0001) (A to C) and a repeated-measures ANOVA with the post hoc Bonferroni test (D). Significant main effects were shown for T. gondii infection [F(1, 70) = 75.90, P < 0.0001] and time [F(5, 350) = 117.5, P < 0.0001], and their interaction was also significant [F(5, 350) = 4.410, P < 0.001]. Freezing was calculated by dividing freezing time into the times for observation (300 s) in the context test, habituation (180 s), tone (180 s) during the tone test, and 5 min for every 5 min in the extinction test. Data represent means ± SEMs. Data are for uninfected mice (n = 32) and T. gondii-infected mice (n = 42) and are summarized from four independent experiments.
Parasite load and pathological examination of brain regions in T. gondii-infected mice.
Because the distribution of the parasite in the brain may be an important factor affecting behavioral changes, we analyzed the expression of the B1 gene by quantitative PCR to compare the parasite counts in eight distinct brain regions: cortex, hippocampus, caudoputamen, amygdala, thalamus, hypothalamus, midbrain, and cerebellum. There were no significant differences in the parasite counts across these brain regions (Fig. 2). To investigate the parasite stages in the brain, the expression levels of SAG1 (a tachyzoite-specific gene), BAG1 (a bradyzoite-specific gene), and GRA1 (a non-stage-specific gene) were measured by real-time PCR (see Fig. S5 in the supplemental material). The expression of BAG1 and GRA1 was detected in each brain region, but the level of expression of SAG1 in the infected mice was very low. There were no significant differences in the levels of expression of these genes among the brain regions. Histopathological analysis showed perivascular cuffs and inflammatory cell infiltration in almost all regions. However, the meningitis in the cortex was significantly more severe than that in the other regions (Fig. 3A). The pathological scores for the perivascular cuffs in the cortex, caudoputamen, thalamus, and hypothalamus were higher than those for the perivascular cuffs in the midbrain (Fig. 3B). In addition to the pathological analysis, the inflammatory infiltrate was assessed by real-time PCR for the expression of the CD4, CD8, and CD11b genes, which are markers of inflammatory cells (see Fig. S6 in the supplemental material). The CD4 levels were higher in the amygdala in the infected mice than in the other regions. The levels of CD8 and CD11b were higher in the amygdala than in the hippocampus. The level of gamma interferon (IFN-γ) was higher in the cortex than in the hippocampus or caudoputamen (see Fig. S6D in the supplemental material).
FIG 2.
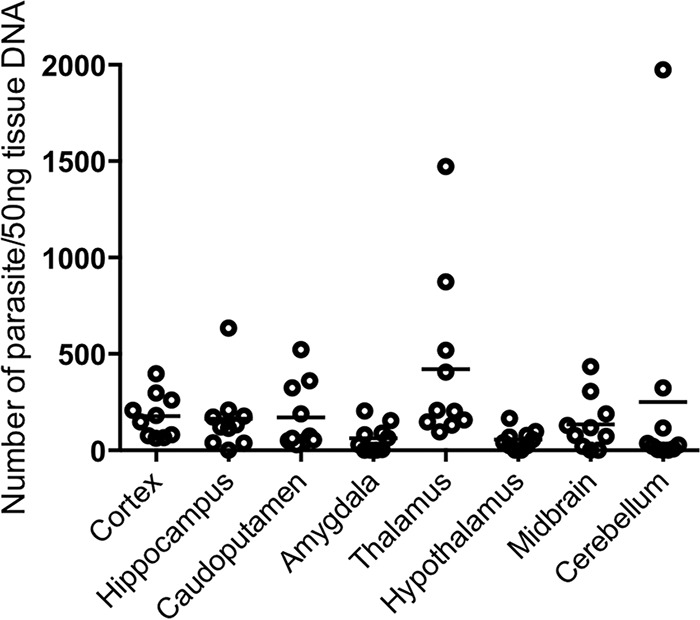
Parasite load in the brains of T. gondii-infected mice. The ordinate shows the parasite number per 50 ng of tissue DNA. Brain samples were collected at 54 days postinfection. Each circle represents the data for one mouse, and bars represent the average value for all data points (T. gondii-infected mice, n = 10). No statistically significant differences were found using one-way ANOVA with Tukey's post hoc test.
FIG 3.

Histopathological changes in the brains of T. gondii-infected mice. The ordinate shows the pathological score for each brain region. Brain samples were collected at 45 days postinfection. Histopathological lesions were scored as follows: 0, no lesion; 1, slight lesion; 2, mild lesion; 3, moderate lesion; and 4, severe lesion. Each circle represents the data for one mouse, and bars represent the average value for all the data points (T. gondii-infected mice, n = 7). Significant differences were determined using one-way ANOVA with Tukey's post hoc test. Different letters (a, b) indicate statistically significant differences among groups (P < 0.05). (A) Data for the hippocampus, caudoputamen, thalamus, and midbrain were excluded because they lack meninges.
Neurotransmitter levels in cortex and amygdala of T. gondii-infected mice.
We analyzed the levels of various neurotransmitters in the cortex and amygdala. Cortical DA levels were not significantly different between uninfected and T. gondii-infected mice at either 40 or 52 dpi (Fig. 4A and E). Amygdalar DA levels were lower in infected mice than in uninfected animals at 40 dpi, but no difference in DA levels was detected at 52 dpi (Fig. 4A and E). We also examined DA metabolism in these animals. Homovanillic acid (HVA) is the primary final DA metabolite produced via the intermediate products 3,4-dihydroxyphenylacetic acid (DOPAC) and 3-methoxytyramine (3-MT) (42). The levels of all DA metabolites in the cortex but not in the amygdala of the infected mice increased compared with those in uninfected animals at 40 and 52 dpi (Fig. 4B to D and F to H). We also determined that the levels of 5-HT in the amygdala but not the cortex of infected mice decreased compared with those in uninfected mice at 40 and 52 dpi (Fig. 5A and C). There was no difference in the levels of the serotonin metabolite 5-hydroxyindoleacetic acid (5-HIAA) in the cortex or amygdala following T. gondii infection (Fig. 5B and D). NE levels in both the cortex and amygdala of infected mice were decreased compared with those in uninfected mice at 40 and 52 dpi (Fig. 6A and B).
FIG 4.

Levels of dopamine and its metabolites in the cortex and amygdala of uninfected and T. gondii-infected mice. The ordinate shows the levels of the neurotransmitter dopamine (DA) and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC), 3-methoxytyramine (3-MT), and homovanillic acid (HVA) at 40 (A to D) and 52 (E to H) days postinfection. Data represent the mean ± SEM. (A to D) Data for uninfected mice (n = 6) and T. gondii-infected mice (n = 8); (E to H) data for uninfected mice (n = 16) and T. gondii-infected mice (n = 19). Significant differences were determined by unpaired t tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 5.

Levels of serotonin and its metabolite in the cortex and amygdala of uninfected and T. gondii-infected mice. The ordinate shows the levels of the neurotransmitter 5-hydroxytryptamine (5-HT, serotonin) and its metabolite 5-hydroxyindoleacetic acid (5-HIAA) in the cortex and amygdala at 40 (A and B) and 52 (C and D) days postinfection. Data represent means ± SEMs. (A and B) Data for uninfected mice (n = 6) and T. gondii-infected mice (n = 8); (C and D) data for uninfected mice (n = 16) and T. gondii-infected mice (n = 19). Significant differences were determined by unpaired t tests (*, P < 0.05; **, P < 0.01).
FIG 6.

Norepinephrine levels in the cortex of uninfected and T. gondii-infected mice. The ordinate shows the levels of the neurotransmitter norepinephrine (NE) in the cortex and amygdala at 40 (A) and 52 (B) days postinfection. Data represent means ± SEMs. (A) Data for uninfected mice (n = 6) and T. gondii-infected mice (n = 8); (B) data for uninfected mice (n = 16) and T. gondii-infected mice (n = 19). Significant differences were determined by unpaired t tests (*, P < 0.05; **, P < 0.01).
The correlations between the percentage of time that the mice spent freezing in the fear-conditioning test and the levels of neurotransmitters in the cortex and amygdala were examined by calculating Pearson correlation coefficients (Fig. 7; see also Table S1 in the supplemental material). We found that the level of HVA in the cortex had a moderately strong negative correlation with the percentage of time spent freezing during the context test (r = −0.613; Fig. 7A). The levels of DOPAC, 3-MT, and 5-HIAA in the cortex showed weak to moderate negative correlations with freezing time during the context test (r = −0.388, −0.378, and −0.447, respectively; Fig. 7B to D). In contrast, the levels of NE displayed weak to moderate positive correlations with freezing time during the context test in both the cortex and amygdala (r = 0.346 and 0.414, respectively; Fig. 7E and F). In addition, amygdalar 5-HT, 5-HIAA, and NE levels were weakly to moderately positively correlated with freezing time during the tone test (r = 0.371, 0.385, and 0.388, respectively; Fig. 7G to I).
FIG 7.

Correlation coefficients between for correlations between neurotransmitter levels and the percentage of time spent freezing during the context test and tone test. After the fear-conditioning test, some mice were used for the correlation analysis (experiments 3 and 4; see Fig. S1 in the supplemental material). The ordinate shows the percentage of time spent freezing during the context test. The abscissa shows the level of each neurotransmitter in the cortex or amygdala at 52 days postinfection. Solid lines, the calculated line of best fit. Correlation coefficients were calculated using Pearson's correlation coefficient: |r| = 0.70, strong correlation; 0.5 < |r| < 0.7, moderately strong correlation; and |r| = 0.3 to 0.5, weak to moderate correlation. Data are for uninfected mice (n = 16) and T. gondii-infected mice (n = 19). HVA, homovanillic acid; DOPAC, 3,4-dihydroxyphenylacetic acid; 3-MT, 3-methoxytyramine; 5-HIAA, 5-hydroxyindoleacetic acid; NE, norepinephrine; 5-HT, 5-hydroxytryptamine.
DISCUSSION
We showed that T. gondii infection in male C57BL/6 mice impaired fear memory consolidation, while extinction remained intact. Vyas et al. observed no obvious deficits in the fear memory of T. gondii-infected rats in the fear-conditioning test (9). However, Witting showed impairment of memory in T. gondii-infected mice (33). In addition, that study demonstrated that T. gondii-infected mice showed a higher sensitivity to learning and greater memory deficits than T. gondii-infected rats (33). Kannan et al. determined that spatial working memory is impaired in mice infected with T. gondii (34). In a recent study, Daniels et al. indicated that spatial memory recall is impaired in rats infected with T. gondii (15). Thus, the effects of T. gondii infection on learning, memory, and emotional behavior have varied widely among different studies, although those studies used different experimental designs, which may have affected the results (36, 37). We are the first to report impaired consolidation of fear memory in T. gondii-infected mice.
Our results showed that T. gondii infection is present throughout the brain without showing a marked tissue tropism. Furthermore, real-time PCR was used to determine the expression of SAG1, BAG1, and GRA1 in the brain tissues and showed that neither the expression of the bradyzoite marker nor the low level of expression of SAG1 was specific to any particular brain region, suggesting that there was no cyst tropism. Consistent with the findings of previous studies in mice, T. gondii had no obvious preference for specific brain regions (12–14). In addition, no other study has reported clear evidence to support the idea that parasite localization plays a critical role in the behavioral changes induced by T. gondii infection (9, 15, 16, 43). Here, our histopathological analysis showed that meningitis in the cortex was more severe than that in other regions. The area of the meninges in the cortex and cerebellum is larger than that in the other brain regions; however, meningitis in the cortex was more severe than that in the cerebellum. Together with the results showing no marked tissue tropism for the parasite, our results suggest that the immune response (indirect effects) may be more brain region specific than the parasitic cyst burden (direct effects). Similarly, our previous study using BALB/c mice showed that the prefrontal cortex is more severely damaged than other brain regions (14). Although the mechanism whereby the T. gondii-induced pathology shows cortical specificity is unclear, these results suggest that T. gondii causes cortical hypofunction independently of the parasite distribution. In addition to the pathological analysis, a real-time PCR analysis of the general markers of inflammatory cells suggested that inflammatory cell infiltration was more severe in the amygdala than in the other brain regions.
Some drug treatments not only reduce the cyst burden but also attenuate the inflammatory response in the brain. Interestingly, Bottari et al. reported that the treatment of T. gondii-infected mice with sulfamethoxazole-trimethoprim partly rescued the behavioral changes associated with T. gondii infection, suggesting that the degree of brain inflammation affects these behavioral changes (44). Therefore, to investigate whether the degree of inflammation in each brain region affected the behavioral changes in the infected mice, we examined the correlation between the expression of IFN-γ and the percentage of time that the mice spent freezing in the fear-conditioning test. The level of IFN-γ was higher in the cortex than in the hippocampus or caudoputamen, but there was no significant correlation between IFN-γ expression and the time spent freezing in the fear-conditioning test (data not shown). Furthermore, the expression levels of CD4, CD8, and CD11b in each brain region did not correlate with the time spent freezing in the fear-condoning test (data not shown). These results suggest that the inflammatory response was more severe in the cortex than in the other brain regions but that the degree of inflammation does not contribute to impaired fear memory consolidation in mice infected with T. gondii. None of the data presented in this study directly correlate the severity of the behavioral deficits with the degree of damage to the cortex and amygdala because the mice used in the fear-conditioning test were different from those used for histopathological analysis. Thus, more direct evidence is required before we can conclude that the degree of brain inflammation affects the behavioral changes. However, because the prefrontal cortex and amygdala are involved in fear memory and emotional behavior (45), our results suggest that cortical and amygdalar lesions, including meningitis or inflammatory infiltration, are related to the impairment of neuronal function in the cortex and amygdala. We also analyzed the cortical and amygdalar levels of DA, 5-HT, NE, and the metabolites of DA and 5-HT, all of which are associated with the expression of emotional behavior, learning, and memory (46). Cortical DA levels were similar in uninfected and T. gondii-infected mice at both 40 and 52 dpi. However, the levels of all DA metabolites increased at both 40 and 52 dpi. Gatkowska et al. reported that dopamine turnover (HVA/DA ratio) is elevated in mice with acute toxoplasmosis but not in mice with chronic toxoplasmosis (18). In contrast, our results indicated that dopamine metabolism activity was upregulated during the chronic stage of T. gondii infection, strongly suggesting that DA metabolites were chronically activated. Increased levels of DA metabolites with unaltered levels of DA itself have been shown to compensate for a deficiency in available DA in the cortex (47), and cortical dysfunction and the dysregulation of dopamine metabolism are involved in schizophrenia (48). Interestingly, T. gondii contains two genes encoding tyrosine hydroxylase, the rate-limiting enzyme of DA biosynthesis (49). Indeed, DA levels are increased in T. gondii-infected neurons and PC12 cells (22). These results suggest that T. gondii may control the host's DA biosynthesis pathway.
In the amygdala, 5-HT levels decreased at 40 and 52 dpi. 5-HT stimulates CORT secretion, and CORT modulates serotonergic activity in the amygdala, suggesting that 5-HT–CORT interactions may be involved in amygdala-dependent emotional behavior (28). T. gondii infection reduces CORT levels (29), suggesting that HPA axis dysfunction is mediated through the amygdala in mice infected with T. gondii. We found that NE levels were decreased in the cortex and amygdala of infected mice at both 40 and 52 dpi. Aversive stimuli enhance the secretion of NE from the locus coeruleus in the cortex and amygdala, resulting in enhanced fear memory consolidation modulated by stress hormone regulation (32). Thus, our results suggest that the decreased NE levels in the infected mice also contributed to the dysfunction of the cortex and amygdala. Moreover, an imbalance in the amygdala serotonergic system has been linked to anxiety and depression (50). Therefore, our results suggest that T. gondii infection causes a highly characteristic brain pathology in these neurological diseases and that lower levels of 5-HT and NE in the cortex and amygdala following T. gondii infection may be associated with neurological dysfunction.
Lastly, we found a negative correlation between the levels of all DA metabolites in the cortex and freezing behavior during the context test, meaning that the higher that the cortical DA metabolite levels were, the less time the animal spent freezing. In other words, fear memory consolidation was impaired in mice showing high levels of DA metabolites. In contrast, levels of 5-HT and 5-HIAA in the amygdala and NE in the cortex and amygdala were positively correlated with freezing behavior, indicating that the lower that the 5-HT, 5-HIAA, and NE levels were, the less time the mouse spent freezing. Auditory stimulus information in the amygdala is regulated by neurotransmitters, including DA, 5-HT, and NE (46). A fear-conditioned tone increases the levels of these neurotransmitters and influences excitatory and inhibitory neuron interactions. Thus, a loss of serotonergic and adrenergic neurons impairs the acquisition of conditioned fear (46), suggesting that the lower levels of brain DA, 5-HT, and NE that we detected in T. gondii-infected mice were associated with diminished fear memory. This is the first report to demonstrate a connection between altered neurotransmitter levels and behavioral changes following T. gondii infection.
Here, we used one behavioral paradigm and experimental model to examine the connection between T. gondii-induced inflammatory and neuronal damage to specific brain regions and subsequent behavioral change. Even though our model using male C57BL/6 mice is commonly used in this field, further investigation is needed to determine whether these findings remain consistent across several rodent models.
In conclusion, T. gondii infection in mice impaired long-term fear memory consolidation through dysfunction of the cortex and amygdala. In the infected mice, the cortex was more severely damaged than other brain regions, with dysfunction likely occurring in the brain. Dopamine metabolism was increased to compensate for a deficiency in available cortical DA in the infected mice due to the hypofunction of the cortex. In addition, we showed imbalances in neurotransmitters associated with modulation of the stress response (5-HT and NE) in the amygdala. These data support the hypothesis that the modification of responsiveness to stress mediated via the limbic-hypothalamic-pituitary-adrenal axis causes behavioral changes following T. gondii infection. Thus, our findings not only provide insight into the mechanisms underlying central nervous system changes during T. gondii infection but also elucidate the underlying mechanism of the relationship between T. gondii infection and the onset of mental disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Toshiaki Ishii for advice on behavioral science. We also thank Youko Matsushita, Megumi Noda, Hikaru Takenaka, and Yoshie Imura for their technical assistance.
This research was supported by the Japan Society for the Promotion of Science through the Funding Program for Next Generation World-Leading Researchers (NEXT Program), initiated by the Council for Science and Technology Policy (2011/LS003) (to Y.N.). This work was also supported by JSPS KAKENHI grant numbers 15K15118 (to Y.N.) and 23117007 (to K.N.). F. Ihara is a research fellow of the Japan Society for the Promotion of Science (15J03171).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
F.I., M.N., and Y.N. conducted the experiments. F.I., M.E.M., N.Y., K.N., and Y.N. designed the experiments. F.I. and Y.N. performed the data analysis. F.I., M.E.M., and Y.N. wrote the manuscript. All authors revised the manuscript and approved the final version.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00217-16.
REFERENCES
- 1.Pappas G, Roussos N, Falagas ME. 2009. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol 39:1385–1394. doi: 10.1016/j.ijpara.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 3.Flegr J. 2013. Influence of latent Toxoplasma infection on human personality, physiology and morphology: pros and cons of the Toxoplasma-human model in studying the manipulation hypothesis. J Exp Biol 216:127–133. doi: 10.1242/jeb.073635. [DOI] [PubMed] [Google Scholar]
- 4.Webster JP, Kaushik M, Bristow GC, McConkey GA. 2013. Toxoplasma gondii infection, from predation to schizophrenia: can animal behaviour help us understand human behaviour? J Exp Biol 216:99–112. doi: 10.1242/jeb.074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torrey EF, Bartko JJ, Yolken RH. 2012. Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr Bull 38:642–647. doi: 10.1093/schbul/sbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berdoy M, Webster JP, Macdonald DW. 2000. Fatal attraction in rats infected with Toxoplasma gondii. Proc Biol Sci 267:1591–1594. doi: 10.1098/rspb.2000.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamberton PHL, Donnelly CA, Webster JP. 2008. Specificity of the Toxoplasma gondii-altered behaviour to definitive versus non-definitive host predation risk. Parasitology 135:1143–1150. doi: 10.1017/S0031182008004666. [DOI] [PubMed] [Google Scholar]
- 8.Webster JP. 2007. The effect of Toxoplasma gondii on animal behavior: playing cat and mouse. Schizophr Bull 33:752–756. doi: 10.1093/schbul/sbl073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vyas A, Kim S-K, Giacomini N, Boothroyd JC, Sapolsky RM. 2007. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci U S A 104:6442–6447. doi: 10.1073/pnas.0608310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubey JP. 2009. History of the discovery of the life cycle of Toxoplasma gondii. Int J Parasitol 39:877–882. doi: 10.1016/j.ijpara.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Ingram WM, Goodrich LM, Robey EA, Eisen MB. 2013. Mice infected with low-virulence strains of Toxoplasma gondii lose their innate aversion to cat urine, even after extensive parasite clearance. PLoS One 8:e75246. doi: 10.1371/journal.pone.0075246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berenreiterová M, Flegr J, Kuběna AA, Němec P. 2011. The distribution of Toxoplasma gondii cysts in the brain of a mouse with latent toxoplasmosis: implications for the behavioral manipulation hypothesis. PLoS One 6:e28925. doi: 10.1371/journal.pone.0028925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatkowska J, Wieczorek M, Dziadek B, Dzitko K, Dlugonska H. 2012. Behavioral changes in mice caused by Toxoplasma gondii invasion of brain. Parasitol Res 111:53–58. doi: 10.1007/s00436-011-2800-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka S, Nishimura M, Ihara F, Yamagishi J, Suzuki Y, Nishikawa Y. 2013. Transcriptome analysis of mouse brain infected with Toxoplasma gondii. Infect Immun 81:3609–3619. doi: 10.1128/IAI.00439-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniels BP, Sestito SR, Rouse ST. 2015. An expanded task battery in the Morris water maze reveals effects of Toxoplasma gondii infection on learning and memory in rats. Parasitol Int 64:5–12. doi: 10.1016/j.parint.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Evans AK, Strassmann PS, Lee I-P, Sapolsky RM. 2014. Patterns of Toxoplasma gondii cyst distribution in the forebrain associate with individual variation in predator odor avoidance and anxiety-related behavior in male Long-Evans rats. Brain Behav Immun 37:122–133. doi: 10.1016/j.bbi.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stibbs HH. 1985. Changes in brain concentrations of catecholamines and indoleamines in Toxoplasma gondii infected mice. Ann Trop Med Parasitol 79:153–157. [DOI] [PubMed] [Google Scholar]
- 18.Gatkowska J, Wieczorek M, Dziadek B, Dzitko K, Dlugonska H. 2013. Sex-dependent neurotransmitter level changes in brains of Toxoplasma gondii infected mice. Exp Parasitol 133:1–7. doi: 10.1016/j.exppara.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Xiao J, Kannan G, Jones-Brando L, Brannock C, Krasnova IN, Cadet JL, Pletnikov M, Yolken RH. 2012. Sex-specific changes in gene expression and behavior induced by chronic Toxoplasma infection in mice. Neuroscience 206:39–48. doi: 10.1016/j.neuroscience.2011.12.051. [DOI] [PubMed] [Google Scholar]
- 20.Xiao J, Li Y, Prandovszky E, Karuppagounder SS, Talbot CC, Dawson VL, Dawson TM, Yolken RH. 2014. MicroRNA-132 dysregulation in Toxoplasma gondii infection has implications for dopamine signaling pathway. Neuroscience 268:128–138. doi: 10.1016/j.neuroscience.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hari Dass SA, Vyas A. 2014. Toxoplasma gondii infection reduces predator aversion in rats through epigenetic modulation in the host medial amygdala. Mol Ecol 23:6114–6122. doi: 10.1111/mec.12888. [DOI] [PubMed] [Google Scholar]
- 22.Prandovszky E, Gaskell E, Martin H, Dubey JP, Webster JP, McConkey GA. 2011. The neurotropic parasite Toxoplasma gondii increases dopamine metabolism. PLoS One 6:e23866. doi: 10.1371/journal.pone.0023866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webster J, Lamberton PH, Donnelly C, Torrey E. 2006. Parasites as causative agents of human affective disorders? The impact of anti-psychotic, mood-stabilizer and anti-parasite medication on Toxoplasma gondii's ability to alter host behaviour. Proc Biol Sci 273:1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pariante CM, Lightman SL. 2008. The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31:464–468. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 25.López JF, Akil H, Watson SJ. 1999. Neural circuits mediating stress. Biol Psychiatry 46:1461–1471. doi: 10.1016/S0006-3223(99)00266-8. [DOI] [PubMed] [Google Scholar]
- 26.Herman JP, Ostrander MM, Mueller NK, Figueiredo H. 2005. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry 29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. 2003. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol 24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Stutzmann GE, McEwen BS, LeDoux JE. 1998. Serotonin modulation of sensory inputs to the lateral amygdala: dependency on corticosterone. J Neurosci 18:9529–9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra R, Sapolsky RM, Vyas A. 2013. Toxoplasma gondii infection induces dendritic retraction in basolateral amygdala accompanied by reduced corticosterone secretion. Dis Model Mech 6:516–520. doi: 10.1242/dmm.009928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harley CW. 2004. Norepinephrine and dopamine as learning signals. Neural Plast 11:191–204. doi: 10.1155/NP.2004.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferry B, Roozendaal B, McGaugh JL. 1999. Role of norepinephrine in mediating stress hormone regulation of long-term memory storage: a critical involvement of the amygdala. Biol Psychiatry 46:1140–1152. doi: 10.1016/S0006-3223(99)00157-2. [DOI] [PubMed] [Google Scholar]
- 32.Sara SJ. 2015. Locus coeruleus in time with the making of memories. Curr Opin Neurobiol 35:87–94. doi: 10.1016/j.conb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Witting PA. 1979. Learning capacity and memory of normal and Toxoplasma-infected laboratory rats and mice. Z Parasitenkd 61:29–51. doi: 10.1007/BF00927085. [DOI] [PubMed] [Google Scholar]
- 34.Kannan G, Moldovan K, Xiao J-C, Yolken RH, Jones-Brando L, Pletnikov MV. 2010. Toxoplasma gondii strain-dependent effects on mouse behaviour. Folia Parasitol (Praha) 57:151–155. doi: 10.14411/fp.2010.019. [DOI] [PubMed] [Google Scholar]
- 35.Gulinello M, Acquarone M, Kim JH, Spray DC, Barbosa HS, Sellers R, Tanowitz HB, Weiss LM. 2010. Acquired infection with Toxoplasma gondii in adult mice results in sensorimotor deficits but normal cognitive behavior despite widespread brain pathology. Microbes Infect 12:528–537. doi: 10.1016/j.micinf.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worth AR, Lymbery AJ, Thompson RCA. 2013. Adaptive host manipulation by Toxoplasma gondii: fact or fiction? Trends Parasitol 29:150–155. doi: 10.1016/j.pt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Kannan G, Pletnikov MV. 2012. Toxoplasma gondii and cognitive deficits in schizophrenia: an animal model perspective. Schizophr Bull 38:1155–1161. doi: 10.1093/schbul/sbs079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 39.Terkawi MA, Kameyama K, Rasul NH, Xuan X, Nishikawa Y. 2013. Development of an immunochromatographic assay based on dense granule protein 7 for serological detection of Toxoplasma gondii infection. Clin Vaccine Immunol 20:596–601. doi: 10.1128/CVI.00747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curzon P, Rustay NR, Browman KE. 2009. Cued and contextual fear conditioning for rodents. CRC Press LLC, Boca Raton, FL. [PubMed] [Google Scholar]
- 41.Grem JL, Danenberg KD, Behan K, Parr A, Young L, Danenberg PV, Nguyen D, Drake J, Monks A, Allegra CJ. 2001. Thymidine kinase, thymidylate synthase, and dihydropyrimidine dehydrogenase profiles of cell lines of the National Cancer Institute's anticancer drug screen. Clin Cancer Res 7:999–1009. [PubMed] [Google Scholar]
- 42.Eisenhofer G, Kopin IJ, Goldstein DS. 2004. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez LE, Rojnik B, Urrea F, Urdaneta H, Petrosino P, Colasante C, Pino S, Hernandez L. 2007. Toxoplasma gondii infection lower anxiety as measured in the plus-maze and social interaction tests in rats: a behavioral analysis. Behav Brain Res 177:70–79. doi: 10.1016/j.bbr.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Bottari NB, Baldissera MD, Tonin AA, Rech VC, Alves CB, D'Avila F, Thomé GR, Guarda NS, Moresco RN, Camillo G, Vogel FF, Luchese C, Schetinger MRC, Morsch VM, Tochetto C, Fighera R, Nishihira VSK, Da Silva AS. 2016. Synergistic effects of resveratrol (free and inclusion complex) and sulfamethoxazole-trimetropim treatment on pathology, oxidant/antioxidant status and behavior of mice infected with Toxoplasma gondii. Microb Pathog 95:166–174. doi: 10.1016/j.micpath.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Rozeske RR, Valerio S, Chaudun F, Herry C. 2015. Prefrontal neuronal circuits of contextual fear conditioning. Genes Brain Behav 14:22–36. doi: 10.1111/gbb.12181. [DOI] [PubMed] [Google Scholar]
- 46.LeDoux J. 2007. The amygdala. Curr Biol 17:R868–R874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Nishi A, Kuroiwa M, Miller DB, O'Callaghan JP, Bateup HS, Shuto T, Sotogaku N, Fukuda T, Heintz N, Greengard P, Snyder GL. 2008. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci 28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laruelle M, Kegeles LS, Abi-Dargham A. 2003. Glutamate, dopamine, and schizophrenia. Ann N Y Acad Sci 1003:138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- 49.Gaskell EA, Smith JE, Pinney JW, Westhead DR, McConkey GA. 2009. A unique dual activity amino acid hydroxylase in Toxoplasma gondii. PLoS One 4:e4801. doi: 10.1371/journal.pone.0004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer EP. 2015. Serotonin in fear conditioning processes. Behav Brain Res 277:68–77. doi: 10.1016/j.bbr.2014.07.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.