

Abstract
The Gram-positive bacterium Listeria monocytogenes transitions from an environmental organism to an intracellular pathogen following its ingestion by susceptible mammalian hosts. Bacterial replication within the cytosol of infected cells requires activation of the central virulence regulator PrfA followed by a PrfA-dependent induction of secreted virulence factors. The PrfA-induced secreted chaperone PrsA2 and the chaperone/protease HtrA contribute to the folding and stability of select proteins translocated across the bacterial membrane. L. monocytogenes strains that lack both prsA2 and htrA exhibit near-normal patterns of growth in broth culture but are severely attenuated in vivo. We hypothesized that, in the absence of PrsA2 and HtrA, the increase in PrfA-dependent protein secretion that occurs following bacterial entry into the cytosol results in misfolded proteins accumulating at the bacterial membrane with a subsequent reduction in intracellular bacterial viability. Consistent with this hypothesis, the introduction of a constitutively activated allele of prfA (prfA*) into ΔprsA2 ΔhtrA strains was found to essentially inhibit bacterial growth at 37°C in broth culture. ΔprsA2 ΔhtrA strains were additionally found to be defective for cell invasion and vacuole escape in selected cell types, steps that precede full PrfA activation. These data establish the essential requirement for PrsA2 and HtrA in maintaining bacterial growth under conditions of PrfA activation. In addition, chaperone function is required for efficient bacterial invasion and rapid vacuole lysis within select host cell types, indicating roles for PrsA2/HtrA prior to cytosolic PrfA activation and the subsequent induction of virulence factor secretion.
INTRODUCTION
The Gram-positive bacterium Listeria monocytogenes habitually exists in soil and decomposing plant matter (1–3) but can cause severe invasive disease in animals and humans following the ingestion of contaminated food (4, 5). The successful transition of L. monocytogenes from the outside environment to life within the mammalian host is dependent upon the activation of PrfA, a transcriptional activator which regulates the majority of the gene products associated with bacterial virulence (6–9). Full activation of PrfA occurs following entry of L. monocytogenes into the cytosol of infected-host cells, with PrfA-dependent gene products facilitating the major steps of L. monocytogenes pathogenesis that include intracellular replication, actin-based bacterial motility, and spread to adjacent cells (10, 11). Following cell entry, the escape of L. monocytogenes from host cell vacuoles is mediated by three PrfA-dependent gene products: the cholesterol-dependent pore-forming cytolysin listeriolysin O (LLO) and two phospholipases (PlcA and PlcB) (12–15). Within the cytosol, L. monocytogenes recruits and polymerizes host cell actin through the expression of a PrfA-dependent bacterial surface protein known as ActA, enabling bacterial movement into adjacent cells (16, 17). Activation of PrfA within the cytosol has been suggested to occur as a result of PrfA binding to glutathione (18); in addition, mutant forms of PrfA have been identified that are constitutively active in the absence of any environmental signal (known as prfA* mutations) (19–24). A number of prfA* mutations have been reported, and some of them (such as the L-to-F change at position 140 encoded by prfA [prfA L140F] and prfA G145S) appear to phenocopy the levels of PrfA activation achieved within the cell cytosol in vivo (18, 24, 25).
Overall, PrfA activation leads to a dramatic increase in the synthesis of numerous secreted proteins that are required for L. monocytogenes pathogenesis (25). These proteins are thought to be translocated in an unfolded state across the cell membrane (26–28), with protein folding occurring within the highly charged and solvent-accessible environment located between the cytoplasmic membrane and the peptidoglycan cell wall (29). Two proteins whose secretion is upregulated following PrfA activation, PrsA2 and HtrA, have been identified as residing in this environment and assist with the folding and stability of secreted proteins as they are translocated across the bacterial membrane (25, 30, 31). PrsA2 is a secreted cis-trans prolyl isomerase and foldase and has been shown to be necessary for the full activity of at least two virulence factors which facilitate phagosome escape during host infection: LLO (encoded by hly) and the broad-spectrum phospholipase PlcB (31, 32). HtrA, also known as DegP in Escherichia coli, is a temperature-regulated serine protease that can also act as a chaperone (33). For E. coli HtrA, the protease function is active at elevated temperatures (37°C), while a conformational change at lower temperature prevents accessibility of the serine in the active site; thus, the chaperone function of HtrA is dominant at low temperatures (33, 34). HtrA is linked to the heat shock response (52°C) and survival at elevated temperatures (44°C) in E. coli, although its expression is not increased upon heat shock in L. monocytogenes (35, 36). HtrA is required for bacterial fitness under conditions such as osmotic, acid, and oxidative stress and for survival following exposure to antibiotic (puromycin, penicillin) (35–37). PrsA2 also contributes to resistance to penicillin and other antibiotics as well as osmotic and pH stress (38, 39), presumably reflecting a role for PrsA2 in cell wall synthesis and/or homeostasis (40), whereas the principal role of HtrA seems to be degradation of proteins under stressful conditions (36). The target proteins for the chaperone function of HtrA in Listeria are unknown; however, activity of LLO appears to be independent of HtrA (37).
Despite the fundamental importance of protein secretion for bacterial virulence, much remains unknown regarding the mechanics of protein folding and the regulation of protein activity at the Gram-positive membrane–cell wall interface. PrsA2 and HtrA have both been shown to contribute to L. monocytogenes pathogenesis (30, 32); however, the identities of their substrates and the mechanisms by which these chaperones contribute to protein folding, activity, and/or degradation remain to be determined. L. monocytogenes mutants containing single deletions of either htrA or prsA2 are significantly compromised for virulence in mice, while the ΔprsA2 ΔhtrA double deletion mutant appeared essentially avirulent with no detectable bacterial colonies recovered from target organs (30). While growth of the ΔprsA2 ΔhtrA deletion mutant was similar to that of wild-type strains in rich broth culture, growth of the mutant in J774 macrophage-like cells was severely restricted (30). These results suggest that HtrA and PrsA2 are dispensable for growth under laboratory conditions but are essential for bacterial life within host cells. Given that PrfA activation within the cytosol of infected-host cells results in dramatic increases in bacterial protein secretion, we hypothesized that the functions of PrsA2 and HtrA are required for secreted protein folding and activity as well as full bacterial viability under conditions of PrfA activation within the cytosol. We therefore took advantage of the constitutively activated PrfA phenotype of prfA* strains to assess the requirement for PrsA2 and HtrA activity in the context of PrfA activation.
MATERIALS AND METHODS
Strains, media, and culture conditions.
All bacterial strains used in this study are listed in Table 1. L. monocytogenes strain 10403S (NF-L100) was used as a wild-type control and parent strain for mutants. All strains were grown in brain heart infusion (BHI) broth (Difco Laboratories, Detroit, MI) or Luria-Bertani (LB) broth (Invitrogen Corp., Carlsbad, CA) with agitation at 37°C unless specifically stated otherwise. The prfA* (prfA L140F) allele was introduced into L. monocytogenes strains by bacteriophage transduction as previously described (24, 41, 42). Briefly, 107 to 108 PFU of Listeria phage U153 lysates (41) prepared from strain NF-L1166 (prfA L140F actA-gus-neo-plcB) (43) were mixed with 108 CFU of mid-log strain NF-L100 (wild type [WT]), NF-L1605 (ΔhtrA), NF-L1651 (ΔprsA2::erm), or NF-L1634 (ΔhtrA ΔprsA2::erm). The transductants were identified by selection for neomycin resistance (10 μg/ml) and a blue colony appearance on BHI agar containing 50 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (X-Gluc). The antibiotic neomycin was added to growth media for all experiments with prfA* mutants at a final concentration of 10 μg/ml.
TABLE 1.
Bacterial strains used in this study
| Strain | Description or relevant genotype or phenotype | Reference or source |
|---|---|---|
| NF-L100 | Wild-type 10403S | 60 |
| NF-L340 | Δhly (DP-L2161) | 61 |
| NF-L1124 | NF-L100 actA-gus-neo-plcB | 43 |
| NF-L1166 | NF-L100 actA-gus-neo-plcB prfA* L140F | 43 |
| NF-L1605 | ΔhtrA | 37 |
| NF-L1651 | ΔprsA2::erm | 32 |
| NF-L1634 | ΔhtrA ΔprsA2::erm | 30 |
| NF-L3611 | NF-L100 prfA* | This work |
| NF-L3612 | NF-L1605 prfA* ΔhtrA | This work |
| NF-L3613 | NF-L1651 prfA* ΔprsA2::erm | This work |
| NF-L3614 | NF-L1634 prfA* ΔhtrA ΔprsA2::erm | This work |
Bacterial growth curves, CFU, colony size determination, and viability assessment.
Bacterial growth was measured in BHI broth beginning with a 1:20 dilution of an overnight culture into fresh BHI broth. The absorbance at an optical density at 600 nm (OD600) was measured in a spectrophotometer to determine growth each hour. Counting of CFU was done by 10-fold dilutions in H2O followed by plating onto LB agar and overnight incubation at 37°C for enumeration. Colony sizes were determined on BHI agar after 24 h of growth at 37°C. Bacterial cell viability was assessed by using the Live/Dead BacLight bacterial viability kit according to the manufacturer's instructions (Invitrogen Corp.). Strains were grown overnight at 30°C, diluted 1:20 into fresh BHI medium, and grown for 6 h at 37°C. Cell cultures (1 ml) were washed twice in phosphate-buffered saline (PBS) before staining. Images were taken using a Zeiss Axio Imager A2 microscope.
Measurement of β-glucuronidase activity.
β-Glucuronidase (GUS) activity was measured as previously described (22) with minor modifications. Briefly, L. monocytogenes cultures grown overnight at 30°C or 37°C in BHI broth were diluted 1:50 and grown with shaking at 30°C or 37°C for 24 h. The OD600 was measured for each time point (3 h, 5 h, 7 h, and 24 h), and two 500-μl culture aliquots were collected. Bacterial cells were recovered by microcentrifugation at 8,000 × g for 3 min, and the supernatants were removed. Bacterial pellets were resuspended in 100 μl (aliquot 1) or 1 ml (aliquot 2) of ABT buffer (0.1 M potassium phosphate [pH 7.0], 0.1 M NaCl, 0.1% Triton X-100). GUS activity was measured as described but with 4-methylumbelliferyl-β-d-glucuronide instead of 4-methylumbilliferyl-β-d-galactoside (Sigma) (44).
Protein extraction, SDS-PAGE, and Western blot analysis.
Secreted proteins were isolated from culture supernatants, and surface-associated fractions were isolated from whole bacterial cells as previously described with minor modifications (25, 38). In brief, 20-ml cultures of each L. monocytogenes strain were grown to mid-log phase (3 to 4 h for Western blot analysis) or late log phase (6.5 h) and stationary phase (24 h) (for Coomassie blue-stained sodium dodecyl sulfate [SDS]-polyacrylamide gels) in BHI broth at 30°C or 37°C with shaking and normalized by adjusting cultures to equivalent OD600 values before protein fractionation. Proteins present in the culture supernatants were precipitated with 10% trichloroacetic acid (TCA) (Fisher Scientific), and the pellets were washed with ice-cold acetone and resuspended in 200 μl of 2× SDS boiling buffer (Bio-Rad). Surface-associated proteins were extracted by boiling of the bacterial pellet in 200 μl of 2× SDS boiling buffer (Bio-Rad). To extract proteins from the cytoplasm, bacterial cells were recovered by centrifugation and resuspended in 1 ml of PBS or 1 ml of buffer (5% SDS, 10% glycerol, 50 mM Tris [pH 6.8]) (24-h samples). Twenty milligrams of lysozyme (Sigma-Aldrich, St. Louis, MO) was added, and samples were incubated at 37°C for 30 min. Prior to sonication with five repeated 1-min bursts and 1-min cooling on ice, 10 μl of 100× protease inhibitor cocktail set III (Calbiochem, Millipore) was added. After sonication, 50 μl of β-mercaptoethanol was added to the 6.5-h samples as well as a toothpick tip full of bromphenol blue (Sigma-Aldrich). One hundred microliters of 2× SDS boiling buffer (Bio-Rad) was added to 100 μl of the 24-h samples. All samples were boiled for 5 min before being subjected to SDS-polyacrylamide gel electrophoresis (PAGE) (45). For detection of PrsA2 and HtrA, 15 μl of the isolated membrane-associated proteins were separated using SDS-PAGE. Protein samples were transferred onto polyvinylidene difluoride (PVDF) membranes. HtrA and PrsA2 were detected using a 1:200 dilution of affinity-purified polyclonal antibodies directed against purified PrsA2- or HtrA-derived peptides in 1× PBST (phosphate-buffered saline solution plus 0.05% Tween 20), followed by incubation with a 1:2,500 dilution of a polyclonal goat anti-rabbit secondary antibody conjugated to alkaline phosphatase (SouthernBiotech, Birmingham, AL). Bands were visualized colorimetrically with the addition of 10 ml of a BCIP/NBT Plus solution (SouthernBiotech, Birmingham, AL) (BCIP stands for 5-bromo-4-chloro-3-indolylphosphate, and NBT stands for nitroblue tetrazolium). ImageJ software was used to determined densitometry (http://imagej.nih.gov/ij/).
Bacterial intracellular growth assays.
Bacterial intracellular growth assays in mouse macrophage-like cells (J774), Potoroo tridactylis kidney epithelial cells (PtK2), human colon epithelium cells (Caco2), human kidney epithelium cells (Henle), and human colon epithelium cells (HTB-38) were performed as previously described (24, 32). In brief, monolayers of mammalian cells were grown on glass coverslips to confluence and infected with bacterial strains with a multiplicity of infection (MOI) of 100:1 except for J774 for which an MOI of 0.1:1 was used. The bacterial cultures were grown without shaking at 37°C overnight, except the ΔhtrA ΔprsA2 prfA* mutant, which was grown at 30°C due to the severe growth defect at 37°C. One hour postinfection (p.i.), monolayers were washed three times in Dulbecco's phosphate-buffered saline (DPBS) (Cellgro Mediatech Inc.), and fresh medium was added, followed by 5 μg/ml of gentamicin to kill extracellular bacteria. At the time points indicated in the figures, coverslips were removed and lysed in 2 ml of sterile H2O to release intracellular bacteria for enumeration of CFU.
Measurement of bacterial association with host cell actin.
For assessment of bacterial association with host cell actin as monitored by fluorescence microscopy, coverslips were prepared as previously described (46) with minor modifications. The cells were fixed by covering the coverslip with 3.2% formaldehyde in PBS followed by 0.1% Triton X-100 treatment. Filamentous host cell actin was stained with Alexa Fluor 488 phalloidin (Invitrogen Corp.). Bacterial cells were stained with Listeria-specific polyclonal antibody (BD Biosciences), followed by a secondary goat anti-rabbit antibody conjugated to rhodamine. DNA-containing nuclei were visualized with 4′,6′-diamidino-2-phenylindole (DAPI) (ProLong Gold antifade reagent; Life Technologies). Images were taken using a Zeiss Axio Imager A2 microscope.
Transfection of PtK2 cells and detection of vacuole perforation.
A mammalian expression vector containing a fusion of the yellow fluorescent protein (YFP) to a cell wall binding domain (CBD) of the phage endolysin Ply118 which binds specifically to the L. monocytogenes cell wall was transfected into PtK2 cells as previously described (47). An MOI of 25:1 was used for infection of PtK2 cells with the wild-type strain, while an MOI of 200:1 was used for the ΔhtrA ΔprsA2::erm and Δhly mutants to increase the chances of detecting rare events of vacuole perforation. Fluorescence staining was conducted as described above except that host cell actin was stained with Alexa Fluor 350 phalloidin (Invitrogen Corp.).
Measurement of LLO-associated hemolysis and PlcB-associated phospholipase activity.
Hemolytic activity was measured as previously described with minor changes (48). Briefly, stationary-phase bacterial cultures were diluted 1:10 into LB medium and grown at 37°C for 5 h with shaking. The OD600 was determined, and 1 ml of each culture was normalized to equivalent OD600 values and centrifuged at 8,000 × g for 5 min. Twofold serial dilutions of the supernatants were incubated with PBS-washed sheep erythrocytes (Cocalico Biologicals Inc., Reamstown, PA) for 30 min at 37°C. After incubation, erythrocytes were recovered by centrifugation to measure 50% lysis by visual inspection.
plcB-dependent phospholipase production was visualized using Brilliance Listeria selective agar plates containing lecithin (Oxoid). Agar was poured into petri dishes, and after solidification of the medium, 5 μl of bacterial overnight cultures (30°C) were spotted onto the surface of the plate and incubated at 30°C for 48 h, and the zones of enzymatic phospholipase activity were measured.
Statistical analyses.
The nonpaired Student t test was used for statistical analyses.
RESULTS
PrfA activation in the absence of the PrsA2 and HtrA secretion chaperones compromises bacterial growth.
High-level activation of PrfA normally occurs following entry of L. monocytogenes into the host cell cytosol, a situation which complicates efforts to directly assess the impact of PrfA activation on bacterial physiology. As an alternative method for assessing the effects of PrfA activation on bacteria lacking PrsA2 and/or HtrA, we took advantage of prfA* mutations that result in the constitutive activation of PrfA to levels that are comparable to those occurring within infected cells (18, 19, 22–25). L. monocytogenes NF-L1166 contains the constitutively activated prfA L140F allele in place of wild-type prfA in the bacterial chromosome together with a closely linked actA transcriptional fusion to both a neomycin resistance gene (neo) and gus encoding β-glucuronidase (GUS), thereby placing the expression of neo and gus under the regulation of the PrfA-dependent actA promoter (43). prfA* actA-neo-gus was introduced into ΔhtrA, ΔprsA2::erm, and ΔhtrA ΔprsA2::erm strains by phage U153 transduction and selection for neomycin-resistant blue transductants on plates containing 10 μg/ml neomycin and 50 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (X-Gluc).
While colonies of the ΔhtrA ΔprsA2::erm double deletion mutant exhibited a noticeable growth defect on BHI plates when grown at 37°C, ΔhtrA ΔprsA2::erm prfA* transductants were profoundly impaired for growth at 37°C and formed tiny neomycin-resistant blue colonies (Fig. 1A). Examination of bacterial growth in broth culture indicated that mutant strains containing wild-type prfA or prfA* exhibited similar growth characteristics at 30°C; however, growth of ΔhtrA ΔprsA2::erm prfA* strains was severely impaired at 37°C (Fig. 1B). Growth of the single chaperone deletion mutants was similar in the presence of wild-type prfA or prfA* at 30°C and 37°C (Fig. 1B). The prfA*-associated growth defect at 37°C was not immediately apparent, as cultures grown first at 30°C for 3 h and then shifted to 37°C did not immediately exhibit reduced growth (Fig. 1C, green line), but when inoculated and allowed to grow overnight at 37°C, the defect became readily apparent (Fig. 1D, red line). ΔhtrA ΔprsA2::erm prfA* cultures grown overnight at 30°C exhibited growth defects following 4 to 5 h of growth at 37°C (Fig. 1D, blue line), and when these cultures were diluted into fresh medium and incubations continued at 37°C, bacterial growth appeared to cease (Fig. 1D, black line). Incubation of ΔhtrA ΔprsA2::erm prfA* cultures overnight at 37°C followed by dilution into fresh medium and growth at 30°C resulted in an extended lag phase before growth was apparent (Fig. 1C, red line). The reductions in optical density observed for ΔhtrA ΔprsA2::erm prfA* cultures incubated at 37°C were consistent with decreased numbers of viable cells as indicated by plating for bacterial CFU (Fig. 1E) and by monitoring bacterial membrane integrity (Fig. 1F and G). Taken together, these results indicate that the introduction of constitutively activated PrfA confers a growth defect to strains lacking the PrsA2 and HtrA chaperones under conditions of rapid cell growth (37°C). The delay observed before the growth defect becomes apparent following the shift to 37°C suggests that, under conditions of rapid growth, the ΔhtrA ΔprsA2::erm prfA* mutant accumulates a toxic substance(s) and/or gradually experiences a cellular modification that impairs bacterial growth. The extended lag phase that occurs before bacterial growth resumes following the shift of the 37°C culture back to 30°C is consistent with the observation that most cells within the 37°C population are not viable (Fig. 1E to G); thus, the outgrowth of a minority of viable cells must occur before an appreciable increase in optical density can be detected.
FIG 1.

L. monocytogenes ΔhtrA ΔprsA2::erm prfA* mutants exhibit pronounced growth defects at 37°C. (A) Colony sizes determined on BHI agar after 24 h of growth at 37°C. At least five colonies were measured from two independent experiments. Values that were statistically significantly different (P < 0.0001) by the t test are indicated by a bar and four asterisks. WT, wild type. (B) Bacterial growth as determined by optical density measurement at 600 nm at 30°C and 37°C at the indicated time points. (C and D) Bacterial growth of ΔhtrA ΔprsA2::erm prfA* at 30°C (C) and 37°C (D). (C) Growth at 30°C from an overnight inoculum culture grown at 37°C or 30°C and a temperature shift from 30°C to 37°C at 3 h. (D) Continuous growth at 37°C from an overnight inoculum culture grown at 37°C or 30°C are shown. The latter culture (blue line) was diluted 1:20 and grown again for 8 h at 37°C (black line). (E) Overnight cultures of the wild-type prfA* strain and the ΔhtrA ΔprsA2::erm prfA* mutant were grown at 30°C or 37°C, diluted 1:20, and grown at 37°C for 6 h. Every hour, a sample was taken, serially diluted, and plated for enumeration of CFU per milliliter. Data are representative of the data from two independent experiments. (F) Live/Dead staining of wild-type prfA* and ΔhtrA ΔprsA2::erm prfA* mutants. Overnight cultures were grown at 30°C, diluted 1:20 into fresh medium, and grown for 6 h at 37°C. Micrographs are representative of at least three independent experiments. (G) Enumeration of bacteria from Live/Dead staining. Data summarize results from at least three independent experiments. For each experiment, at least 30 bacteria were counted from 5 to 10 independent fields.
Gross examination of proteins associated with the surface of bacterial cells by SDS-PAGE indicated that the ΔhtrA ΔprsA2::erm prfA* mutant exhibited changes in the abundance and diversity of protein species associated with the cell surface in comparison to the wild-type strain following growth at 37°C (Fig. 2A). Noticeable were both the absence of some prominent species as well as an increase in protein species spanning a variety of molecular masses (Fig. 2A). These changes could reflect protein released and associated with the bacterial membrane as a result of cell lysis and/or an accumulation of proteins reflecting folding and secretion defects due to the absence of the secretion chaperones.
FIG 2.

Changes in surface-associated proteins in the absence of the secretion chaperones and assessment of activation of PrfA at 37°C and 30°C. (A) Proteins isolated from lysed bacterial cells (sonicated), surface-associated proteins, and secreted proteins from supernatants. All cultures were normalized to an equal volume of a solution with an OD600 of 0.5 before treatment. The cultures were then subjected to fractionation, separation of proteins by SDS-PAGE, and staining with Coomassie blue. Overnight cultures were grown at 30°C, diluted 1:20 into fresh medium, and grown for 6.5 h at 37°C; the growth defect of the ΔhtrA ΔprsA2::erm prfA* mutant at 37°C prevented the use of this temperature for the growth of overnight cultures for these assays. Images shown are representative of at least three independent experiments. WT, wild type; Mut, ΔhtrA ΔprsA2::erm prfA* mutant. (B) PrfA activity was assessed based on the expression of a transcriptional fusion reporter gene (gus, encoding β-glucuronidase [GUS]) located between actA and plcB. Both actA and plcB are directly dependent on PrfA for expression, and thus, GUS activity serves as a readout of PrfA activity. The assay was performed with the wild-type (WT) and prfA L140F (WT prfA*) strains grown at 30°C and 37°C at the time points indicated. Two independent experiments were performed, and means are shown with error bars representing the standard deviations.
The 37°C growth defect associated with prfA* strains lacking PrsA2 and HtrA does not reflect temperature-dependent changes in PrfA* activity.
It has been previously demonstrated that the expression of prfA is subject to temperature-dependent regulation based on the formation of secondary structure that inhibits translation in the prfA mRNA directed by the prfA P1 promoter at 30°C (49, 50). Transcripts directed by the prfA P2 promoter do not contain the mRNA thermosensor, and it is not clear whether transcripts initiating from the upstream plcA promoter exhibit temperature-dependent translation (50). To determine whether temperature-dependent regulation of PrfA* synthesis and activity contributed to the growth defect of ΔhtrA ΔprsA2::erm prfA* strains observed at 37°C but not at 30°C, activity of PrfA* was assessed by measuring PrfA*-dependent GUS activity associated with the actA-neo-gus transcriptional reporter gene fusion (43). Strains containing wild-type prfA exhibited a modest induction of GUS activity during bacterial growth at 37°C in comparison to growth at 30°C; however, no difference in GUS activity was observed for prfA* cultures grown at either 30°C or 37°C (Fig. 2B), indicating that temperature-dependent posttranscriptional control of prfA* does not play a major role in regulating PrfA* activity. Temperature-dependent differences in PrfA* activity are therefore not responsible for the growth defects of the ΔhtrA ΔprsA2::erm prfA* mutants grown at 37°C versus 30°C. Instead, it would appear that PrfA*-dependent high-level expression of secreted proteins under conditions of rapid bacterial growth act together to confer the growth defect observed for strains lacking the PrsA2/HtrA chaperones.
Loss of PrsA2 increases HtrA secretion during growth in broth culture and in the presence of PrfA activation.
L. monocytogenes mutants that lack both PrsA2 and HtrA secretion chaperones exhibit dramatic reductions in bacterial intracellular growth within tissue culture cells and a greater than 107-fold reduction in bacterial burdens in target organs in mouse models of infection (30); these defects are more extensive than those observed for bacterial mutants that lack only one of the two chaperones (compared to the ∼104-fold target organ burden reduction for the ΔprsA2 mutant and 103-fold burden reduction for the ΔhtrA mutant). Similarly, introduction of the constitutively activated prfA* allele into single chaperone deletion mutants had much less of a negative impact on bacterial growth than the introduction of prfA* into the double mutant (Fig. 1). These results suggest that each secreted chaperone may exhibit some degree of functional overlap with respect to protein folding and secretion under conditions of PrfA activation. HtrA protein levels have been shown to increase in response to a variety of stress conditions (35); thus, we speculated that the loss of either HtrA or PrsA2 might induce a stress that would increase the expression of the other chaperone. Western blot analysis using antibodies directed against HtrA and PrsA2 in the presence and absence of prfA* indicated no significant increase in the levels of PrsA2 in the absence of functional HtrA (Fig. 3). In contrast, the levels of HtrA protein increased severalfold in strains lacking functional PrsA2 (Fig. 3). These results indicate that L. monocytogenes responds to the lack of PrsA2 by increasing levels of secreted HtrA; however, the loss of functional HtrA does not result in a complementary increase in PrsA2 secretion, suggesting distinct mechanisms of chaperone regulation. Alternatively, it is possible that the loss of HtrA induces less of a stress response than the loss of PrsA2.
FIG 3.
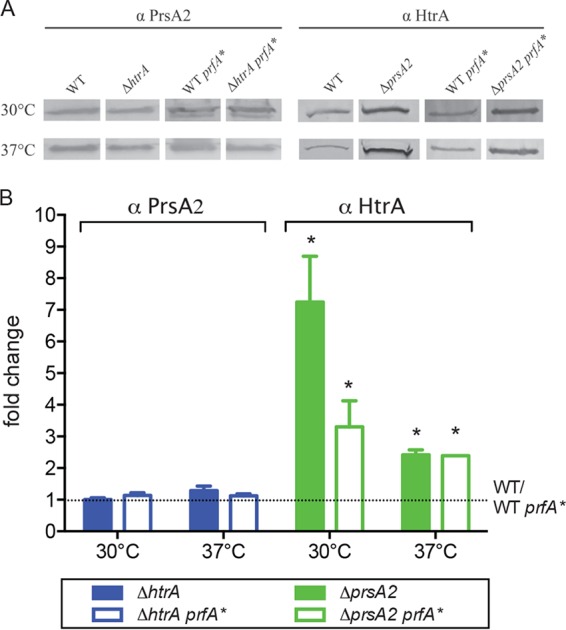
HtrA abundance increases in strains lacking PrsA2. (A) Western blot analysis of surface-associated HtrA and PrsA2 proteins from ΔprsA2 and ΔhtrA mutants, respectively. Proteins were isolated from mid-log-phase cultures grown for 3 to 4 h with shaking at 30°C or 37°C. Bacterial cultures were normalized to equal volumes of a solution with an OD600 of 0.5 before protein extraction. The HtrA and PrsA2 proteins were detected with antibodies against PrsA2 (α PrsA2) or HtrA (α HtrA). Images shown in panel A are representative of at least three independent experiments. The density of protein bands was measured using ImageJ software. (B) Fold change of HtrA and PrsA2 expression in mutants compared to the wild type (WT) or prfA* (WT prfA*). The values for the mutants that were significantly different (P < 0.05) from the values for the wild type are indicated by an asterisk.
ΔhtrA ΔprsA::erm strains exhibit cell type-dependent defects in vacuole escape.
Previous experiments using mouse J774 macrophage-like cell lines indicated that the double deletion chaperone mutant ΔhtrA ΔprsA2::erm strain was severely impaired for intracellular replication (30). It was unclear from these experiments whether bacteria were delayed and/or defective for escape from host cell vacuoles, or alternatively if the growth defect occurred as a result of PrfA activation following bacterial entry into the cytosol. ΔhtrA ΔprsA2::erm prfA* strains were defective for growth in broth culture at 37°C (Fig. 1); however, this growth defect became evident only after several hours of growth (>5 h) after the cultures were shifted from 30°C to 37°C. Interestingly, ΔhtrA ΔprsA2::erm prfA* cultures grown overnight at 30°C and then used to infect J774 cells exhibited an immediate intracellular growth defect, consistent with either a vacuole escape defect or defective replication within the cytosol (Fig. 4A). The ΔhtrA ΔprsA2::erm double mutant was found to be similarly compromised for growth within a variety of different cell types, including Potoroo tridactylis rat kidney epithelial cells (PtK2) and human kidney epithelial cells (Henle) (Fig. 4B and D). The significantly compromised intracellular growth defect of the ΔhtrA ΔprsA2::erm double mutant with prfA* was also evident when the wild type and single mutants with prfA* were grown at 30°C before infection, the temperature required for successful propagation of ΔhtrA ΔprsA2::erm prfA* strains in broth culture (see Fig. S1 in the supplemental material). Interestingly, comparable numbers of the ΔhtrA ΔprsA2::erm mutant with or without prfA* were observed in comparison to the wild type and single mutants in human colonic epithelial cells (Caco2 cells) with a very modest defect in invasion and intracellular replication evident for the ΔhtrA ΔprsA2::erm mutant with prfA* (Fig. 4C).
FIG 4.

L. monocytogenes strains lacking htrA and prsA2 are deficient for cell invasion and bacterial replication in assorted tissue culture cell lines. (A to D) Intracellular growth of the wild type (WT) and the indicated mutants was assessed in J774 (A), PtK2 (B), Caco2 (C), and Henle (D) cell lines. The cell lines were grown as monolayers on glass coverslips and infected with an MOI of 0.1:1 for J774 cells and an MOI 100:1 for all other cells. Gentamicin was added 1 h postinfection to kill extracellular bacteria. Three coverslips were removed at each of the indicated time points, host cells were lysed, and the CFU of intracellular bacteria were enumerated. Data are representative of at least three independent experiments. The values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Entry of L. monocytogenes into the cytosol can be detected based on the ability of the bacterium to associate with host cell actin and stimulate actin polymerization (51). Examination of infected cells using fluorescence-based microscopy at 5 h postinfection indicated that the ΔhtrA ΔprsA2::erm double mutant failed to form any detectable actin tails in J774, PtK2, or Henle cells (Fig. 5A, B, and D), even after extended periods of incubation in PtK2 cells of up to 24 h (Fig. 6A and B). In contrast, mutants lacking both PrsA2 and HtrA chaperones were fully capable of cell invasion and vacuole escape as indicated by actin association in human Caco2 cells (Fig. 5C), although actin association and assembly were delayed, as indicated by the reduction in actin tails, but an increased number of bacteria associated with actin clouds, the precursor of actin-based motility (Fig. 6C). The ability of the ΔhtrA ΔprsA2::erm mutant to invade host cells, escape from host cell vacuoles, and polymerize host cell actin in Caco2 cells did not appear to reflect properties of colonic cell lines in general, as the mutant exhibited reduced invasion of another human colon cell line (HTB-38) (Fig. 6D) and failed to polymerize actin (Fig. 6E).
FIG 5.

L. monocytogenes mutants lacking HtrA and PrsA2 are defective for recruitment of host cell actin in multiple cell lines. (A to D) Intracellular growth of the wild type (WT) and ΔhtrA ΔprsA2::erm mutant in J774 (A), PtK2 (B), Caco2 (C) and Henle (D) cell lines was visualized by fluorescence microscopy at 5 h postinfection. Pictures shown are representative of at least two experiments. In the pictures, Listeria is shown in red, host cell actin is shown in green, and DNA is shown in blue.
FIG 6.

L. monocytogenes ΔhtrA ΔprsA2::erm mutants exhibit delayed actin assembly in a cell type-dependent manner even following prolonged incubation. (A) Fluorescence-based microscopy of PtK2 cell monolayers after 24-h infection with the wild type and the ΔhtrA ΔprsA2::erm mutant. In the micrographs, Listeria is shown in red, host cell filamentous actin is shown in green, and DNA is shown in blue. Micrographs shown are representative of images obtained from at least two experiments. (B) Enumeration of bacteria inside PtK2 host cells after 24 h of infection. Bacteria were counted from three coverslips per experiment from at least five independent fields, and experiments were repeated four times. The values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.005. (C) Quantification of host cell actin tails and clouds associated with L. monocytogenes wild-type and ΔhtrA ΔprsA2::erm mutant in Caco2 and PtK2 cell lines at 5 h postinfection. Between 50 and 500 bacteria were enumerated in 10 to 30 independent fields. Experiments were repeated twice. *, P < 0.05; **, P < 0.005. (D) Intracellular growth of the wild type and ΔhtrA ΔprsA2::erm mutant was assessed in HTB-38 cells, a human colon cell line. Host cells were grown to monolayers on glass coverslips and infected with an MOI of 100:1. Gentamicin was added 1 h postinfection to kill extracellular bacteria. Three coverslips were removed at each of the indicated time points, host cells were lysed, and the numbers of intracellular bacteria were enumerated. The experiment was repeated three times. For comparison reasons, intracellular growth in Caco2 cells is shown in gray (Caco2 data is the same data as in Fig. 4C). (E) Fluorescence-based microscopy of HTB-38 cell monolayers after 5 h of infection with the wild type or the ΔhtrA ΔprsA2::erm mutant. In the micrographs, Listeria is shown in red, host cell filamentous actin is shown in green, and DNA is shown in blue. Pictures shown are representative of at least two independent experiments.
The failure of the ΔhtrA ΔprsA2::erm mutant to polymerize actin in most cell types examined could reflect either a defect in actin assembly, a defect in vacuole escape, or defects in both activities. PrsA2 function has been associated with stability and activity of the pore-forming hemolysin LLO as well as with efficient processing of the PlcB phospholipase, of which both contribute to vacuole escape (12–15). Significant levels of secreted LLO were detected in the supernatant fractions of ΔhtrA ΔprsA2::erm mutants with and without prfA* (Fig. 7E). PlcB phospholipase activity was greatly reduced on Brilliance Listeria agar plates for the ΔhtrA ΔprsA2::erm mutant but only modestly affected for the ΔhtrA ΔprsA2::erm mutant with prfA* (Fig. 7F). As both single prsA2 and htrA mutants are capable of efficient vacuole escape based on their kinetics of intracellular replication (Fig. 4), we examined whether the presence of at least one of the secretion chaperones is required for vacuole escape by examining the ability of ΔhtrA ΔprsA2::erm double mutant to perforate host cell vacuoles. PtK2 cells were transfected with a mammalian expression vector containing a fusion of the yellow fluorescent protein (YFP) to a cell wall binding domain (CBD) of the phage endolysin Ply118 that binds specifically to the L. monocytogenes cell wall (47). Henry et al. have previously shown that the YFP-CBD fusion protein is constitutively expressed in the host cytosol and nuclei of transfected tissue culture cells and colocalizes with L. monocytogenes only after the bacteria have perforated the vacuole (47). For a negative control for vacuole perforation, we included a Δhly mutant that lacks LLO and fails to perforate host vacuoles (52). Similar to the Δhly mutant, strains lacking the PrsA2 and HtrA secretion chaperones were unable to perforate the vacuole by up to 5 h postinfection (Fig. 7A to D). Thus, although the ΔhtrA ΔprsA2::erm double chaperone mutant exhibits detectable levels of LLO and PlcB activity when grown in broth culture and remains viable for several hours at 37°C, the mutant is still defective for vacuole perforation and escape within most infected cells, thereby indicating a requirement for at least one of the two secretion chaperones to promote lysis of the host cell vacuole in many, but not all, cell types.
FIG 7.

Loss of HtrA and PrsA2 inhibits membrane perforation of host cell vacuoles. PtK2 cells were transfected with a mammalian expression vector expressing a fusion of the yellow fluorescent protein (YFP) to a cell wall binding domain of the phage endolysin Ply118 that binds specifically to the L. monocytogenes cell wall. (A to C) Five hours postinfection of transfected cells with the wild type (A), ΔhtrA ΔprsA2::erm mutant (B), or Δhly mutant (C), the coverslips were removed and stained for fluorescence-based microscopy. Note that the MOI used for the ΔhtrA ΔprsA2::erm and Δhly mutants was 8-fold higher than that for the wild type to increase the numbers of intracellular bacteria for which vacuole perforation might be observed. The increased number of Δhly bacteria seen in association with cells in comparison to ΔhtrA ΔprsA2::erm reflects the increased invasive capacity of the Δhly mutant for PtK2 cells compared to the ΔhtrA ΔprsA2::erm mutant. In the micrographs, Listeria is shown in red, phage cell wall binding domain fused to YFP is shown in green, and host cell filamentous actin is shown in blue. The images shown are representative of two experiments. (D) Quantitation of bacteria in PtK2 host cells with (green) and without (red) colocalization with phage cell wall binding protein from the host cytosol. (E) Measurement of LLO-associated bacterial hemolytic activity. Dilutions of bacterial culture supernatants were assessed for their ability to lyse sheep's red blood cells (RBCs) in vitro. The reciprocal of the supernatant dilution that resulted in 50% lysis of RBCs (hemolytic units) was determined in a minimum of three independent experiments. The values are averages plus standard deviations (error bars). (F) The production of PlcB-dependent phospholipase was assessed on Brilliance selective agar plates. Bacteria were spotted onto agar plates and incubated overnight at 30°C. The halo or zone of opacity surrounding bacterial growth is indicative of PlcB activity. The experiment was repeated independently three times. The values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001.
DISCUSSION
The HtrA and PrsA2 secretion chaperones have both been thought to be involved in stress resistance and virulence; however, their roles have often appeared to be nonoverlapping and/or nonequivalent (30, 32, 35, 36, 38). L. monocytogenes strains lacking prsA2 are at least 100-fold-less virulent than htrA deletion strains are, and the mutants differ in secreted protein profiles as well as levels of resistance to various stresses (30, 35, 36, 39). Our studies of single and combined prsA2 and htrA deletion mutations indicate that both chaperones contribute to bacterial fitness under conditions of rapid growth in the presence of PrfA activation, such as would be anticipated to occur during host cell infection. The growth of the ΔhtrA ΔprsA2::erm double chaperone mutant was only slightly impaired at 37°C in the absence of PrfA activation, indicating that it is PrfA activation and the associated increase in the translocation of secreted proteins across the bacterial cell membrane that compromises bacterial fitness. The activities of PrsA2 and HtrA thus play crucial and complementary roles to promote L. monocytogenes survival during host infection.
The 37°C temperature-associated growth defect for ΔhtrA ΔprsA2::erm prfA* strains was not immediately apparent following the shift from 30°C to 37°C but rather developed over several hours of continued incubation. The expression levels of PrfA-dependent gene products by constitutively active PrfA* appears to be similar at either 30°C or 37°C (Fig. 2B); thus, we hypothesize that it is the increase in protein secretion combined with conditions of rapid bacterial growth that compromises L. monocytogenes fitness. Intriguingly, a similar temperature-dependent growth defect at 37°C has been observed for E. coli strains lacking both DegP (HtrA) and a periplasmic protein chaperone known as Skp (53). In E. coli, DegP has increased protease activity at 37°C (33), and it has been suggested that the absence of the chaperone Skp leads to the accumulation of unfolded proteins in the periplasm, which normally would be degraded by DegP (53). We hypothesize that a similar accumulation of misfolded or unfolded proteins at the membrane-cell wall interface becomes toxic for L. monocytogenes, given that the decrease in growth rate observed for strains lacking both secretion chaperones occurs only after several hours (Fig. 1D). Likewise, ΔhtrA ΔprsA2::erm prfA* cultures that have been incubated overnight at 37°C exhibit a prolonged lag phase before growth initiates, suggesting that cells must either overcome conditions of growth inhibition before growth can resume and/or alternatively that only a small minority of cells remain viable. Our results are thus consistent with a model in which PrsA2 contributes to the folding and stabilization of proteins as they translocate across the bacterial membrane and that HtrA assists in this process so as to avoid the accumulation of misfolded proteins at the membrane-cell wall interface.
HtrA and PrsA2 activities are indispensable for bacterial infection of host cells, conditions under which PrfA activation normally occurs. The contributions of the chaperones to bacterial intracellular growth appear to extend beyond the requirement for viability under conditions of PrfA activation, given that the growth defect takes several hours to manifest, while the mutants appear almost immediately compromised for intracellular growth. The intracellular replication defect of the ΔhtrA ΔprsA2::erm mutant appears to result largely, at least initially, from the failure of the mutant to mediate escape from the host cell vacuole. The apparent severity of the vacuole escape defect in several cell types is unexpected, given that reduced but still substantial levels of active LLO and phosphatidylcholine-preferring phospholipase C (PC-PLC) are secreted by the mutant (Fig. 7E and F). It has been previously shown that mutants expressing ∼20% of wild-type LLO activity are still capable of efficient vacuole escape (50). The failure to perforate vacuoles despite substantial LLO and PC-PLC enzymatic activity in broth (∼40% of wild type) suggests that the environment of the vacuole may be more stringent for protein folding and activity of LLO and PC-PLC than conditions of broth culture. Interestingly, the ΔhtrA ΔprsA2::erm mutant remains capable of escape from the vacuoles of the human intestinal cell line Caco2 with only a slight delay compared to wild-type bacteria. This suggests that the vacuoles of these cells may be more permissive for bacterial escape; if so, this does not appear to be a feature common to other human cell types or to other intestine- or colon-derived cell lines (Fig. 5 and 6). Cell type-dependent differences in vacuole escape have been previously observed for other cell lines, such as the ability of L. monocytogenes mutants lacking LLO to escape from the vacuoles of human Henle epithelial cell lines (54) while remaining trapped in other cell types (15, 48, 52). It is intriguing to speculate that perhaps the permissiveness of a cell type for vacuole escape contributes to some degree to L. monocytogenes cell and/or tissue tropism during infection.
PrsA2 functions as a foldase and catalyzes the cis-trans isomerization of peptide bonds N terminal to polypeptide proline residues (38), and several target proteins have been identified (32), while HtrA appears to be a broad-spectrum chaperone/protease with specific protein targets yet to be identified in Listeria. Our data revealed that L. monocytogenes increases HtrA abundance in response to a lack of PrsA2 but that a similar increase in PrsA2 is not detected in cells lacking HtrA. Similarly, it has been observed that a reduction in prsA expression in Bacillus subtilis triggers an increase in expression of htrA (55). In B. subtilis, htrA is regulated by the CssR-CssS two-component system which senses the presence of extracytoplasmic misfolded proteins. In Listeria, the LisRK two-component system has been linked to the transcriptional activation of htrA (35), and the histidine kinase sensor LisK was found to respond to membrane stress (56–58). The upregulation of htrA in the ΔprsA2::erm mutant is independent of temperature or PrfA activation and therefore might occur as a result of a general membrane stress due to the absence of PrsA2. We speculate that the combined chaperone-protease function of HtrA makes it a more broadly functional stress response protein, whereas the more substrate-specific chaperone functions of PrsA2 restrict its overall utility as a stress-responsive chaperone. How prsA2 expression is regulated remains to be defined; it has been shown that PrfA contributes to prsA2 expression; however, this regulation is dispensable in vivo (59), suggesting that other mechanism exist for the induction of prsA2 expression.
PrsA homologues are present in single and multiple copies in a variety of Gram-positive bacteria, and HtrA can be found in both Gram-positive and Gram-negative bacteria. It is possible that these two secretion chaperones have evolved complementary and perhaps interconnected roles in other Gram-positive bacteria. The essential requirement for at least one of the two secretion chaperones to promote L. monocytogenes intracellular growth serves to emphasize the demands placed upon the bacterial cell as a result of increased virulence factor secretion and the degree to which PrsA2 and HtrA contribute to protein folding and stability and bacterial surface integrity during the process of mammalian host infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joel Swanson for the gift of the CBD-YFP plasmid and members of the Freitag laboratory and Positive Thinking group for helpful discussions.
Nancy E. Freitag has a consulting relationship with and a financial interest in Advaxis Immunotherapies, and both she and the company stand to benefit from the commercialization of the results of this research.
This work was supported by NIH grant R01 AI083241 to N.E.F.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the funding source. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Statement
This work was supported by NIH grant R01 AI083241 to NEF. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding source. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00312-16.
REFERENCES
- 1.Sauders BD, Overdevest J, Fortes E, Windham K, Schukken Y, Lembo A, Wiedmann M. 2012. Diversity of Listeria species in urban and natural environments. Appl Environ Microbiol 78:4420–4433. doi: 10.1128/AEM.00282-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sauders BD, Durak MZ, Fortes E, Windham K, Schukken Y, Lembo AJ Jr, Akey B, Nightingale KK, Wiedmann M. 2006. Molecular characterization of Listeria monocytogenes from natural and urban environments. J Food Prot 69:93–105. [DOI] [PubMed] [Google Scholar]
- 3.Weis J, Seeliger HP. 1975. Incidence of Listeria monocytogenes in nature. Appl Microbiol 30:29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2011. Multistate outbreak of listeriosis associated with Jensen Farms cantaloupe–United States, August-September 2011. MMWR Morb Mortal Wkly Rep 60:1357–1358. [PubMed] [Google Scholar]
- 5.Olsen SJ, Patrick M, Hunter SB, Reddy V, Kornstein L, MacKenzie WR, Lane K, Bidol S, Stoltman GA, Frye DM, Lee I, Hurd S, Jones TF, LaPorte TN, Dewitt W, Graves L, Wiedmann M, Schoonmaker-Bopp DJ, Huang AJ, Vincent C, Bugenhagen A, Corby J, Carloni ER, Holcomb ME, Woron RF, Zansky SM, Dowdle G, Smith F, Ahrabi-Fard S, Ong AR, Tucker N, Hynes NA, Mead P. 2005. Multistate outbreak of Listeria monocytogenes infection linked to delicatessen turkey meat. Clin Infect Dis 40:962–967. doi: 10.1086/428575. [DOI] [PubMed] [Google Scholar]
- 6.Freitag NE, Port GC, Miner MD. 2009. Listeria monocytogenes - from saprophyte to intracellular pathogen. Nat Rev Microbiol 7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de las Heras A, Cain RJ, Bielecka MK, Vazquez-Boland JA. 2011. Regulation of Listeria virulence: PrfA master and commander. Curr Opin Microbiol 14:118–127. doi: 10.1016/j.mib.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. 2007. The PrfA virulence regulon. Microbes Infect 9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 9.O'Neil HS, Marquis H. 2006. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun 74:6675–6681. doi: 10.1128/IAI.00886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camejo A, Carvalho F, Reis O, Leitao E, Sousa S, Cabanes D. 2011. The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2:379–394. doi: 10.4161/viru.2.5.17703. [DOI] [PubMed] [Google Scholar]
- 11.Mostowy S, Cossart P. 2012. Virulence factors that modulate the cell biology of Listeria infection and the host response. Adv Immunol 113:19–32. doi: 10.1016/B978-0-12-394590-7.00007-5. [DOI] [PubMed] [Google Scholar]
- 12.Vazquez-Boland JA, Kocks C, Dramsi S, Ohayon H, Geoffroy C, Mengaud J, Cossart P. 1992. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect Immun 60:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mengaud J, Braun-Breton C, Cossart P. 1991. Identification of phosphatidylinositol-specific phospholipase C activity in Listeria monocytogenes: a novel type of virulence factor? Mol Microbiol 5:367–372. doi: 10.1111/j.1365-2958.1991.tb02118.x. [DOI] [PubMed] [Google Scholar]
- 14.Bielecki J, Youngman P, Connelly P, Portnoy DA. 1990. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- 15.Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. 1987. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect Immun 55:2822–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531. [DOI] [PubMed] [Google Scholar]
- 17.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. 1993. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A 90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, Portnoy DA. 2015. Glutathione activates virulence gene expression of an intracellular pathogen. Nature 517:170–173. doi: 10.1038/nature14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ripio MT, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland JA. 1997. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol 179:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miner MD, Port GC, Bouwer HG, Chang JC, Freitag NE. 2008. A novel prfA mutation that promotes Listeria monocytogenes cytosol entry but reduces bacterial spread and cytotoxicity. Microb Pathog 45:273–281. doi: 10.1016/j.micpath.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mueller KJ, Freitag NE. 2005. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect Immun 73:1917–1926. doi: 10.1128/IAI.73.4.1917-1926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shetron-Rama LM, Mueller K, Bravo JM, Bouwer HG, Way SS, Freitag NE. 2003. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol Microbiol 48:1537–1551. doi: 10.1046/j.1365-2958.2003.03534.x. [DOI] [PubMed] [Google Scholar]
- 23.Vega Y, Rauch M, Banfield MJ, Ermolaeva S, Scortti M, Goebel W, Vazquez-Boland JA. 2004. New Listeria monocytogenes prfA* mutants, transcriptional properties of PrfA* proteins and structure-function of the virulence regulator PrfA. Mol Microbiol 52:1553–1565. doi: 10.1111/j.1365-2958.2004.04052.x. [DOI] [PubMed] [Google Scholar]
- 24.Wong KK, Freitag NE. 2004. A novel mutation within the central Listeria monocytogenes regulator PrfA that results in constitutive expression of virulence gene products. J Bacteriol 186:6265–6276. doi: 10.1128/JB.186.18.6265-6276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Port GC, Freitag NE. 2007. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect Immun 75:5886–5897. doi: 10.1128/IAI.00845-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Randall LL. 1992. Peptide binding by chaperone SecB: implications for recognition of nonnative structure. Science 257:241–245. doi: 10.1126/science.1631545. [DOI] [PubMed] [Google Scholar]
- 27.Wolff N, Sapriel G, Bodenreider C, Chaffotte A, Delepelaire P. 2003. Antifolding activity of the SecB chaperone is essential for secretion of HasA, a quickly folding ABC pathway substrate. J Biol Chem 278:38247–38253. doi: 10.1074/jbc.M302322200. [DOI] [PubMed] [Google Scholar]
- 28.Schneewind O, Missiakas DM. 2012. Protein secretion and surface display in Gram-positive bacteria. Philos Trans R Soc Lond B Biol Sci 367:1123–1139. doi: 10.1098/rstb.2011.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matias VR, Beveridge TJ. 2005. Cryo-electron microscopy reveals native polymeric cell wall structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol Microbiol 56:240–251. doi: 10.1111/j.1365-2958.2005.04535.x. [DOI] [PubMed] [Google Scholar]
- 30.Alonzo F III, Freitag NE. 2010. Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect Immun 78:4944–4957. doi: 10.1128/IAI.00532-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forster BM, Zemansky J, Portnoy DA, Marquis H. 2011. Posttranslocation chaperone PrsA2 regulates the maturation and secretion of Listeria monocytogenes proprotein virulence factors. J Bacteriol 193:5961–5970. doi: 10.1128/JB.05307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonzo F III, Port GC, Cao M, Freitag NE. 2009. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect Immun 77:2612–2623. doi: 10.1128/IAI.00280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spiess C, Beil A, Ehrmann M. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97:339–347. doi: 10.1016/S0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- 34.Singh N, Kuppili RR, Bose K. 2011. The structural basis of mode of activation and functional diversity: a case study with HtrA family of serine proteases. Arch Biochem Biophys 516:85–96. doi: 10.1016/j.abb.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Stack HM, Sleator RD, Bowers M, Hill C, Gahan CG. 2005. Role for HtrA in stress induction and virulence potential in Listeria monocytogenes. Appl Environ Microbiol 71:4241–4247. doi: 10.1128/AEM.71.8.4241-4247.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wonderling LD, Wilkinson BJ, Bayles DO. 2004. The htrA (degP) gene of Listeria monocytogenes 10403S is essential for optimal growth under stress conditions. Appl Environ Microbiol 70:1935–1943. doi: 10.1128/AEM.70.4.1935-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson RL, Brown LL, Kirkwood-Watts D, Warren TK, Lund SA, King DS, Jones KF, Hruby DE. 2006. Listeria monocytogenes 10403S HtrA is necessary for resistance to cellular stress and virulence. Infect Immun 74:765–768. doi: 10.1128/IAI.74.1.765-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alonzo F III, Xayarath B, Whisstock JC, Freitag NE. 2011. Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol Microbiol 80:1530–1548. doi: 10.1111/j.1365-2958.2011.07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cahoon LA, Freitag NE. 2015. Identification of conserved and species-specific functions of the Listeria monocytogenes PrsA2 secretion chaperone. Infect Immun 83:4028–4041. doi: 10.1128/IAI.00504-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cahoon LA, Freitag NE. 2014. Listeria monocytogenes virulence factor secretion: don't leave the cell without a chaperone. Front Cell Infect Microbiol 4:13. doi: 10.3389/fcimb.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodgson DA. 2000. Generalized transduction of serotype 1/2 and serotype 4b strains of Listeria monocytogenes. Mol Microbiol 35:312–323. doi: 10.1046/j.1365-2958.2000.01643.x. [DOI] [PubMed] [Google Scholar]
- 42.Lennox ES. 1955. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- 43.Miner MD, Port GC, Freitag NE. 2008. Functional impact of mutational activation on the Listeria monocytogenes central virulence regulator PrfA. Microbiology 154:3579–3589. doi: 10.1099/mic.0.2008/021063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youngman P. 1987. Plasmid vectors recovering and exploiting Tn917 transposons in Bacillus and other Gram-positive bacteria, p 79–103. In Hardy KG. (ed), Plasmids: a practical approach. IRL Press, Oxford, United Kingdom. [Google Scholar]
- 45.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 46.Freitag NE, Jacobs KE. 1999. Examination of Listeria monocytogenes intracellular gene expression by using the green fluorescent protein of Aequorea victoria. Infect Immun 67:1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry R, Shaughnessy L, Loessner MJ, Alberti-Segui C, Higgins DE, Swanson JA. 2006. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell Microbiol 8:107–119. doi: 10.1111/j.1462-5822.2005.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camilli A, Paynton CR, Portnoy DA. 1989. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci U S A 86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P. 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110:551–561. doi: 10.1016/S0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 50.Freitag NE, Rong L, Portnoy DA. 1993. Regulation of the prfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect Immun 61:2537–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Portnoy DA, Jacks PS, Hinrichs DJ. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schafer U, Beck K, Muller M. 1999. Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem 274:24567–24574. doi: 10.1074/jbc.274.35.24567. [DOI] [PubMed] [Google Scholar]
- 54.Marquis H, Doshi V, Portnoy DA. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immun 63:4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hyyrylainen HL, Bolhuis A, Darmon E, Muukkonen L, Koski P, Vitikainen M, Sarvas M, Pragai Z, Bron S, van Dijl JM, Kontinen VP. 2001. A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol Microbiol 41:1159–1172. [DOI] [PubMed] [Google Scholar]
- 56.Cotter PD, Guinane CM, Hill C. 2002. The LisRK signal transduction system determines the sensitivity of Listeria monocytogenes to nisin and cephalosporins. Antimicrob Agents Chemother 46:2784–2790. doi: 10.1128/AAC.46.9.2784-2790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cotter PD, Emerson N, Gahan CG, Hill C. 1999. Identification and disruption of lisRK, a genetic locus encoding a two-component signal transduction system involved in stress tolerance and virulence in Listeria monocytogenes. J Bacteriol 181:6840–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kallipolitis BH, Ingmer H. 2001. Listeria monocytogenes response regulators important for stress tolerance and pathogenesis. FEMS Microbiol Lett 204:111–115. doi: 10.1111/j.1574-6968.2001.tb10872.x. [DOI] [PubMed] [Google Scholar]
- 59.Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J Bacteriol 191:3950–3964. doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bishop DK, Hinrichs DJ. 1987. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol 139:2005–2009. [PubMed] [Google Scholar]
- 61.Jones S, Portnoy DA. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun 62:5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.