

Abstract
As the major cause of antibiotic-associated diarrhea, Clostridium difficile is a serious problem in health care facilities worldwide. C. difficile produces two large toxins, TcdA and TcdB, which are the primary virulence factors in disease. The respective functions of these toxins have been difficult to discern, in part because the cytotoxicity profiles for these toxins differ with concentration and cell type. The goal of this study was to develop a cell culture model that would allow a side-by-side mechanistic comparison of the toxins. Conditionally immortalized, young adult mouse colonic (YAMC) epithelial cells demonstrate an exquisite sensitivity to both toxins with phenotypes that agree with observations in tissue explants. TcdA intoxication results in an apoptotic cell death that is dependent on the glucosyltransferase activity of the toxin. In contrast, TcdB has a bimodal mechanism; it induces apoptosis in a glucosyltransferase-dependent manner at lower concentrations and glucosyltransferase-independent necrotic death at higher concentrations. The direct comparison of the responses to TcdA and TcdB in cells and colonic explants provides the opportunity to unify a large body of observations made by many independent investigators.
INTRODUCTION
Clostridium difficile is the most common cause of antibiotic-associated diarrhea in the United States, and C. difficile infection (CDI) has been steadily increasing in prevalence and severity over the last 15 years (1–3). Symptoms of CDI can range from mild diarrhea to pseudomembranous colitis, and hallmarks of the disease include neutrophil infiltration, fluid release, and necrotic lesions in the colonic epithelium (4, 5). The bacteria produce two main virulence factors, large toxins called TcdA and TcdB (6, 7).
The respective function and relative importance of each toxin in pathogenesis have been active topics of investigation. Genetic knockout experiments in C. difficile have shown that both toxins are important for disease pathology, although TcdB alone is sufficient to cause death in both the hamster and mouse models (6–8). For many years, TcdA and TcdB have been thought to act synergistically, with TcdA acting as an enterotoxin and TcdB acting as a cytotoxin (9, 10). The general term enterotoxin refers to the capacity of TcdA to induce inflammation, cytokine release, and fluid secretion in animal intoxication models (11–13). While TcdB does not always induce these same phenotypes in models, such as the ileal loop model, it has been shown to disrupt the integrity of the epithelial structure in human explant and xenograft models (14, 15). TcdB is also notably more potent as a cytotoxin in cell culture models (9, 10, 16).
The toxins have an N-terminal glucosyltransferase domain (GTD) that is delivered into the host cytosol by the C-terminal portion of the protein (17, 18). The GTD has been shown to target and inactivate a number of Rho-family GTPases (19, 20). This inactivation has been linked to a cell rounding or cytopathic effect (CPE) (21–24) and to an apoptotic cytotoxic effect (25–33). In tissue culture models, the apoptotic effects of TcdA and TcdB occur at toxin concentrations of picomolar or lower and are evident at 24 to 48 h postintoxication (26, 28, 29, 34–36). TcdB also induces a glucosyltransferase-independent necrosis that is mediated by the assembly and activation of the NADPH oxidase (NOX) complex, subsequently producing high levels of reactive oxygen species (ROS) (34, 37–40). Indicators of necrosis are apparent within 2 to 4 h using nanomolar concentrations of TcdB in both tissue culture and colonic explant models.
Most published reports discuss the effects of a single toxin; it is rare to see the effects of the toxins compared side-by-side in colonic cell and tissue models. In this study, we wanted to investigate the mechanisms and pathological outcomes associated with TcdA and TcdB intoxication under comparable conditions. We reasoned that the antiapoptotic mutations associated with transformed cell lines were preventing TcdA-induced cell death pathways. Young adult mouse colonic (YAMC) epithelial cells are derived from the Immortomouse, which expresses a temperature-sensitive simian virus 40 (SV40) T antigen that suppresses p53 (41). The cells can be carried as an antiapoptotic cell line at the permissive temperature of 33°C, and then, when they are shifted to the nonpermissive temperature of 37°C, YAMC cells behave as primary cells with an intact p53 pathway able to undergo normal apoptosis. Using this tool, we were able to investigate the effects of TcdA and TcdB side-by-side using the same time points and assay readouts. Our observations provide an opportunity to unify the many, seemingly conflicting reports describing the mechanisms by which TcdA and TcdB cause cell death in epithelial cells.
MATERIALS AND METHODS
Recombinant protein expression and purification.
The glucosyltransferase domain double point mutation (TcdA D285/287N and TcdB D286/288N) plasmids (pBL764 and pBL765, respectively) were made using the TcdA and TcdB parent plasmids (42) according to the QuikChange protocol (Stratagene). Recombinant TcdA, TcdA D285/287N, TcdB, and TcdB D286/288N proteins were expressed in Bacillus megaterium and purified as previously described (42).
YAMC cell culture and viability assays.
YAMC cells were maintained in RPMI 1640 supplemented with 5% fetal bovine serum, 1 mg/ml insulin, 10 μM alpha-thioglycerol, 1 μM hydrocortisone, and 5 U/ml mouse interferon gamma. Cells were maintained at 33°C with 5% CO2. For assays performed at 37°C, cells were plated and incubated at 37°C with 5% CO2 overnight prior to intoxication. Viability was measured at the concentrations and time points indicated below using the CellTiter Glo luminescent cell viability assay (catalog number G7573; Promega). Lactate dehydrogenase (LDH) release was quantified using the CytoTox Glo assay (catalog number G9290; Promega). Apoptosis was assessed by measuring active caspase-3 and -7 levels using the Apo-ONE homogeneous caspase-3/7 assay (catalog number Promega, G7792). ROS production was assayed with carboxy-2′,7′-dichlorodihydrofluorescein diacetate (catalog number C400; Life Technologies) as previously described (37).
Colonic explants.
Animal husbandry and experimental procedures related to the porcine colonic explants were performed in accordance with the Vanderbilt University Institutional Animal Care and Use Committee (IACUC) policy. Discarded colon tissues were obtained from pigs following euthanization at the end of IACUC-approved animal use protocols and prepared for intoxication as previously described (34). Tissue was challenged with 10 nM TcdA, TcdB, TcdA D285/287N, or TcdB D286/288N for 5 h at 37°C. The sections were cut by the Vanderbilt University Translational Pathology Shared Resource Core. Staining of tissues for detection of caspase-3 and ROS was done as previously described (34, 37). All slides stained with fluorescent markers were analyzed with an LSM 510 confocal microscope.
Statistical analysis.
Statistical analysis was performed using a two-way analysis of variance (ANOVA) and post hoc test in GraphPad Prism software. Two-tailed, paired Student's t tests were performed using Excel software.
RESULTS
TcdA induces a robust cell death in conditionally immortalized cells.
We hypothesized that the lack of rapid TcdA-induced cell death in typical tissue culture models was due to mutations in the apoptotic pathways of many transformed cell lines. To test our hypothesis, we obtained YAMC cells, a conditionally immortalized cell line with temperature-sensitive large T antigen (ts58) that can affect p53 function and, more broadly, drive cells into S phase to promote cell growth (43). YAMC cells were challenged with the TcdA and TcdB toxins at both the permissive (33°C) and nonpermissive (37°C) temperatures, and cell death was quantified using CellTiter Glo, an ATP-sensitive viability indicator. Consistent with what is observed in transformed cell lines, TcdA does not induce appreciable cell death at 33°C, where p53 is inactivated (Fig. 1A). Consistent with our previous observations in transformed HeLa and Caco2 cell lines (34), TcdB kills cells efficiently at concentrations of ≥100 pM but does not cause cell death at concentrations of ≤10 pM (Fig. 1A). Upon shifting to 37°C, where large T antigen is no longer expressed, we detected a drastically different phenotype. TcdA caused cytotoxicity in YAMC cells across a wide concentration range (10 fM to 100 nM; Fig. 1B), and these changes were significant compared to the responses at 33°C (P < 0.0001, as judged by a two-way ANOVA). The switch to 37°C also allowed TcdB-induced cell death at concentrations of ≤10 pM (Fig. 1B). These changes were also significant relative to the responses at 33°C (P < 0.0001).
FIG 1.
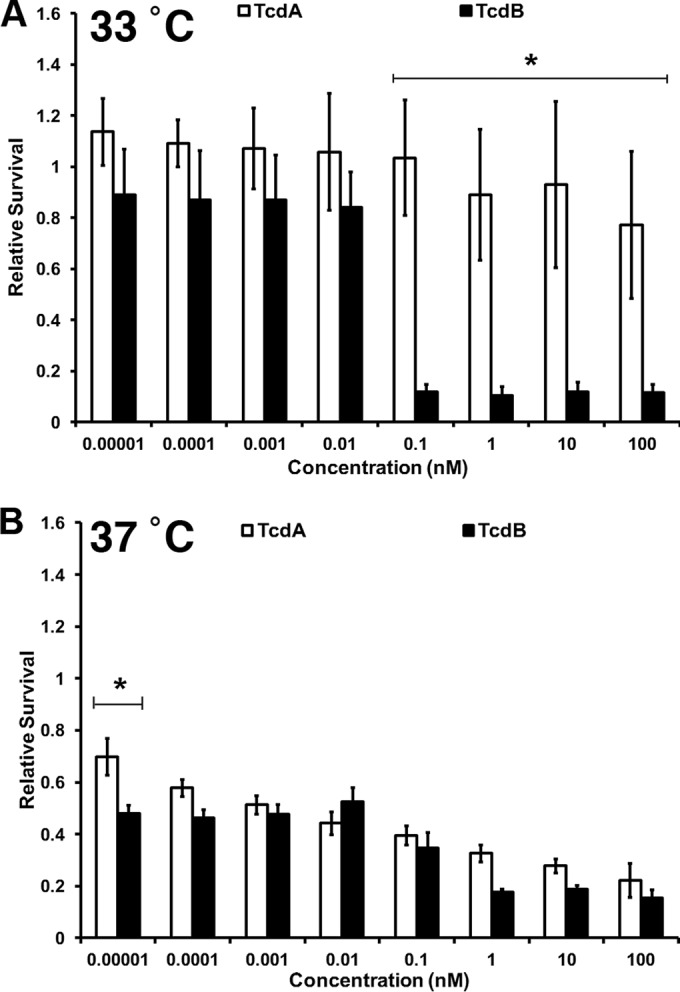
TcdA is cytotoxic when applied to conditionally immortalized epithelial cells. YAMCs were incubated overnight at either 33°C or 37°C. Cells were intoxicated as indicated and incubated at the respective temperature for 18 h. Cell viability was determined using CellTiter Glo. Relative survival was calculated by normalizing the survival of treated samples to that of untreated samples. (A) At 33°C, there was no statistically significant difference between TcdA and TcdB at concentrations of between 10 fM and 10 pM. There was a statistically significant difference in the cell death induced by TcdA and TcdB at concentrations of 100 pM to 100 nM, as determined by a two-way ANOVA (*, P < 0.0001). (B) At 37°C, there was no statistically significant difference in cell death induced by the toxins at concentrations ranging from 100 fM to 100 nM by two-way ANOVA. At 10 fM, a significant difference in potency became apparent when analyzed by a two-tailed, paired Student's t test (*, P < 0.0001). The viability of cells treated with TcdA was statistically significantly different between that at 33°C and that at 37°C (P < 0.0001). The viability of cells treated with TcdB was statistically significantly different between the two temperatures at concentrations of ≤10 pM (P < 0.0001). At concentrations of ≥100 nM, the differences were not statistically significant. Data represent the averages from three experiments performed in triplicate. Error bars represent the standard deviations of the means.
Both TcdA and TcdB bind YAMC cells and glucosylate Rac1 with similar efficiencies at 33°C and 37°C (see Fig. S1 in the supplemental material), suggesting that the increased cytotoxicity of the toxins at 37°C is not the result of different binding, entry, or glucosylating activities. Similar results were observed in HeLa cells, which are p53 null (although we were unable to detect the TcdA binding by Western blotting in these cells) (see Fig. S2 in the supplemental material). As another control experiment, we used a live/dead indicator to assess the toxin-induced CPE and cell death in HeLa cells at the two different temperatures. The cells rounded in response to 100 pM or 10 nM concentrations of TcdA at both temperatures, but as has been observed in previous studies (34, 37), there was very little cell death (see Fig. S3 in the supplemental material). With TcdB applied at a 10 pM or 10 nM concentration, the cells were either round at both temperatures or dead, respectively. These experiments support the hypothesis that the different effects of the toxins at different temperatures are not a result of different binding and glucosylating activities but, rather, are a result of a cell death mechanism that depends on the expression of ts58.
TcdB causes a loss of membrane integrity faster than TcdA.
The extent of the temperature effect was less pronounced for the cells treated with TcdB at concentrations of ≥100 pM (Fig. 1). This led us to hypothesize that TcdA and TcdB kill cells through distinct mechanisms at concentrations of ≥100 pM. To evaluate this further, we assessed the impact of TcdA and TcdB on cell membrane integrity over time using an LDH indicator (Fig. 2). We detected LDH release as early as 2 h after cells were challenged with ≥0.1 nM TcdB (Fig. 2A), consistent with a necrotic mechanism of cytotoxicity. The LDH signal was high in cells treated with high TcdB concentrations (100 pM to 100 nM) for up to 8 h postintoxication (Fig. 2A to C). Concentrations below 100 pM did not demonstrate an appreciable rise in LDH levels at the 2-, 4-, or 8-h time points, and the LDH signal was not significantly different from the signal produced from cells intoxicated with TcdA (Fig. 2A to C). However, an increase in the LDH signal at lower concentrations was detected at 24 h postintoxication with TcdB (Fig. 2D). LDH release was detected in response to all concentrations of TcdA but only after 24 h of intoxication (Fig. 2D). At the 24-h time point, there was no significant difference in the LDH signals from cells treated with either TcdA or TcdB.
FIG 2.

TcdB causes a loss of membrane integrity faster than TcdA. YAMC cells at 37°C were intoxicated with the indicated concentrations of TcdA or TcdB and incubated at 37°C for 2 h (A), 4 h (B), 8 h (C), and 24 h (D). LDH release was measured using CytoTox Glo and normalized to that for the unintoxicated controls. Data were analyzed by two-way ANOVA at each time point. At 2, 4, and 8 h, there was a significant difference between TcdA and TcdB at concentrations of ≥100 pM (*, P < 0.0001). There was no significant difference at concentrations of ≤10 pM. At 24 h, two-way ANOVA revealed no significant difference between TcdA and TcdB at any concentration tested. Data represent the averages from three experiments performed in triplicate. Error bars represent the standard deviations of the means.
TcdA induces cell death by a mechanism distinct from that for TcdB at higher toxin concentrations.
The difference in the rate at which TcdA and TcdB induce a loss of membrane integrity further supported our hypothesis that the toxins kill cells by different mechanisms at higher concentrations. We expected TcdB to induce ROS-driven necrosis at these higher concentrations based on previous work in HeLa and Caco-2 cells (37) and therefore measured ROS production in response to each toxin in YAMC cells (Fig. 3A). TcdB induced the production of high levels of ROS at higher concentrations (≥100 pM), where we have previously observed necrosis, but not at toxin concentrations below 100 pM. This observation is consistent with the mechanistic switch noted in Fig. 1A and 2A to C. The correlation of ROS production and LDH release in response to higher concentrations (≥100 pM) of TcdB indicates a necrotic cell death. In cells treated with higher concentrations (≥100 pM) of TcdB D286/288N, a mutant deficient in glucosylation (40), we detected ROS production and cell death at levels that were indistinguishable from those for cells treated with TcdB (see Fig. S4 in the supplemental material). These results are consistent with the hypothesis that TcdB-induced necrosis is glucosyltransferase independent. The absence of ROS and the delayed LDH signal in response to TcdA intoxication further suggest that TcdA and TcdB at concentrations of 100 pM and above cause cell death by different mechanisms. We did not observe an additive or synergistic effect when TcdA and TcdB were added to YAMC cells simultaneously (see Fig. S5 in the supplemental material).
FIG 3.
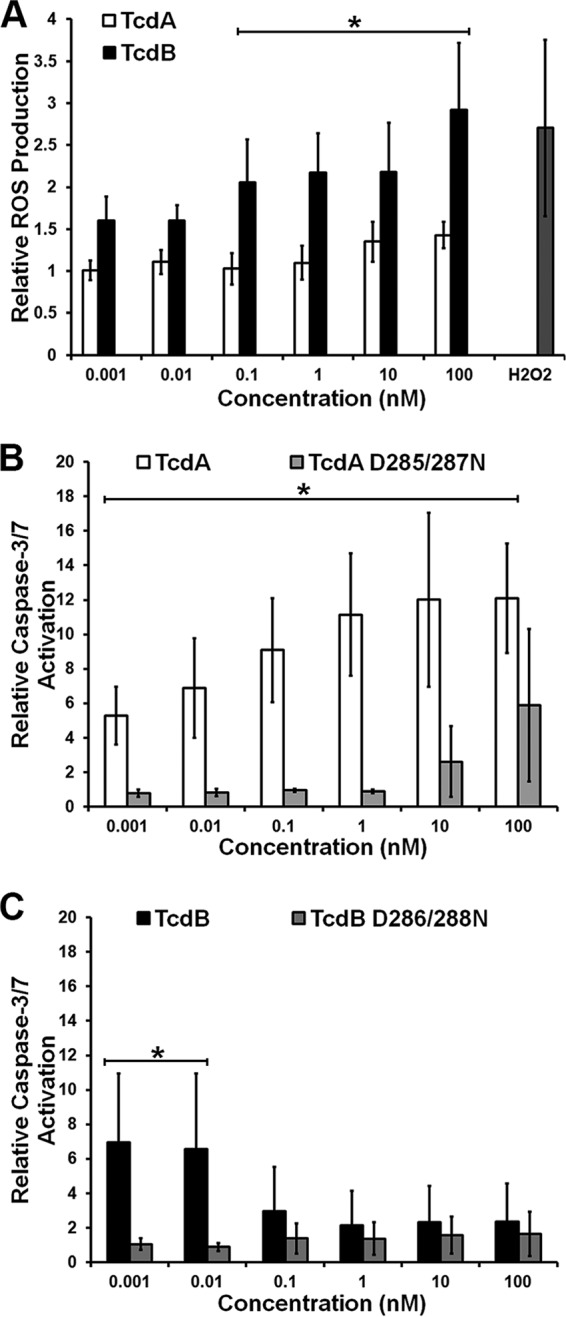
TcdA and TcdB induce cell death by different mechanisms. (A) YAMC cells were tested for ROS production in response to TcdA (24 h) or TcdB (6 h) using a fluorescent ROS reporter. Statistical analysis by two-way ANOVA revealed that the results for TcdA and TcdB were significantly different from each other at concentrations of ≥100 pM (*, P < 0.0001). (B) YAMC cells were also tested for activated caspase-3/7 using Apo-ONE in response to TcdA and TcdA D285/287N. A two-way ANOVA revealed a significant difference between TcdA and TcdA D285/287N at all concentrations (*, P < 0.0001). (C) Activated caspase was measured in response to TcdB or TcdB D286/288N, as described in the legend to panel B. A two-way ANOVA showed no significant difference between TcdB and TcdB D286/288N at concentrations of ≥100 pM. At concentrations of ≤10 pM, there was a significant difference between TcdB and TcdB D286/288N (*, P < 0.01). In all panels, data represent the averages from three experiments performed in triplicate, and error bars represent the standard deviations of the means.
The importance of ts58 expression (Fig. 1B), the observation that TcdA and low concentrations of TcdB do not induce early LDH release (Fig. 2), and the lack of significant ROS production under similar conditions (Fig. 3A) are consistent with reports that TcdA and TcdB can trigger apoptotic cell death. Apoptosis in response to the toxins is thought to be dependent upon glucosyltransferase activity and subsequent GTPase inactivation (19, 24, 29). To test if this is true in YAMC cells, we challenged cells with TcdA, TcdB, and their respective glucosyltransferase-deficient mutants and assayed for caspase-3/7 activation (Fig. 3B and C). We detected robust and concentration-dependent caspase-3/7 activation in response to TcdA (Fig. 3B). The concentration dependence of caspase-3/7 activation correlates with the concentration-dependent loss in cell viability that we observed at 37°C (Fig. 1B). The glucosyltransferase mutant TcdA D285/287N caused some activation of caspase-3/7 at 100 nM, but the result was not statistically significant (Fig. 3B). We presume that the residual activity of the mutant seen in the caspase-3/7 activation assay is a result of residual glucosylating activity, the effects of which are detectable only at the highest concentration tested (34, 44). When the cells were challenged with higher concentrations (≥100 pM) of TcdB, very little caspase activation was detected (Fig. 3C). At concentrations of 10 pM and lower, however, we observed caspase-3/7 activation at a level comparable to what was observed with TcdA. The TcdB glucosyltransferase mutant TcdB D286/288N did not activate caspase-3/7 at any concentration tested (Fig. 3C). The observed ROS and caspase-3/7 activation of YAMC cells challenged with the glucosylation-deficient mutants (Fig. 3) is consistent with the viability data (see Fig. S4B in the supplemental material). There was some cell death at high concentrations (≥10 nM) of TcdA D285/287N, consistent with the residual caspase-3/7 activity at these concentrations (Fig. 3B). In cells treated with TcdB D286/288N, cell death was induced at higher concentrations (≥10 pM), where ROS is produced, similar to what is observed with wild-type TcdB. At lower concentrations (≤1 pM), less cell death was observed (see Fig. S4B in the supplemental material), as TcdB D286/288N did not induce caspase-3/7 activation (Fig. 3C). These data suggest that TcdA induces a glucosyltransferase-dependent, apoptotic cell death. TcdB can induce a glucosyltransferase-dependent, apoptotic cell death at lower concentrations (≤10 pM), where necrosis is not observed. The concentration-dependent switch in caspase-3/7 activation in response to TcdB further supports the concept of a concentration-dependent mechanistic switch for TcdB-induced cytotoxicity.
TcdA induces glucosyltransferase-dependent caspase-3 activation, while TcdB induces glucosyltransferase-independent ROS production in colonic tissue.
We next wanted to test whether our observations that TcdA and TcdB induced cell death by different mechanisms at higher concentrations in YAMC cells could also be observed in colonic explants. We intoxicated tissue with 10 nM TcdA, TcdB, or their corresponding glucosyltransferase-deficient mutants and assessed ROS production using a fluorescent indicator and apoptosis using an antibody specific for active caspase-3. While the ROS signal in TcdA-treated tissue was minor (Fig. 4A), we detected a robust signal for active caspase-3 that was attenuated with the TcdA D285/287N mutant (Fig. 4B). Consistent with previous observations (37), we detected significant ROS in colonic explants treated with both TcdB and TcdB D286/288N (Fig. 4A). There was very little active caspase-3 signal in these samples (Fig. 4B). Together, these data support our observations in YAMC cells and suggest that TcdA and TcdB induce cell death by different mechanisms at concentrations of ≥100 pM.
FIG 4.

TcdA induces glucosyltransferase-dependent caspase-3 activation, while TcdB induces glucosyltransferase-independent ROS production in colonic tissue. (A) Porcine colonic explants were treated with an ROS detection agent and incubated at 37°C for 1 h. Samples were then treated with 10 nM TcdA, TcdA D285/287N, TcdB, or TcdB D286/288N at 37°C for 5 h, after which the tissue was flash frozen. Slides were prepared and immediately imaged. (B) Porcine colonic explants were treated with toxins and incubated at 37°C for 5 h before being fixed and stained with an active caspase-3 antibody (red). The fluorescent and bright-field images in panels A and B were captured using confocal microscopy.
DISCUSSION
To more fully elucidate the mechanisms of TcdA- and TcdB-induced cytotoxicity, we designed a study that allowed the analysis of comparable concentrations of toxins at identical time points in the same tissue or cell type. The comparison permits an understanding of the different cellular processes engaged by each toxin, the concentration at which the toxin is capable of injuring the cell or tissue, and the relative contribution of the glucosyltransferase activity of the toxins.
Using YAMC cells, we could readily detect TcdA-induced cell death over a wide range of concentrations (10 fM to 100 nM) in 18 h (Fig. 1B). TcdB-induced cell death was also readily detectable down to 10 fM at the same time point. The sensitivity of YAMC cells to TcdA- and TcdB-induced cell death at low concentrations is dependent on maintenance of the cells at 37°C, a temperature that prevents the expression of ts58. The large T antigen binds several cellular factors, including p53, a tumor suppressor known to regulate cell cycle, mitotic division, and apoptotic cell death pathways. There are conflicting reports regarding the function of p53 in TcdA-induced apoptosis (35, 45). One study using a nontransformed colonic cell line concluded that TcdA-induced apoptosis is dependent upon p53 function (45). Another study used two cell lines with different p53 expression profiles and directly compared the lines in the context of p53 presence or absence to conclude that TcdA-induced apoptosis is independent of p53 (35). In the second study, the cell line that expressed p53 was mutated in various other components of the apoptotic pathway, however. The data in this study are consistent with a view where p53 function is important for TcdA- and TcdB-induced apoptosis, although we acknowledge that p53 is just one of many ts58 targets.
The temperature-dependent TcdB-induced cytotoxicity profiles observed in YAMC cells (Fig. 1) are consistent with the disparity in cell death events reported by many different groups over the years. Studies where TcdB was used at high picomolar to nanomolar concentrations report necrosis (34, 37–39), while subpicomolar intoxication results in an apoptotic event (30–33, 46). Notably, TcdB does not require p53 function at concentrations of ≥100 pM that result in necrotic cell death (Fig. 1A). We have previously shown that TcdB induces a glucosyltransferase-independent necrotic cell death as a result of aberrant ROS production through the NADPH oxidase (NOX) complex (34, 37). To further investigate the possibility that TcdB induces a concentration-dependent mechanism that switches from apoptosis to necrosis, we used cellular indicators to determine the death pathway activated at a given concentration. We observed that TcdA induces an apoptotic cell death at all concentrations (Fig. 3B) and the induction of apoptosis by TcdA is completely dependent on a fully active glucosyltransferase (Fig. 3B), and we interpret the LDH release at 24 h (Fig. 2D) to be the result of necrosis secondary to apoptosis. Most interestingly, we could observe two distinct cell death mechanisms occurring in response to TcdB. At higher concentrations (≥100 pM), we saw clear indications of a necrotic cell death, including ROS production (Fig. 3A), rapid LDH release (Fig. 2A), and minimal caspase-3/7 activation (Fig. 3C). At lower concentrations (≤10 pM), where there were no indications of necrosis, we saw a rise in caspase-3/7 activation (Fig. 3C), indicating apoptotic cell death. Also as clearly demonstrated in Fig. 3C, the activation of the apoptotic cell death pathway at lower concentrations of TcdB mirrors that seen at lower concentrations of TcdA, in that it requires glucosyltransferase activity. This is the first report of TcdB inducing a bimodal cell death mechanism, dependent upon the concentration of toxin, allowing the unification of the observations described by groups with seemingly opposing data.
We were able to extrapolate and confirm our findings in the colonic explant model. At a concentration of 10 nM toxin, TcdA induces glucosyltransferase-dependent apoptotic cell death, while TcdB induces the glucosyltransferase-independent production of ROS (Fig. 4). While our current explant model system does not allow observation for longer times, we anticipate that at lower concentrations (≤10 pM) both toxins will cause damage through a glucosyltransferase-dependent apoptotic mechanism.
The next question is, how can TcdA and TcdB cause cell death by different mechanisms? While differences in GTPase substrates between TcdA and TcdB have been reported, we would not expect substrate differences to account for differences in apoptosis and necrosis since the mechanism of necrosis is glucosyltransferase independent. Rather, we think that the most likely reason is that the toxins engage different receptors. While the human receptor for TcdA has yet to be identified, two receptors for TcdB have been described: poliovirus receptor-like protein 3 (PVRL3) and chondroitin sulfate proteoglycan 4 (CSPG4) (47, 48). PVRL3 seems to account for the cell death occurring at high concentrations in HeLa cells (where CSPG4 is also highly expressed), but it can mediate both necrotic and apoptotic mechanisms in Caco2 cells (47, 48). We hypothesize that TcdB binding to PVRL3 initiates the assembly and activation of the NOX complex and that the lack of PVRL3 binding by TcdA accounts for the differences in the cell death responses of the two toxins. What is unclear, however, is how PVRL3 can be involved in both apoptotic and necrotic mechanisms depending only on the TcdB concentration. This is a topic of ongoing investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Phil Williams in the Vanderbilt Light Surgical Laboratory for providing porcine tissue samples. We also thank Fang Yan (Vanderbilt) for the gift of YAMC cells and the Novel Cell Line Development Subcore.
Core services performed through Vanderbilt University Medical Center's Digestive Disease Research Center were supported by NIH grant P30DK058404.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00583-16.
REFERENCES
- 1.Voelker R. 2010. Increased Clostridium difficile virulence demands new treatment approach. JAMA 303:2017–2019. doi: 10.1001/jama.2010.647. [DOI] [PubMed] [Google Scholar]
- 2.Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N Engl J Med 359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 3.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly CP, Pothoulakis C, LaMont JT. 1994. Clostridium difficile colitis. N Engl J Med 330:257–262. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett JG, Gerding DN. 2008. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis 46(Suppl 1):S12–S18. doi: 10.1086/521863. [DOI] [PubMed] [Google Scholar]
- 6.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 8.Carter GP, Chakravorty A, Pham Nguyen TA, Mileto S, Schreiber F, Li L, Howarth P, Clare S, Cunningham B, Sambol SP, Cheknis A, Figueroa I, Johnson S, Gerding D, Rood JI, Dougan G, Lawley TD, Lyras D. 2015. Defining the roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile infections. mBio 6:e00551-15. doi: 10.1128/mBio.00551-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donta ST, Sullivan N, Wilkins TD. 1982. Differential effects of Clostridium difficile toxins on tissue-cultured cells. J Clin Microbiol 15:1157–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaves-Olarte E, Weidmann M, Eichel-Streiber C, Thelestam M. 1997. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest 100:1734–1741. doi: 10.1172/JCI119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell TJ, Ketley JM, Haslam SC, Stephen J, Burdon DW, Candy DC, Daniel R. 1986. Effect of toxin A and B of Clostridium difficile on rabbit ileum and colon. Gut 27:78–85. doi: 10.1136/gut.27.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyerly DM, Lockwood DE, Richardson SH, Wilkins TD. 1982. Biological activities of toxins A and B of Clostridium difficile. Infect Immun 35:1147–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. 1985. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 47:349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. 2003. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 125:413–420. doi: 10.1016/S0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 15.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT. 1995. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest 95:2004–2011. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aktories K. 1997. Bacterial toxins that target Rho proteins. J Clin Invest 99:827–829. doi: 10.1172/JCI119245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofmann F, Busch C, Prepens U, Just I, Aktories K. 1997. Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem 272:11074–11078. doi: 10.1074/jbc.272.17.11074. [DOI] [PubMed] [Google Scholar]
- 18.Rupnik M, Pabst S, Rupnik M, von Eichel-Streiber C, Urlaub H, Soling HD. 2005. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 151:199–208. doi: 10.1099/mic.0.27474-0. [DOI] [PubMed] [Google Scholar]
- 19.Just I, Fritz G, Aktories K, Giry M, Popoff MR, Boquet P, Hegenbarth S, von Eichel-Streiber C. 1994. Clostridium difficile toxin B acts on the GTP-binding protein Rho. J Biol Chem 269:10706–10712. [PubMed] [Google Scholar]
- 20.Aktories K, Just I. 1995. Monoglucosylation of low-molecular-mass GTP-binding Rho proteins by clostridial cytotoxins. Trends Cell Biol 5:441–443. doi: 10.1016/S0962-8924(00)89107-2. [DOI] [PubMed] [Google Scholar]
- 21.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. 1995. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 22.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 23.Giesemann T, Egerer M, Jank T, Aktories K. 2008. Processing of Clostridium difficile toxins. J Med Microbiol 57:690–696. doi: 10.1099/jmm.0.47742-0. [DOI] [PubMed] [Google Scholar]
- 24.Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95:1026–1031. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brito GA, Carneiro-Filho B, Oria RB, Destura RV, Lima AA, Guerrant RL. 2005. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: role of Gln and Ala-Gln in toxin A effects. Dig Dis Sci 50:1271–1278. doi: 10.1007/s10620-005-2771-x. [DOI] [PubMed] [Google Scholar]
- 26.Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. 2002. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis 186:1438–1447. doi: 10.1086/344729. [DOI] [PubMed] [Google Scholar]
- 27.Carneiro BA, Fujii J, Brito GA, Alcantara C, Oria RB, Lima AA, Obrig T, Guerrant RL. 2006. Caspase and Bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect Immun 74:81–87. doi: 10.1128/IAI.74.1.81-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matte I, Lane D, Cote E, Asselin AE, Fortier LC, Asselin C, Piche A. 2009. Antiapoptotic proteins Bcl-2 and Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect Immun 77:5400–5410. doi: 10.1128/IAI.00485-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerhard R, Nottrott S, Schoentaube J, Tatge H, Olling A, Just I. 2008. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J Med Microbiol 57:765–770. doi: 10.1099/jmm.0.47769-0. [DOI] [PubMed] [Google Scholar]
- 30.Lica M, Schulz F, Schelle I, May M, Just I, Genth H. 2011. Difference in the biological effects of Clostridium difficile toxin B in proliferating and non-proliferating cells. Naunyn Schmiedebergs Arch Pharmacol 383:275–283. doi: 10.1007/s00210-010-0595-5. [DOI] [PubMed] [Google Scholar]
- 31.Qa'Dan M, Ramsey M, Daniel J, Spyres LM, Safiejko-Mroczka B, Ortiz-Leduc W, Ballard JD. 2002. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol 4:425–434. doi: 10.1046/j.1462-5822.2002.00201.x. [DOI] [PubMed] [Google Scholar]
- 32.Fiorentini C, Fabbri A, Falzano L, Fattorossi A, Matarrese P, Rivabene R, Donelli G. 1998. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect Immun 66:2660–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huelsenbeck J, Dreger S, Gerhard R, Barth H, Just I, Genth H. 2007. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect Immun 75:801–809. doi: 10.1128/IAI.01705-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Lacy DB. 2012. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nottrott S, Schoentaube J, Genth H, Just I, Gerhard R. 2007. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis 12:1443–1453. doi: 10.1007/s10495-007-0074-8. [DOI] [PubMed] [Google Scholar]
- 36.Kreimeyer I, Euler F, Marckscheffel A, Tatge H, Pich A, Olling A, Schwarz J, Just I, Gerhard R. 2011. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn Schmiedebergs Arch Pharmacol 383:253–262. doi: 10.1007/s00210-010-0574-x. [DOI] [PubMed] [Google Scholar]
- 37.Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. 2013. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110:18674–18679. doi: 10.1073/pnas.1313658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Park M, Tam J, Auger A, Beilhartz GL, Lacy DB, Melnyk RA. 2014. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci U S A 111:3721–3726. doi: 10.1073/pnas.1400680111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D'Auria KM, Bloom MJ, Reyes Y, Gray MC, van Opstal EJ, Papin JA, Hewlett EL. 2015. High temporal resolution of glucosyltransferase dependent and independent effects of Clostridium difficile toxins across multiple cell types. BMC Microbiol 15:7. doi: 10.1186/s12866-015-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wohlan K, Goy S, Olling A, Srivaratharajan S, Tatge H, Genth H, Gerhard R. 2014. Pyknotic cell death induced by Clostridium difficile TcdB: chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol 16:1678–1692. doi: 10.1111/cmi.12317. [DOI] [PubMed] [Google Scholar]
- 41.Whitehead RH, Joseph JL. 1994. Derivation of conditionally immortalized cell lines containing the Min mutation from the normal colonic mucosa and other tissues of an “Immortomouse”/Min hybrid. Epithelial Cell Biol 3:119–125. [PubMed] [Google Scholar]
- 42.Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB. 2010. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107:13467–13472. doi: 10.1073/pnas.1002199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.An P, Saenz Robles MT, Pipas JM. 2012. Large T antigens of polyomaviruses: amazing molecular machines. Annu Rev Microbiol 66:213–236. doi: 10.1146/annurev-micro-092611-150154. [DOI] [PubMed] [Google Scholar]
- 44.Chumbler NM, Rutherford SA, Zhang Z, Farrow MA, Lisher JP, Farquhar E, Giedroc DP, Spiller BW, Melnyk RA, Lacy DB. 2016. Crystal structure of Clostridium difficile toxin A. Nat Microbiol 1:15002. doi: 10.1038/nmicrobiol.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim H, Kokkotou E, Na X, Rhee SH, Moyer MP, Pothoulakis C, Lamont JT. 2005. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 129:1875–1888. doi: 10.1053/j.gastro.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 46.Matarrese P, Falzano L, Fabbri A, Gambardella L, Frank C, Geny B, Popoff MR, Malorni W, Fiorentini C. 2007. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J Biol Chem 282:9029–9041. doi: 10.1074/jbc.M607614200. [DOI] [PubMed] [Google Scholar]
- 47.LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB. 2015. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc Natl Acad Sci U S A 112:7073–7078. doi: 10.1073/pnas.1500791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, He R, Li C, Guo S, Li S, Huang T, Perez-Cordon G, Feng H, Wei W. 2015. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25:157–168. doi: 10.1038/cr.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.