

Abstract
Yersinia pestis is a Gram-negative bacterium that is the causative agent of bubonic and pneumonic plague. It is commonly acquired by mammals such as rodents and humans via the bite of an infected flea. We previously reported that multiple substrains of the 129 mouse background are resistant to pigmentation locus-negative (pgm−) Yersinia pestis and that this phenotype maps to a 30-centimorgan (cM) region located on chromosome 1. In this study, we have further delineated this plague resistance locus to a region of less than 20 cM through the creation and phenotyping of recombinant offspring arising from novel crossovers in this region. Furthermore, our experiments have revealed that there are at least two alleles in this initial locus, both of which are required for resistance on a susceptible C57BL/6 background. These two alleles work in trans since resistance is restored in offspring possessing one allele contributed by each parent. Our studies also indicated that the Slc11a1 gene (formerly known as Nramp1) located within the chromosome1 locus is not responsible for conferring resistance to 129 mice.
INTRODUCTION
The Gram-negative bacterium Yersinia pestis is the causative agent of bubonic and pneumonic plague. Responsible for several global pandemics and millions of deaths, including the “Black Death” of the Middle Ages, Yersinia pestis is still an active global threat. While present-day outbreaks involve far fewer cases, they are still of concern, especially since around 10% of bubonic plague cases proceed to the pneumonic form, which both spreads more easily and is much harder to treat given its rapidly manifested severity (1). Indeed, a recent outbreak in Madagascar is associated with 263 cases and 71 deaths, and a previous outbreak in 2011 involved the pneumonic form (2). In addition, antibiotic resistance to Y. pestis has been documented and there is no vaccine currently licensed for the prevention of plague in the Western world (3).
Historically, the majority of studies of Y. pestis have focused on chromosomal genes or virulence plasmids of the bacteria that contribute to pathogenicity. Many of these so-called virulence factors have also been considered potential vaccine targets. For example, antibodies to the capsular F1 antigen and the low-calcium-response V antigen (LcrV) have been shown to confer immunity against plague (4–6). More recently, several groups have begun to focus on the interaction between the bacteria and specific host cell types. It has been shown that the bacteria can survive and replicate in both macrophages and neutrophils (7, 8) and that they can disseminate to the draining lymph node nearest the site of flea bite within an hour (9). However, relatively few studies have investigated host factors and responses that may confer resistance to infection.
It would be expected that a pathogen as lethal as that causing plague would exert a high evolutionary pressure on its host populations. Indeed, rats in areas of plague endemicity have been shown to resist infection, and this resistance has been shown to be heritable (10). We therefore chose to use inbred mice as a model to identify genes that may confer resistance against plague. We and others subsequently identified several inbred strains of mice that are resistant (11–14). Resistance of the 129 line was first identified by Congleton et al. in a pigmentation locus-negative (pgm−) infection model of plague (11) but not for a fully virulent Y. pestis strain. The resistance is observed in multiple substrains of this line, and further studies by our laboratory have localized a major portion of the resistance phenotype to chromosome 1. This locus was initially discovered while looking into reports that interleukin-10 (IL-10) knockout mice were resistant to plague (15). We found that these mice possess a large portion of chromosome 1 from the 129 background and that it was this, in addition to the loss of the IL-10 allele, that contributed to improved survival (13). The goal of this study was to determine which genes on chromosome 1 might be involved in the resistance phenotype. To this end, we continued to backcross this locus onto the susceptible C57BL/6 background in order to identify offspring with novel crossovers that could then be tested for resistance. We found that the initial locus segregates into two separate loci, both of which are required to confer resistance.
MATERIALS AND METHODS
Bacteria.
The bacterial strain used in this study was Y. pestis KIM D27 (pgm−), cultured as described previously (13). Bacteria were grown overnight in Luria broth (LB) at 25°C. After overnight growth, bacteria were diluted 1:10 in LB and grown to mid-exponential phase (optical density at 600 nm of ∼0.4). Bacteria were collected by centrifugation, washed twice, and serially diluted in phosphate-buffered saline (PBS) to the appropriate concentration. For all experiments, mice were intravenously infected via the caudal tail vein with a 200-μl inoculum. The actual dose given was ascertained by plating the inoculum on heart infusion agar plates incubated at 25°C.
Mice.
All experimental mice were between 6 and 12 weeks of age, and only female mice were used. All intercrosses were performed in our Animal Research Facility at the University of Illinois at Urbana—Champaign. C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were bred with our previously described 129 congenic strains (13). 129/Sv Slc11a1−/− mice (also known as 129S6-Slc11a1tm1Mcg) were obtained from P. Gros (McGill University, Montreal, Quebec, Canada). All congenic strains were backcrossed a minimum of 10 times to C57BL/6J. All experiments were approved by the University of Illinois at Urbana-Champaign Institutional Animal Care and Use Committee. For survival experiments, animals were monitored at least twice daily and culled in a timely fashion upon reaching a moribund state.
Genotyping.
For congenic strains, mice were genotyped using microsatellite markers on DNA isolated from ear punches. PCRs were run on 3.3% agarose gels. Markers used included D1Mit303, D1Mit251, D1Mit19, D1Mit7, D1Mit46, D1Mit253, D1Mit134, D1Mit439, D1Mit49, D1Mit48, D1Mit305, D1Mit10, D1Mit45, D1Mit187, D1Mit54, D1Mit135, D1Mit157, D1Mit94, D1Mit494, D1mit495, and D1Mit286. The maximum interval between markers used was less than 3 centimorgans (cM) and was chosen in an effort to avoid confounding due to double crossovers. The exact marker sequences can be found online through the MGI database.
Enumeration of bacterial titer.
Infected mice were sacrificed 60 h postinfection by2 asphyxiation. Spleens were removed aseptically and weighed. Organs were homogenized in Stomacher bags and serially diluted in PBS. Bacterial burden was determined by plating the dilutions on LB agar plates and incubating them at 23°C for 3 days.
Flow cytometry.
The protocol followed has been previously described (16). Briefly, spleens were removed aseptically approximately 60 h postinfection following CO2 asphyxiation. They were homogenized in 10 ml of RPMI 1640 and then centrifuged, followed by red blood cell lysis performed using 2 ml of 150 mM NH4Cl. The unlysed cells were then resuspended in a PBS-based buffer containing bovine serum albumin (BSA) and sodium azide; cell numbers were enumerated and found to be similar across all samples. The following antibodies from Fisher/BD were then used for staining: allophycocyanin-conjugated anti-CD45 for all hematopoietic cells, fluorescein isothiocyanate-conjugated anti-CD4 and phycoerythrin-conjugated anti-CD8 for T cells, fluorescein isothiocyanate-conjugated anti-CD19 for B cells, phycoerythrin-conjugated anti-NK1.1 for NK cells, and phycoerythrin-conjugated anti-Ly-6G for neutrophils. Phycoerythrin-conjugated anti-F4/80 for macrophages was purchased from BioLegend. Cells were incubated on ice for 1 h with appropriate antibodies and then washed and resuspended in a PBS-paraformaldehyde mixture. For each sample, at least 20,000 events were counted. After an initial gating on CD45+ cells, samples were analyzed by forward and side scatter, followed by analysis of positive cell populations for each stain.
Statistics.
Survival curves were generated using GraphPad Prism 6, and reported significance was measured by a log rank (Mantel-Cox) test. Results were also assessed for significance using either the log-rank test for trend or the GBW (Gehan-Breslow-Wilcoxon) test, and the results matched the findings of the Mantel-Cox test in each case. For splenic bacterial load experiments and comparisons of flow cytometry data, P values were calculated using an unpaired t test in GraphPad Prism 6.
RESULTS
The region between D1Mit251 and D1Mit494 is required for resistance.
We previously identified a plague resistance locus on chromosome 1 of mice of the 129 background. This locus resides between markers D1Mit303 and D1Mit286 and covers approximately 25 cM. Resistant mouse lines that had been previously challenged intravenously with a high dose of Yersinia pestis showed a survival rate of about 60% (13). In an attempt to further define the genomic region required for plague resistance, we backcrossed resistant mice with C57BL/6 mice and screened for offspring with novel crossovers that maintained only a portion of the resistant region. These mice served as founders to generate new lines that were then tested in our model (Fig. 1). The first of these sublines, containing the greatest amount of parental 129 DNA, was line 4, which possesses 129 DNA between markers D1Mit251 and D1Mit19 proximally and between markers D1Mit94 and D1Mit494 distally, thereby spanning a region of 16.7 to 19.7 cM. Challenged intravenously with a dose of 103 Y. pestis KIM D27, line 4 mice exhibited a survival rate of around 60%, whereas littermate control C57BL/6 mice generally all succumbed to the infection (Fig. 2; P < 0.005 for each experiment). These results are consistent with those determined with the resistant mice profiled in our initial study (13).
FIG 1.

Schematic diagram of congenic lines used in this study. For each congenic strain, the thick black lines represent DNA from the 129 background and thin black lines represent DNA from the C57BL/6 background. White lines demarcate boundaries within which a crossover event has occurred as determined using chromosomal markers (shown). Physical and linkage distances are also shown for chromosome 1. Mbp, megabase pairs; cM, centimorgans.
FIG 2.

A region of chromosome 1, defined by congenic line 4, confers resistance to Y. pestis KIM D27. Line 4 mice and C57BL/6 littermate controls were infected intravenously (i.v.) with 103 Y. pestis bacteria and monitored for survival for 15 days. Panels A and B are representative of the results of multiple experiments. For panel A, both groups contained 12 mice; for panel B, groups of 8 C57BL/6 mice and 12 line 4 mice were used. Data in both graphs are statistically significant (P < 0.005) as determined by either log-rank analysis or overall survival.
Congenic lines maintaining the distal end of the region do not exhibit resistance.
We next attempted to examine the contribution of the proximal side of the region to resistance to Y. pestis KIM D27 by generating novel crossovers in offspring from our resistant lines. These new lines were denoted lines 7, 5, 8, and 240 (Fig. 1). Line 7 possesses 3.2 to 5.5 cM of 129 DNA between markers D1Mit187 and D1Mit54 proximally and between markers D1Mit495 and DMit286 distally. Line 5 harbors 11.8 to 14.5 cM of 129 DNA between markers D1Mit439 and D1Mit49 proximally and between markers D1Mit495 and D1Mit286 distally, whereas line 8 has 14.6 to 16.5 cM of 129 DNA between markers D1Mit253 and D1Mit134 proximally and between D1Mit495 and D1Mit286 distally. As shown in Fig. 3, none of these new lines are resistant in our infection model, as neither the overall survival results nor time-to-death results were significantly different from those determined for the littermate C57BL/6 control mice in these experiments.
FIG 3.

Congenic mice which possess 129 DNA from the distal end of the region are not resistant to Y. pestis infection. Mice of line 7 (A and B), line 5 (C and D), line 8 (E and F), or line 240 (G and H), along with C57BL/6 mice, were infected with 103 Y. pestis KIM D27 bacteria and monitored for survival for 15 days. For each panel, the following mouse numbers were used: for panel A, 6 C57BL/6 and 10 line 7 mice; for panel B, 5 C57BL/6 and 25 line 7 mice; for panel C, 6 C57BL/6 and 13 line 5 mice; for panel D, 6 C57BL/6 and 19 line 5 mice; for panel E, 9 C57BL/6 and 7 line 8 mice; for panel F, 11 C57BL/6 and 9 line 8 mice; for panel G, 10 C57BL/6 and 6 line 240 mice; and for panel H, 14 C57BL/6 and 7 line 240 mice. Compared to littermate C57BL/6 mice, none of the lines were resistant to Y. pestis as determined by either log-rank analysis or overall survival.
Line 240 mice harbor 129 DNA between markers D1Mit253 to D1Mit134 proximally and D1Mit94 to D1Mit494 distally, which spans a region of 13.5 to 15.2 cM. As shown in Fig. 3G and H, these mice also failed to exhibit significant resistance in our infection model. Taken together, these results imply that there is a region in the proximal side of our locus, somewhere between D1Mit251 and D1Mit134, that is required for conferring resistance to Y. pestis.
Congenic lines maintaining the proximal end of the region do not exhibit resistance.
Since the distal portion of the region was insufficient by itself to confer resistance to Y. pestis, we therefore screened and generated lines that would enable us to examine the contribution of the proximal region to the resistance phenotype. Line 46 harbors 1.8 to 5.9 cM of 129 DNA between markers D1Mit251 and D1Mit19 proximally and between markers D1Mit253 and D1Mit134 distally, while line 232 possesses 6.7 to 9.7 cM of 129 DNA between markers D1Mit251 and D1Mit19 proximally and between markers D1Mit48 and D1Mit305 distally. As shown in Fig. 4, neither of these lines demonstrated resistance in our infection model. This result implies that a chromosomal region required for resistance exists between D1Mit48 and D1Mit494. Combined with the data from the congenic lines presented in Fig. 3, these results indicate that there are multiple genes required for the resistance phenotype of our parental mice.
FIG 4.

Congenic mice which possess 129 DNA from the proximal end of the region are not resistant to Y. pestis infection. Mice from either line 232 (A and B) or line 46 (C and D), along with C57BL/6 littermate controls, were infected with 103 Y. pestis KIM D27 bacteria and monitored for survival for 15 days. For each panel, the following mouse numbers were used: for panel A, 10 C57BL/6 and 8 line 232 mice; for panel B, 7 C57BL/6 and 10 line 232 mice; for panel C, 15 C57BL/6 and 10 line 46 mice; and for panel D, 10 C57BL/6 and 5 line 46 mice. Compared to littermate C57BL/6 mice, none of the lines were resistant to Y. pestis as determined by either log-rank analysis or overall survival.
Slc11a1 is not required for resistance in a 129 background.
In a previous study, we had attempted to determine whether the resistance of 129 mice might be due to Slc11a1 (formerly known as Nramp1), since this gene resides at one end of the resistance locus and is known to play a role in resistance to infection by intracellular bacteria (13). Importantly, Slc11a1 is known to be nonfunctional in several inbred mouse strains such as C57BL/6J mice, but 129 mice harbor the resistant version of the allele (17). Since the resistance in our model acts in a dominant fashion, a functional allele of Slc11a1 was crossed into the nonresistant C57BL/6J mouse background and these mice were then tested in our KIM D27 infection model. These mice exhibited no resistance, so it was concluded that a functional version of Slc11a1 was not associated with resistance (13). Our recent finding that at least two separate loci are required to confer resistance means that Slc11a1 could still be involved despite not being sufficient by itself. In order to address this, we obtained 129 mice that had had their copy of Slc11a1 knocked out (a generous gift from Philippe Gros, McGill University) in order to mimic the situation in C57BL/6 mice (18). Tested in our model of Y. pestis KIM D27 infection, these 129/Sv Slc11a1−/− mice were fully resistant to infection (P < 0.005), indicating that the active allele of Slc11a1 is not required for plague resistance on the 129 background (Fig. 5). Since B6129SF1/J mice are more resistant than line 4 mice, which harbor the region determined by us, it is possible that Slc11a1 is still important in our congenic mice but is not required in the full 129 background due to other genetic factors that influence resistance.
FIG 5.

Slc11a1 is not required for resistance to Y. pestis KIM D27 in mice of the 129 background. 129 mice with the Slc11a1 gene knocked out (129/Sv Slc11a1−/−), along with C57BL/6 mice, were infected with 103 Y. pestis bacteria and monitored for survival for 15 days. Panel A represents 10 mice per group, while panel B represents 7 mice per group (P < 0.005 for both experiments).
The proximal and distal regions of the locus can act in trans to confer resistance.
We next sought to determine whether the two subregions of the larger locus could act in trans to restore resistance or whether they needed to act in cis through colocalization on the same chromosome. Mice carrying the proximal region (line 46) were bred with lines carrying the distal region (line 240; see Fig. 1). The resulting offspring were then tested in our KIM D27 infection model. As shown in Fig. 6, mice carrying both portions of the region were resistant whereas those that harbored only one portion did not demonstrate resistance. This result was statistically significant for both experiments (P < 0.005). The parental origin of each region did not make any difference with respect to the observed result (data not shown). These results demonstrate that two independent subregions of the original locus can act in trans to confer resistance to Y. pestis.
FIG 6.

Two subregions of 129 DNA act in trans to restore resistance to Y. pestis infection. Various mouse lines, along with C57BL/6 mice, were infected with 103 Y. pestis KIM D27 bacteria and monitored for survival for 15 days. Data in panels A and B represent 10 C57BL/6, 9 line 4, 13 46 × 240, 10 line 46, and 10 line 240 mice. Data in panels C and D represent 13 C57BL/6, 13 line 4, 15 46 × 240, 11 line 46, and 15 line 240 mice. Panels A and B represent a single experiment, while C and D represent a second single experiment. Compared to C57BL/6 mice, both line 4 and line 46 × 240 mice were resistant to Y. pestis by either log-rank analysis or by overall survival (P < 0.005). As before, neither line 46 nor line 240 mice were resistant.
Splenic bacterial load correlates with survival.
In an attempt to find a surrogate endpoint to survival, as well as to get an idea of the kinetics of the infection, mice were sacrificed at a given time point postinfection with Y. pestis and splenic CFU counts were calculated. Our initial study indicated that there was not a significant difference in splenic CFU counts between resistant 129 mice and susceptible C57BL/6 mice until 2 days postinfection. In order to maximize our ability to detect a difference while protecting mice from unnecessary suffering or death, we chose 2.5 days postinfection for this study. Livers were not tested since there was no significant difference in bacterial load in this organ observed at any time point in our previous study (13). Since our resistant lines generally exhibit around 60% resistance, we expected several line 4 mice to have lower bacterial counts than the C57BL/6 controls, although some might be expected to exhibit levels similar to those seen with susceptible mice and would represent those mice not expected to have survived had this experiment been continued (for survival determinations) to death as an endpoint. As shown in Fig. 7A, this is exactly what was observed. In this experiment, 5 line 4 mice were tested, and while 2 had bacterial loads similar to those of the C57BL/6 control, the bacterial loads in 3 mice were drastically lower. The difference in the means between the levels determined for the C57BL/6 mice and the line 4 mice was significant (P < 0.05), and this result is consistent with those of multiple repeat experiments.
FIG 7.

Resistance to infection correlates to reduced bacterial load in the spleen. Various mouse lines, along with C57BL/6 mice, were infected with 103 Y. pestis KIM D27 bacteria, and spleens were harvested 60 h postinfection. The following mouse numbers were used: for panel A, 4 C57BL/6 and 5 line 4 mice; for panel B, 5 C57BL/6 and 5 line 46 mice; for panel C, 4 C57BL/6 and 5 line 240 mice; and for panel D, 4 C57BL/6 and 7 46 × 240 mice. Data in panels C and D represent parts of the same experiment, and thus only 4 C57BL/6 mice in total were used for those two panels. Results are representative of 3 independent experiments. Horizontal lines represent the mean for each group. Asterisks indicate significant differences between groups as determined by Student's t test where P is <0.05; NS = not significant.
We next tested some of the sublines for their ability to control bacterial load. Mice from lines 240 and 46 were tested in our Y. pestis model and did not exhibit a significant difference in bacterial load from the C57BL/6 mice (Fig. 7B and C). While they both tended to show a trend toward reduced colony counts, this was not necessarily unexpected, as we believe each region contains at least 1 factor that helps the parental 129 mice achieve resistance to Y. pestis infection. In repeat experiments, line 46 appeared to confer an ability to maintain lower bacterial loads than line 240. This potentially indicates that the smaller line 46 locus makes a greater contribution to control of Y. pestis growth than the larger, more distal line 240 locus. The survival results indicate that neither line confers enough of a benefit in the control of bacterial growth to improve survival in this Y. pestis model.
Mice possessing both portions of the region in trans, with each contributed by a different parent, did exhibit a significant reduction in bacterial load (Fig. 7D; P < 0.05). Importantly, variations in the data show that some of the line 46 × 240 mice exhibited the higher bacterial loads which would be expected to correlate with mice that succumb to Y. pestis in the survival assay.
Assessment of splenic leukocyte infiltrates.
We next sought to determine if differences in the numbers of infiltrating splenic leukocytes might be the cause of the resistance phenotype. To assess this, mice were infected with Y. pestis KIM5 and spleens were harvested 2.5 days postinfection. As shown by Fig. 8, flow cytometry of splenic cells revealed no significant differences in the percentage of B cells, T cells, macrophages, neutrophils, or NK cells between resistant line 4 mice and susceptible C57BL/6 mice. The total numbers of leukocytes in the spleens of infected mice were also similar (data not shown). It therefore appears that the resistance phenotype is not due to gross differences in the immune cell infiltration of the spleen during infection.
FIG 8.
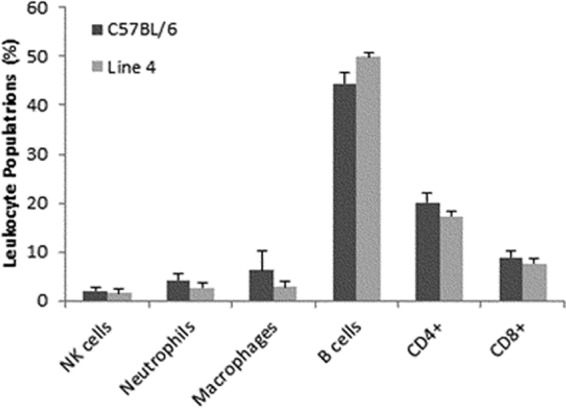
Immune cell populations in the spleen following infection are not different. C57BL/6 and line 4 mice were infected with 103 Y. pestis KIM D27 bacteria, and spleens were harvested 60 h postinfection. Leukocyte populations were analyzed by flow cytometry using specific markers for NK cells, neutrophils, macrophages, B cells, CD4+ T cells, and CD8+ T cells. The data represent average results determined for three individual mice, and the error bars are standard deviations. An unpaired t test found no differences between any of the cell populations analyzed.
DISCUSSION
The application of forward genetic methods in mouse models of infection has proven to be a powerful approach for the identification of novel genes involved in immune defense. For example, this approach has enabled the identification of the Slc11a1 and TLR4 genes, which were originally linked to the Ity-Lsh-Bcg locus and the lipopolysaccharide (LPS) locus, respectively (19–22). In the context of mouse models of infection, the main advantages of a forward-genetics approach in comparison to reverse genetics are that there is an established phenotype and that any changes seen are known to be physiologically relevant. The greatest drawback is that infection phenotypes are often caused by multiple genetic changes which are either lost or greatly reduced with respect to the magnitude of the phenotypes in attempting to ascribe them to a single gene. Our work here illustrates both these advantages and disadvantages.
The initial investigation of our Y. pestis resistance locus revealed a broad linkage association that spanned most of chromosome 1 (13). The lack of a sharp peak in the linkage analysis implied either the use of an insufficient number of mice in the study or the involvement of multiple genes spanning the region in the resistance phenotype. In order to address this, we narrowed our analysis further by testing the resistance phenotype of new congenic lines harboring novel genetic crossovers. In this study, we found that multiple genetic loci are required for the plague resistance observed in mice of the 129 background. This result is not surprising given the fact that Yersinia pestis survives both intra- and extracellularly and utilizes a variety of mechanisms to subvert the immune response. Resistance appears to be an early event, with multiple genes working in concert to prevent the infection from getting out of control. In comparison to the results seen with C57BL/6 mice, the course of infection did not change in mice harboring any smaller portion of the region, indicating that the presence of each subregion alone was insufficient to provide a survival benefit.
We have observed statistically significant differences in bacterial load in the spleen as early as 2 days postinfection (13). These early differences in bacterial load suggest that the resistance phenotype is due to differences in innate immunity but not in adaptive immunity. Interestingly, an earlier study by Congleton et al., also using pgm− Yersinia pestis, showed that 129 mice have a greater influx of NK cells to the spleen than C57BL/6 mice (11, 16) as well as an overall higher level of systemic NK cells during infection. Here we report that we found no differences in NK cell levels between the resistant and susceptible mice, suggesting that this is not the basis of the resistance phenotype. Additionally, no significant differences were observed in the numbers of any other immune cell types, implying that the recruitment or survival of these cells is not the basis of the resistance conferred by the locus on chromosome 1. We cannot exclude the possibility that differences in immune cell function, rather than number, provide the basis of the resistance phenotype.
In their study on the role of the Yersinia pgm locus in pneumonic plague, Lee-Lewis and Anderson theorized that the resistance of 129 mice may be due to the Slc11a1 (also known as Nramp1) gene (23). The Slc11a1 gene is located at one end of the resistance locus and encodes a divalent metal iron transporter that is recruited to macrophage phagosomes, where it serves to promote killing of intracellular pathogens. Since one of the major ways in which the pgm− strains of Y. pestis are attenuated is through their loss of the Ybt locus which impacts their ability to acquire iron (24, 25), a difference in a gene with the potential to regulate iron availability in the body, such as Slc11a1, could explain the differences in susceptibility to pgm− plague. Previously, Slc11a1 has been demonstrated to play a role in resistance to pathogens such as Mycobacterium bovis, Leishmania donovani, and Salmonella enterica serovar Typhimurium (20, 26–29). Susceptibility of mouse strains to certain infections has been shown to depend on whether that strain carries the naturally occurring R (resistant) or S (susceptible) form of the Slc11a1 allele. For example, Lee-Lewis and Anderson had found that C57BL/6 and BALB/c mice, which harbor the S allele, are more sensitive to the KIM D27 strain of Y. pestis than either the 129 strain or the C3H strain, each of which carries the R allele (23). In our earlier published study, we did not observe any increased resistance to Y. pestis when the R allele of Slc11a1, from C3H mice, was crossed into the C57BL/6 background (13). This finding is consistent with our current observation that 129 mice harboring the S version of Slc11a1 retain full resistance against plague. The concept that the R allele of Slc11a1 is not required to confer resistance is also supported by our broader finding that multiple genes in the chromosome 1 locus, including a region distinct from that containing Slc11a1, contribute to resistance. Both the parental 129 strain and B6129SF1/J mice are more resistant than any of our C57BL/6 congenic lines which possess 129 DNA, suggesting that there are other genetic factors, outside the 129 chromosome 1 locus, that contribute to Y. pestis resistance. Since these other genetic factors may function to make the Slc11a1 status irrelevant, due to the improved resistance of these mice, we cannot rule out a role for the Slc11a1 allele in the resistance of our congenic mice possessing regions of 129 chromosome 1.
Despite this shortcoming, results from other studies lend support to the theory that Slc11a1 does not play a role in the resistance of mice to pgm− strains of Y. pestis. During Salmonella Typhimurium infection, the R allele of Slc11a1, but not the S allele, was shown to directly modulate the expression of genes on Salmonella pathogenicity island 6 (SPI-6) (30). While Y. pestis has homologues of these genes in the ripCBA operon, this operon is part of the pgm locus which is lacking in the bacterial strain that we used in this study. Pujol et al. demonstrated that intracellular Y. pestis can evade killing by macrophages that are exposed to interferon (IFN) gamma and identified a potential virulence gene present in the pgm locus that is required for this activity (31). These results suggest that examining the role of Slc11a1 allele in host resistance to fully virulent Y. pestis may be a worthwhile venture.
Several studies have examined the resistance of different mouse strains to plague. A recent study using fully virulent plague pathogens found that multiple loci were required for mice to survive in a Y. pestis subcutaneous infection model (32–34). To date, the Mus spretus SEG/Pas mouse strain has been the only strain to have demonstrated resistance to fully virulent plague pathogens. While working with a pgm− plague model, our laboratory and those of others have demonstrated resistance against plague in several inbred strains of mice, including BALB/cJ mice (14), 129 mice (13), B10(T6R) mice (12), and DBA2/J mice (M. Tencati and R. Tapping, unpublished data). The resistance in BALB/cJ mice has been localized to chromosome 17, whereas at least part of the resistance in 129 mice localizes to chromosome 1. Neither of these two loci overlaps those found in the SEG/Pas mice. Likely due to differences in dose and route of infection, the course of infection in resistant SEG/Pas mice and susceptible mice differs from that which was seen here; SEG/Pas mice clear the infection from blood by day 4 and show decreases in bacterial load in the liver and spleen at that time, whereas our resistant lines appear to show a tapering off of bacterial growth between days 2 and 4 but no decrease in overall levels until later time points (13, 34).
Aside from Slc11a1, there are many other candidate genes in this chromosome 1 region that could contribute to resistance and play a role in immune defense. Several genes in this region are known to be involved in neutrophil recruitment, survival of immune cells, and ion transport. Looking for genetic differences between the resistant and susceptible C57BL/6 mice using the Wellcome Trust Sanger Institute's Mouse Genome Project database (release REL-1505) helps to narrow the list of possible candidates. Cxcr2 is involved in neutrophil recruitment following exposure to LPS (35) and has single nucleotide polymorphisms (SNPs) within both the 5′ and 3′ untranscribed regions (UTRs), while Cxcr1, which is also involved in neutrophil recruitment, has a missense variant. Bcl2 may be involved in blocking the apoptotic death of lymphocytes and has variations in the 5′ and 3′ UTRs as well as throughout the intronic regions. Atg4b is involved in autophagy and has two missense mutations along with variations in the 3′ UTR. Genes such as C1ql2, Irs1, Lrrfip1, Rab17, Slco4c1, and Slco6d1 exhibit variations in either the 5′ or 3′ UTRs. Other genes in the region with immune function are unlikely candidates due to lack of genetic variations. Atg9a, Ccl20, Marco, Slc23a3, and Slc35f5 exhibit no SNP differences, while Atg16l1, Traf3ip1, Slc16a14, Slc19a3, and Slco6c1 all contain genetic differences but only in intronic regions that are not known to be involved in splice variant formation.
Overall, the resistance locus described here contains 319 protein coding genes, 115 unclassified genes, 34 microRNAs (miRNAs), 236 long intervening noncoding RNAs (lincRNAs) or long noncoding RNAs (lncRNAs), and 16 unclassified noncoding RNA genes. Since there are currently 130,721 SNP differences between the two parental strains across the resistance locus in the Mouse Genome Project database, defining the specific SNPs responsible for conferring resistance to Y. pestis is inherently difficult. Recent reports suggest that 93% of disease-associated SNPs in humans are located in gene-regulatory regions or intergenic regions, as opposed to the 7% which are located in protein-coding (36, 37). While transcriptome sequencing (RNA-seq) or microarray analysis could be used to assess expression differences in protein coding genes within the region and thereby potentially to identify cis-acting gene regulatory changes, this would still not address the issue of the many lncRNAs and lincRNAs which could be acting on genes either inside or outside the quantitative trait locus (QTL) region.
Assessing noncoding RNAs is currently complicated by the findings that RNA-seq does not perform well on these low-abundance transcripts, their expression is very often tissue specific, and they remain poorly annotated. Improvements in both proteomic techniques and genomic databases may be required to eventually identify the SNP changes which are responsible for the resistance of the 129 mice to Yersinia infection.
ACKNOWLEDGMENTS
This work was generously supported by the NIH-NIAID under grant number 5R21AI097969. During part of the studies, M.T. was supported under NIH training grant T32 AI078876.
We thank Joshua Turner for technical assistance and Philippe Gros for the generous gift of the Slc11a1tm1Mcg mice.
REFERENCES
- 1.Kool JL. 2005. Risk of person-to-person transmission of pneumonic plague. Clin Infect Dis 40:1166–1172. doi: 10.1086/428617. [DOI] [PubMed] [Google Scholar]
- 2.Richard V, Riehm JM, Herindrainy P, Soanandrasana R, Ratsitoharina M, Rakotomanana F, Andrianalimanana S, Scholz HC, Rajerison M. 2015. Pneumonic plague outbreak, Northern Madagascar, 2011. Emerg Infect Dis 21:8–15. doi: 10.3201/eid2101.131828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feodorova VA, Motin VL. 2012. Plague vaccines: current developments and future perspectives. Emerg Microbes Infect 1:e36. doi: 10.1038/emi.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quenee LE, Schneewind O. 2009. Plague vaccines and the molecular basis of immunity against Yersinia pestis. Hum Vaccin 5:817–823. doi: 10.4161/hv.9866. [DOI] [PubMed] [Google Scholar]
- 5.Baker EE, Sommer H, Foster LE, Meyer E, Meyer KF. 1952. Studies on immunization against plague. I. The isolation and characterization of the soluble antigen of Pasteurella pestis. J Immunol 68:131–145. [PubMed] [Google Scholar]
- 6.Burrows TW. 1956. An antigen determining virulence in Pasteurella pestis. Nature 177:426–427. [DOI] [PubMed] [Google Scholar]
- 7.Pujol C, Bliska JB. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun 71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinner JL, Winfree S, Starr T, Shannon JG, Nair V, Steele-Mortimer O, Hinnebusch BJ. 2014. Yersinia pestis survival and replication within human neutrophil phagosomes and uptake of infected neutrophils by macrophages. J Leukoc Biol 95:389–398. doi: 10.1189/jlb.1112551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shannon JG, Bosio CF, Hinnebusch BJ. 2015. Dermal neutrophil, macrophage and dendritic cell responses to Yersinia pestis transmitted by fleas. PLoS Pathog 11:e1004734. doi: 10.1371/journal.ppat.1004734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahalison L, Ranjalahy M, Duplantier JM, Duchemin JB, Ravelosaona J, Ratsifasoamanana L, Chanteau S. 2003. Susceptibility to plague of the rodents in Antananarivo, Madagascar. Adv Exp Med Biol 529:439–442. doi: 10.1007/0-306-48416-1_87. [DOI] [PubMed] [Google Scholar]
- 11.Congleton YH, Wulff CR, Kerschen EJ, Straley SC. 2006. Mice naturally resistant to Yersinia pestis Delta pgm strains commonly used in pathogenicity studies. Infect Immun 74:6501–6504. doi: 10.1128/IAI.00597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert ND, Langfitt DM, Nilles ML, Bradley DS. 2011. Resistance to Yersinia pestis infection decreases with age in B10.T(6R) mice. Infect Immun 79:4438–4436. doi: 10.1128/IAI.05267-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner JK, Xu JL, Tapping RI. 2009. Substrains of 129 mice are resistant to Yersinia pestis KIM5: implications for interleukin-10-deficient mice. Infect Immun 77:367–373. doi: 10.1128/IAI.01057-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner JK, McAllister MM, Xu JL, Tapping RI. 2008. The resistance of BALB/cJ mice to Yersinia pestis maps to the major histocompatibility complex of chromosome 17. Infect Immun 76:4092–4099. doi: 10.1128/IAI.00488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philipovskiy AV, Cowan C, Wulff-Strobel CR, Burnett SH, Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. 2005. Antibody against V antigen prevents Yop-dependent growth of Yersinia pestis. Infect Immun 73:1532–1542. doi: 10.1128/IAI.73.3.1532-1542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. 2004. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun 72:4589–4602. doi: 10.1128/IAI.72.8.4589-4602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malo D, Vogan K, Vidal S, Hu J, Cellier M, Schurr E, Fuks A, Bumstead N, Morgan K, Gros P. 1994. Haplotype mapping and sequence analysis of the mouse Nramp gene predict susceptibility to infection with intracellular parasites. Genomics 23:51–61. doi: 10.1006/geno.1994.1458. [DOI] [PubMed] [Google Scholar]
- 18.Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, Skamene E, Olivier M, Jothy S, Gros P. 1995. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J Exp Med 182:655–666. doi: 10.1084/jem.182.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidal SM, Malo D, Marquis JF, Gros P. 2008. Forward genetic dissection of immunity to infection in the mouse. Annu Rev Immunol 26:81–132. doi: 10.1146/annurev.immunol.26.021607.090304. [DOI] [PubMed] [Google Scholar]
- 20.Vidal SM, Malo D, Vogan K, Skamene E, Gros P. 1993. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73:469–485. doi: 10.1016/0092-8674(93)90135-D. [DOI] [PubMed] [Google Scholar]
- 21.Govoni G, Vidal S, Gauthier S, Skamene E, Malo D, Gros P. 1996. The Bcg/Ity/Lsh locus: genetic transfer of resistance to infections in C57BL/6J mice transgenic for the Nramp1 Gly169 allele. Infect Immun 64:2923–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 23.Lee-Lewis H, Anderson DM. 2010. Absence of inflammation and pneumonia during infection with nonpigmented Yersinia pestis reveals a new role for the pgm locus in pathogenesis. Infect Immun 78:220–230. doi: 10.1128/IAI.00559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fetherston JD, Kirillina O, Bobrov AG, Paulley JT, Perry RD. 2010. The yersiniabactin transport system is critical for the pathogenesis of bubonic and pneumonic plague. Infect Immun 78:2045–2052. doi: 10.1128/IAI.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sebbane F, Jarrett C, Gardner D, Long D, Hinnebusch BJ. 2010. Role of the Yersinia pestis yersiniabactin iron acquisition system in the incidence of flea-borne plague. PLoS One 5:e14379. doi: 10.1371/journal.pone.0014379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plant J, Glynn AA. 1976. Genetics of resistance to infection with Salmonella typhimurium in mice. J Infect Dis 133:72–78. doi: 10.1093/infdis/133.1.72. [DOI] [PubMed] [Google Scholar]
- 27.Bradley DJ, Taylor BA, Blackwell J, Evans EP, Freeman J. 1979. Regulation of Leishmania populations within the host. III. Mapping of the locus controlling susceptibility to visceral leishmaniasis in the mouse. Clin Exp Immunol 37:7–14. [PMC free article] [PubMed] [Google Scholar]
- 28.Gros P, Skamene E, Forget A. 1981. Genetic control of natural resistance to Mycobacterium bovis (BCG) in mice. J Immunol 127:2417–2421. [PubMed] [Google Scholar]
- 29.Fortier A, Min-Oo G, Forbes J, Lam-Yuk-Tseung S, Gros P. 2005. Single gene effects in mouse models of host: pathogen interactions. J Leukoc Biol 77:868–877. doi: 10.1189/jlb.1004616. [DOI] [PubMed] [Google Scholar]
- 30.Shi L, Adkins JN, Coleman JR, Schepmoes AA, Dohnkova A, Mottaz HM, Norbeck AD, Purvine SO, Manes NP, Smallwood HS, Wang H, Forbes J, Gros P, Uzzau S, Rodland KD, Heffron F, Smith RD, Squier TC. 2006. Proteomic analysis of Salmonella enterica serovar typhimurium isolated from RAW 264.7 macrophages: identification of a novel protein that contributes to the replication of serovar typhimurium inside macrophages. J Biol Chem 281:29131–29140. doi: 10.1074/jbc.M604640200. [DOI] [PubMed] [Google Scholar]
- 31.Pujol C, Grabenstein JP, Perry RD, Bliska JB. 2005. Replication of Yersinia pestis in interferon gamma-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc Natl Acad Sci U S A 102:12909–12914. doi: 10.1073/pnas.0502849102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blanchet C, Jaubert J, Carniel E, Fayolle C, Milon G, Szatanik M, Panthier JJ, Montagutelli X. 2011. Mus spretus SEG/Pas mice resist virulent Yersinia pestis, under multigenic control. Genes Immun 12:23–30. doi: 10.1038/gene.2010.45. [DOI] [PubMed] [Google Scholar]
- 33.Chevallier L, Blanchet C, Jaubert J, Pachulec E, Demeure C, Carniel E, Panthier JJ, Montagutelli X. 2013. Resistance to plague of Mus spretus SEG/Pas mice requires the combined action of at least four genetic factors. Genes Immun 14:35–41. doi: 10.1038/gene.2012.50. [DOI] [PubMed] [Google Scholar]
- 34.Demeure CE, Blanchet C, Fitting C, Fayolle C, Khun H, Szatanik M, Milon G, Panthier JJ, Jaubert J, Montagutelli X, Huerre M, Cavaillon JM, Carniel E. 2012. Early systemic bacterial dissemination and a rapid innate immune response characterize genetic resistance to plague of SEG mice. J Infect Dis 205:134–143. doi: 10.1093/infdis/jir696. [DOI] [PubMed] [Google Scholar]
- 35.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. 2006. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest 116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pennisi E. 2011. The biology of genomes. Disease risk links to gene regulation. Science 332:1031. doi: 10.1126/science.332.6033.1031. [DOI] [PubMed] [Google Scholar]
- 37.Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zhernakova A, Reinmaa E, Vosa U, Hofker MH, Fehrmann RS, Fu J, Withoff S, Metspalu A, Franke L, Wijmenga C. 2013. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet 9:e1003201. doi: 10.1371/journal.pgen.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]