

Abstract
Intestinal segmented filamentous bacteria (SFB) protect from ameba infection, and protection is transferable with bone marrow dendritic cells (BMDCs). SFB cause an increase in serum amyloid A (SAA), suggesting that SAA might mediate SFB's effects on BMDCs. Here we further explored the role of bone marrow in SFB-mediated protection. Transient gut colonization with SFB or SAA administration alone transiently increased the H3K27 histone demethylase Jmjd3, persistently increased bone marrow Csf2ra expression and granulocyte monocyte precursors (GMPs), and protected from ameba infection. Pharmacologic inhibition of Jmjd3 H3K27 demethylase activity during SAA treatment or blockade of granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling in SFB-colonized mice prevented GMP expansion, decreased gut neutrophils, and blocked protection from ameba infection. These results indicate that alteration of the microbiota and systemic exposure to SAA can influence myelopoiesis and susceptibility to amebiasis via epigenetic mechanisms. Gut microbiota-marrow communication is a previously unrecognized mechanism of innate protection from infection.
INTRODUCTION
Several studies have suggested that intestinal infection with one organism may persistently alter innate immune populations to provide protection from infection with unrelated pathogens (1–3). This idea has been referred to as innate trained immunity (4). However, the mechanism of how unrelated organisms might generate this protective yet nonspecific memory is not currently well described. Some studies have suggested that epigenetic modification of inflammatory genes in innate immune cells might underlie this effect (5, 6). Recent studies have also suggested that the microbiome might have long-term epigenetic effects on the immune system (7). Indeed, microbiota-produced serum soluble mediators and pathogen-associated molecular patterns (PAMPs) can impact myelopoiesis and hematopoiesis (8–10). We hypothesize that host-derived factors such as damage-associated molecular patterns (DAMPs) that are systemically induced by the microbiota may also have a role in altering hematopoiesis and susceptibility to infection.
We have previously demonstrated that alteration of the microbiota via introduction of segmented filamentous bacteria (SFB) (50) can alter bone marrow-derived cells and protect from infection with the protozoan parasite Entamoeba histolytica (11). This protection was associated with increased gut neutrophils following amebic infection. This suggested that alteration of the microbiota might have a persistent influence on the bone marrow and myelopoiesis. To address this possibility further, we explored changes in bone marrow hematopoiesis during introduction of SFB to the gut microbiota. Serum amyloid A (SAA), a DAMP, was upregulated during SFB colonization (11, 12) and is known to induce Jmjd3 (13), an epigenetic mediator and H3K27 demethylase linked to inflammation (14). SAA induced by the microbiota has also been shown to be important in neutrophil infiltration in a nonmammalian model (15). Hematopoiesis and myelopoiesis have been shown to be influenced by granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling (16). Therefore, we also assessed the ability of SAA, Jmjd3, and GM-CSF signaling to alter bone marrow hematopoiesis and susceptibility to subsequent amebic colitis.
Here we demonstrate that addition of SFB to the gut induced persistent expansion of bone marrow granulocyte monocyte precursors (GMPs) and an increase in GM-CSF alpha receptor gene (Csf2ra) expression, which persisted after SFB infection was cleared. Blockade of GM-CSF signaling in SFB-colonized mice prevented GMP expansion as well as gut neutrophils and mucosal protection from E. histolytica. Treatment of mice with SAA led to similar increases in Jmjd3 and Csf2ra and expansion of bone marrow GMPs, as well as protection from E. histolytica infection. This protection was associated with increased neutrophils. Inhibition of H3K27 demethylase (Jmjd3) activity during SAA treatment, both in vivo and in vitro, prevented these effects and reversed the phenotype of protection from the ameba. These results suggest that alteration of the microbiota and systemic exposure to SAA can epigenetically alter myelopoiesis and susceptibility to amebiasis via alteration of GM-CSF signaling.
MATERIALS AND METHODS
Mice.
Five-week-old male CBA/J mice (Jackson Laboratories), which are susceptible to E. histolytica infection (17), were housed in a specific-pathogen-free facility in microisolator cages and provided autoclaved food (lab diet 5010) and water ad libitum. Specific-pathogen-free status was monitored quarterly. A sentinel mouse was removed from each room and was humanely euthanized for serologic evaluation, examination of pelage for fur mites, and examination of cecal contents for pinworms. The serologic assays, conducted in-house using CRL reagents, were for mouse hepatitis virus (MHV), epizootic diarrhea of infant mice (EDIM) virus, GD-7, minute virus of mice (MVM), mouse parvovirus (MPV), murine norovirus (MNV), Sendai virus, pneumonia virus of mice (PVM), RPV/Kilham's rat virus (KRV)/H-1, and Mycoplasma pulmonis. In the final quarter, a comprehensive serologic examination was performed, which included the above-mentioned agents plus K virus, mouse cytomegalovirus (MCMV), mouse thymic virus (MTV), lymphocytic choriomeningitis (LCM) virus, ectromelia virus, polyomavirus, reovirus 3, and mouse adenoviruses (K87 and FL). All procedures were approved by the Institutional Animal Care and Use Committee of the University of Virginia.
SFB colonization.
Mice were colonized with SFB by a single oro-gastric gavage with SFB-monoassociated feces at 5 weeks of age (monoassociated feces was a gift from Yakult, Tokyo, Japan). SFB-monoassociated feces was independently confirmed to be monoassociated with SFB by quantitative PCR (qPCR), Sanger sequencing, and culture in anaerobic and aerobic media for the presence of non-SFB bacteria (11).
GM-CSF blockade.
Mice were treated with 400 μg of anti-GM-CSF antibody (Bioxcell, BE0259) or an isotype control antibody (cMAB; Bioxcell, BE0089) 1 day before and 12 days after SFB colonization and then were challenged with E. histolytica trophozoites on day 21.
In vitro and in vivo SAA and Jmjd3 (H3K27 demethylase) inhibitor treatment.
Bone marrow cells were isolated as previously described (18). Five hundred thousand mouse bone marrow cells were treated for 16 h with 200 μl of growth medium (RPMI 1640, 5% fetal bovine serum [FBS], 1% penicillin-streptomycin; Sigma) or medium containing ultrapure lipopolysaccharide (LPS) (10 ng/ml; InvivoGen), Pam3CSK4 (10 ng/ml; InvivoGen), SAA (10 μg; PeproTech, 300-13), or SAA with GSK-J5 or GSK-J4 (2.5 mg; Cayman Chemical, 12073 and 12074) and then harvested in RNAlater (Ambion, Austin, TX). Mice were treated intraperitoneally on days 1, 3, and 5 with 200 μl of PBS, SAA (15 μg; PeproTech, 300-13), or SAA with GSK-J5 or GSK-J4 (100 mg/kg; Cayman Chemical, 12073 and 12074) before E. histolytica infection on day 14.
E. histolytica culture and intracecal injection.
Animal-passaged HM1:IMSS E. histolytica trophozoites were cultured from cecal contents of infected mice in complete trypsin-yeast-iron (TYI-33) medium supplemented with Diamond vitamin mixture (JRH Biosciences), 100 U/ml of both penicillin and streptomycin, and 5% heat-inactivated bovine serum (Sigma-Aldrich). Prior to injection, trophozoites were grown to log phase, and 2 ×106 parasites were suspended in 150 μl culture medium and injected intracecally (11).
Quantitative real-time reverse transcription-PCR.
SFB and E. histolytica colonization and mouse gene expression were measured by real-time PCR in stool and cecal lysate. For SFB colonization, qPCR with Sybr green was performed, and data were normalized to expression of a conserved eubacterial 16S RNA gene (EUB) (19). Primer concentrations, annealing temperatures, and cycle numbers were optimized for each primer pair. For each primer pair (Table 1), a dilution curve of a positive cDNA sample was included to enable calculation of the efficiency of the amplification. The relative message levels of each target gene were then normalized to that of the mouse housekeeping gene S14 using a method described and utilized previously (11, 20, 21). Data are presented as relative expression. For E. histolytica quantification, qPCR with a labeled probe and a standard curve of trophozoites (22) was utilized. Data are presented as trophozoites per microliter of cecal lysate. Primers and probes were purchased from Integrated DNA Technologies, Coralville, IA, USA.
TABLE 1.
Primer sequences
| Primer | Sequence |
|---|---|
| EUB forward | 5′-ACTCCTACGGGAGGCAGCAGT-3′ |
| EUB reverse | 5′-ATTACCGCGGCTGCTGGC-3′ |
| SFB forward | 5′-GACGCTGAGGCATGAGAGCAT-3′ |
| SFB reverse | 5′-GACGGCACGGATTGTTATTCA-3′ |
| E. histolytica probe | 5′-YYTATTAGTACAAAATGGCCAATTCATTCA-Dark Quencher-3′ |
| E. histolytica forward | 5′-AACAGTAATAGTTTCTTTGGTTAGTAAAA-3′ |
| E. histolytica reverse | 5′-CTTAGAATGTCATTTCTCAATTCAT-3′ |
| S14 forward | 5′-TGGTGTCTGCCACATCTTTGCATC-3′ |
| S14 reverse | 5′-AGTCACTCGGCAGATGGTTTCCTT-3′ |
| Jmjd3 forward | 5′-CTCTGGAACTTTCATGCCGG-3′ |
| Jmjd3 reverse | 5′-CTTAGCCCCATAGTTCCGTTTG-3′ |
| Gm-csf forward | 5′-ACCACCTATGCGGATTTCAT-3′ |
| Gm-csf reverse | 5′-TCATTACGCAGGCACAAAAG-3′ |
| Csf2ra forward | 5′-GCTGGTTCAGGAGGATGATG-3′ |
| Csf2ra reverse | 5′-CTTTCGTTGACGAAGCTCAG-3′ |
| Csf2rb forward | 5′-CTGTCGCCCAAGCACAGA-3′ |
| Csf2rb forward | 5′-ATTGACCCGGGGTTCTGTAT-3′ |
Colony formation assay for determination of bone marrow hematopoietic precursors.
Bone marrow cells were isolated (18) and then cultured in methylcellulose-based medium that included 3 units/ml erythropoietin (Epo), 10 ng/ml mouse recombinant interleukin-3 (IL-3), 10 ng/ml human recombinant IL-6, and 50 ng/ml mouse recombinant stem cell factor per the manufacturer's procedures (M3434; StemCell Technologies, Vancouver, British Columbia, Canada). Colony formation of burst-forming unit erythroid cells (BFU-Es), CFU granulocytes/monocytes (CFU-GMs), and CFU granulocytes/erythrocytes/monocytes/macrophages (CFU-GEMMs) was analyzed after 7 days.
Determination of total H3K27me3 in bone marrow.
An EpiQuik global histone H3K27 methylation assay kit (Epigentek, P-3020-48) was utilized to quantify total H3K27me3 in bone marrow from mice that had been treated with SAA and Jmjd3 inhibitors as described above.
Flow cytometry of intestinal cells.
Minced intestinal tissue was digested in Liberase TL (0.17 mg/ml; Roche) and DNase (0.5 mg/ml; Sigma) for 45 min at 37°C and processed into a single-cell suspension following washing with a buffer containing EDTA. A total of 1 × 106 cells per mouse were stained with antibodies from BioLegend (CD11c-BV421, CD4-BV605, Ly6c-fluorescein isothiocyanate [FITC], CD3e-peridinin chlorophyll protein [PerCP]-Cy 5.5, Siglec F-phycoerythrin [PE], Ly6G-PE-Cy7, CD11b-allophycocyanin [APC], CD8a-AF700, and CD45-APC-Cy7). Flow cytometric analysis was performed on an LSR Fortessa (BD Biosciences) and data analyzed via FlowJo (Tree Star Inc.). All gates were set based on fluorescence-minus-one (FMO) controls.
Statistical analysis.
Analysis of variance (ANOVA) followed by the Tukey-Kramer test was used for analysis of differences among multiple groups. Student's t test was used for comparisons between two groups. P values of less than 0.05 were considered significant. Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). All results are representative of at least two independent replicates.
RESULTS
Alteration of bone marrow granulocyte monocyte precursors persists after clearance of introduced commensal bacteria.
Prior work has suggested that the presence of segmented filamentous bacteria (SFB) in the gut can cause bone marrow changes that may mediate protection against subsequent infection with E. histolytica (11). In order to determine if alteration of the microbiota via SFB colonization would have a persistent influence on hematopoietic precursors in the bone marrow, we colonized mice with SFB at 5 weeks of age and followed colonization until SFB were no longer shed at 6 months of age (Fig. 1A). We then waited until 2 months after the last positive stool, at 8 months of age, to measure bone marrow precursor cells. Clearance of SFB in the ileum was also confirmed via Gram staining and qPCR upon euthanasia (not shown). Colonization of mice with SFB increased the CFU of GMPs in the bone marrow (Fig. 1B), and this increase in GMPs remained after the SFB were cleared (Fig. 1B). Expansion of GMPs was confirmed via flow cytometry (see Fig. S1A and B in the supplemental material). We concluded that SFB resulted in a persistent increase in GMPs.
FIG 1.

Expansion of bone marrow granulocyte monocyte precursors in mice with an altered microbiota persists after the introduced bacteria are cleared. (A) Male CBA/J mice were colonized with SFB at 5 weeks of age and followed until colonization was cleared. (B) Bone marrow precursors, including granulocyte-monocyte progenitors (CFU-GM), erythroid progenitors (BFU-E), and multipotential granulocyte, erythroid, macrophage, and megakaryocyte progenitors (CFU-GEMM), were measured during acute SFB colonization, 2 weeks after SFB treatment (7 weeks of age), and 2 months after SFB was cleared (8 months of age). (C) Serum levels of SAA were also measured at these time points. (D and E) Bone marrow expression of Jmjd3 (D) and Csf2ra (E) was also determined at these time points. Results are means ± standard errors of the means (SEM) from a representative experiment of two independent experiments with five mice per group. *, P < 0.05.
Alteration of the gut microbiota systemically increases SAA levels as well as bone marrow Jmjd3 and Csf2ra expression.
Systemic DAMP signaling can alter hematopoiesis and may have long-term effects on myeloid lineages via epigenetic mechanisms (10, 23, 24). We thus hypothesized that a serum soluble mediator that was increased during colonization with SFB might correlate with changes in the bone marrow that could support increased GMP expansion. In confirmation of our previously published data (11) we observed elevated serum amyloid A (SAA) levels in the sera of SFB-colonized mice (Fig. 1C); however, this increase did not persist after colonization was cleared (Fig. 1C). We also observed significantly, but transiently, elevated Jmjd3 expression in the bone marrow of SFB-colonized mice (Fig. 1D) There was no difference in Toll-like receptor 2 (TLR2), TLR4, M-CSF (Csf1), or M-CSF receptor (M-CSFR) (Csf1r) expression in the bone marrow at either time point (data not shown), factors which have been shown to have a role in regulating hematopoiesis (9, 25). GM-CSF is also important in hematopoiesis and generation of GMPs (26–28). Therefore, to assess the possible contribution of GM-CSF in this model, expression of GM-CSF and the receptor subunits Csf2ra and Csf2rb was measured (29). The GM-CSF alpha receptor gene (Csf2ra) was upregulated in the bone marrow of SFB-colonized mice, as well as after SFB were cleared (Fig. 1E). We concluded that SFB colonization resulted in transient increases in SAA and bone marrow Jmjd3 expression and a longer-term increase in Csf2ra expression.
Inhibition of GM-CSF signaling blocks both microbiota-mediated alteration of myelopoiesis and protection from ameba infection.
Antibody neutralization of GM-CSF in SFB-colonized mice prevented GMP expansion (Fig. 2A), subsequent increases in intestinal neutrophils (Fig. 2B), and mucosal protection from amebic infection (Fig. 2C). We concluded that GM-CSF signaling mediated SFB-induced GMP expansion and downstream protection from E. histolytica.
FIG 2.

GM-CSF signaling is necessary for microbiota-mediated expansion of GMPs and protection from amebiasis. Mice were treated with anti-GM-CSF antibody or an isotype control antibody 1 day before and 12 days after they were colonized with SFB or treated with PBS and then challenged with E. histolytica trophozoites on day 21. (A) A CFU assay was utilized to determine the composition of hematopoietic precursors in the bone marrow of SFB colonized mice. (B and C) Intestinal neutrophils (B) and amebic burden (C) in the cecum were determined after 6 days via flow cytometry and qPCR. Results are Means ± SEM from a representative experiment of two independent experiments with 12 mice per group. *, P < 0.05.
SAA increases bone marrow cell Csf2ra expression in a Jmjd3-dependent manner.
We postulated that exposure of bone marrow cells to increased SAA might increase Csf2ra expression in a Jmjd3-dependent manner. SAA treatment of bone marrow cells in culture increased Jmjd3 expression and Csf2ra expression (Fig. 3A). Blockade of Jmjd3 activity with an inhibitor of H3K27 demethylase activity, GSK-J4, but not the inactive control, GSK-J5, (30, 31) reversed the increase in Csf2ra expression (Fig. 3B). Treatment of bone marrow cells with TLR2 and TLR4 agonists (pam3csk4 and LPS) did not have a significant effect on Csf2ra or Jmjd3 expression at this time point, suggesting that the effect was not due to PAMPs potentially contaminating the SAA preparation.
FIG 3.
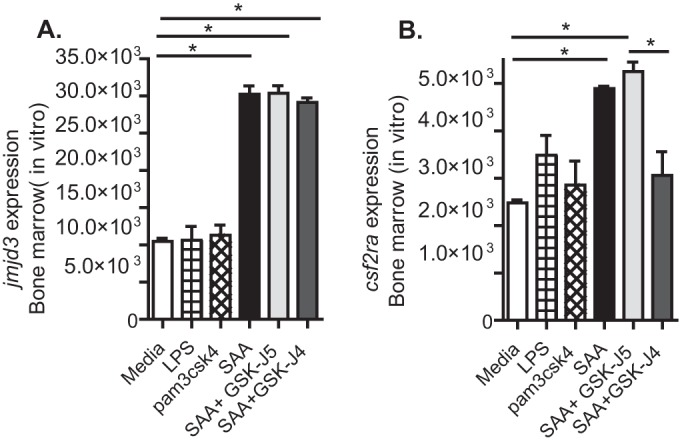
Serum amyloid A (SAA) increases isolated bone marrow cell Csf2ra expression in a Jmjd3-dependent manner. Isolated total bone marrow cells in growth medium were treated with recombinant SAA with or without an inhibitor of Jmjd3, GSK-J4, or its inactive control GSK-J5. Cells were collected at 16 h posttreatment and Jmjd3 (A) and Csf2ra (B) expression determined. Results are means ± SEM from a representative experiment of three independent experiments with 3 mice per group. *, P < 0.05.
Treatment of mice with SAA increases bone marrow Csf2ra expression and alters hematopoiesis in an H3K27 demethylase-dependent manner.
Because SAA increased Jmjd3 and Csf2ra expression in bone marrow cells, we hypothesized that treatment of mice with SAA might alter hematopoiesis in an H3K27 demethylase-dependent manner. Treatment of mice with recombinant SAA transiently and significantly increased serum levels of SAA and bone marrow expression of the H3K27 demethylase Jmjd3 (Fig. 4A and B). SAA treatment decreased total bone marrow H3K27me3 and increased Csf2ra expression in a Jmjd3-dependent manner (Fig. 4C and D, day 14). Treatment of mice with SAA also caused expansion of GMPs, which was reversed with the Jmjd3 inhibitor (Fig. 4E, day 14; see Fig. S1C in the supplemental material). We concluded that SAA treatment in vivo increased bone marrow Csf2ra expression and GMP expansion in an H3K27 demethylase-dependent manner.
FIG 4.

Treatment of mice with serum amyloid A (SAA) increases bone marrow Csf2ra expression and granulocyte-monocyte precursors in an H3K27 demethylase-dependent manner. Mice were injected intraperitoneally with recombinant SAA (days 1, 3, and 5) with or without an inhibitor of Jmjd3, GSK-J4, or its inactive control GSK-J5. (A and B) Serum and bone marrow were collected at days 7 and 14, and SAA and Jmjd3 expression were measured. (C to E) Bone marrow was collected at day 14, and total H3K27me3 (C), Csf2ra (D), and bone marrow precursors (E) were determined. Results are means ± SEM from a representative experiment of three independent experiments with 3 (A and B) or 5 (C to E) mice per group. *, P < 0.05.
SAA provides protection from E. histolytica infection in an H3K27 demethylase-dependent manner.
We hypothesized that SAA would protect from E. histolytica infection as it increased GMP expansion in an H3K27 demethylase-dependent manner. Bone marrow GMPs remained elevated in SAA-treated mice that were not administered a Jmjd3 inhibitor after ameba infection (Fig. 5A). There was no significant change in intestinal expression of IL-17A (Fig. 5B), IL-23 (Fig. 5C), or IL-22 (Fig. 5D) or a significant difference in CD4+ T cells in SAA-treated mice (Fig. 5E). As neutrophils arise from bone marrow GMPs (32), we postulated that they would be increased in the intestine following SAA administration and E. histolytica infection. An increase in neutrophil frequency (Fig. 5F) and number (data not shown) in the cecal lamina propria in SAA-treated mice was observed during, but not prior to, challenge with E. histolytica. There was no increase in eosinophils or inflammatory monocytes, which also arise from GMPs (32), at either time point (not shown). SAA administration prior to infection provided protection from E. histolytica infection, and this protection, along with increased gut neutrophils and bone marrow GMPs, was dependent on H3K27 demethylase activity (Fig. 5A, F, and G, day 21). We concluded that SAA provided protection from E. histolytica in an H3K27 demethylase-dependent manner.
FIG 5.

Serum amyloid A (SAA) provides protection from E. histolytica infection in an H3K27 demethylase-dependent manner. Mice were injected intraperitoneally with recombinant SAA (days 1, 3, 5) with or without an inhibitor of Jmjd3, GSK-J4, or its inactive control GSK-J5. Mice were then infected with E. histolytica 9 days after the final injection of SAA (day 14) and sacrificed 7 days later (day 21). (A) Bone marrow was collected at day 21, and bone marrow precursors were determined. (B to G) Intestinal expression of il17a (B) Il23p19 (C), and Il22 (D), CD4+ T cells (E), neutrophils (F), and infection burden (G) were measured (day 21). Results are means ± SEM from a representative experiment of three independent experiments with 5 (A) or 12 (B to G) mice per group. *, P < 0.05.
DISCUSSION
The key finding of this study is that addition of SFB to the gut microbiota epigenetically induced an increase in GM-CSF alpha receptor expression and expansion of bone marrow GMPs, which persisted after SFB infection was cleared. Blockade of GM-CSF signaling in SFB-colonized mice prevented GMP expansion as well as gut neutrophils and mucosal protection from E. histolytica. Therefore, microbiota-marrow signaling via GM-CSF resulted in GMP expansion, gut neutrophil infiltration, and protection from enteric infection.
In young mice after weaning, SFB colonization is known to follow distinct kinetics of colonization, with intestinal levels of the bacteria increasing dramatically shortly after birth and then decreasing significantly as the mice age (33). In adult mice, we also observed a short-term period of high levels of SFB colonization, followed by a longer period of lower-level colonization, followed by a cessation of shedding. As GMPs remained elevated even after clearance of SFB, we hypothesized that transient intestinal SFB colonization may have induced epigenetic changes that influenced bone marrow hematopoiesis. Consistent with this hypothesis, we observed increased expression of Jmjd3, encoding an H3K27 demethylase, in the bone marrow of SFB-colonized mice during acute colonization. Our studies have not yet identified the precise mechanism by which SFB colonization increases bone marrow GMPs, which may be multifactorial in nature. Previous studies have linked changes in M-CSF to alteration of hematopoiesis and protection from infection (34). We did not observe changes in M-CSF cytokine or receptor (Csf1r) expression in the bone marrow with SFB treatment. This was not entirely unexpected, however, as it has been shown that Jmjd3 has little effect on M-CSF-mediated inflammatory monocyte generation (35). However, GM-CSF signaling is also known to be important in hematopoiesis and generation of GMPs, and the magnitude and duration of signaling influence outcomes, with weak signaling favoring cell survival and stronger signaling promoting both survival and proliferation (26–28). The GM-CSF alpha receptor gene (Csf2ra) was upregulated in the bone marrow of acutely SFB colonized mice, as well as after SFB was cleared. Blockade of GM-CSF during SFB colonization prevented GMP expansion, increased intestinal neutrophils, and protection from ameba infection. These data demonstrated that GM-CSF signaling during SFB colonization is important in protection from amebiasis.
In addition to GM-CSF signaling, we explored the potential role of SAA in SFB-induced GMP expansion. Our laboratory has previously observed that SFB colonization can increase serum levels of SAA (11), but this had not been explored beyond acute colonization with the bacteria. During acute colonization with SFB, serum levels of the DAMP SAA were significantly increased, as was bone marrow expression of Jmjd3, encoding an H3K27 demethylase (36). However, SAA and Jmjd3 did not remain elevated after SFB was cleared. From these data, we surmised that transiently increased serum SAA levels might have influenced bone marrow GMP expansion in an epigenetic manner. Treatment of mice with SAA was able to momentarily increase serum levels of SAA to a magnitude comparable to that observed during SFB colonization. SAA administration, both in vitro and in vivo, increased expression of the GM-CSF alpha receptor gene (Csf2ra) in bone marrow in a manner similar to that observed during SFB colonization. SAA treatment led to a significant decrease in bone marrow H3K27me3 levels and expansion of GMPs. Blockade of Jmjd3 activity in SAA-treated mice prevented changes in H3K27me3 levels, Csf2ra expression, and GMP expansion. Hematopoiesis, including expansion of GMPs and their differentiation into myeloid subsets, is known to be associated with global changes in histone methylation (24). Our results demonstrated that SAA treatment specifically could increase bone marrow GMPs in an H3K27 demethylase-dependent manner. SAA treatment therefore skewed bone marrow development toward the generation of myeloid-derived cells, which might at least partially explain how SFB in the gut resulted in expansion of GMPs. Inflammatory stimuli induced by E. histolytica infection alone may also induce epigenetic changes in the bone marrow, and this possibility will be examined in future studies.
SAA administration prior to ameba infection provided protection from amebiasis. Protection was associated with increased intestinal neutrophils following ameba infection. There was no increase in eosinophils or inflammatory monocytes, which can also arise from GMPs (32), either before or after ameba infection. This protection, along with intestinal neutrophil infiltration during infection, was dependent on H3K27 demethylase activity. The lack of change in inflammatory monocytes was somewhat expected, as Jmjd3 has been shown to be dispensable for inflammatory monocyte generation (35). It is not yet clear why eosinophils were unaffected. However, as they have a unique precursor population downstream of GMPs, they might behave differently than neutrophils (37). We previously have demonstrated that neutrophil infiltration is protective during E. histolytica infection in murine models (38) and that increased neutrophil infiltration correlated with protection during SFB colonization (11). The observed increase in intestinal neutrophils after SAA treatment and ameba infection is consistent with these studies. This increase in neutrophils during ameba infection and following SAA treatment is likely a consequence of the expanded bone marrow GMPs. Blockade of H3K27 demethylase activity prevented both SAA-mediated GMP expansion and increases in intestinal neutrophils. This suggests that systemic exposure to SAA can influence myelopoiesis and susceptibility to amebiasis, potentially via epigenetic mechanisms. However, the specific cause of increased intestinal neutrophils during SAA treatment is not yet known.
Our previous work suggested that IL-17A induction after adoptive transfer of bone marrow dendritic cells (BMDCs) from SFB-colonized mice may be important in induction of neutrophils and protection from amebiasis (11). It has also been demonstrated that SAA administration can induce a Th17 response and increased neutrophilia in the lung (39). It has further been recognized that SAA-mediated Th17 induction during SFB colonization is most robust in the small intestine and that Th17 responses are SFB specific (40–43). IL-22, associated with Th17 induction, has also recently been shown to provide colonization resistance against intra-abdominal bacterial infection and to induce SAA (44). In this study, we did not observe a significant increase in IL-17A, IL-23, IL-22, or CD4+ T cells with SAA treatment. This suggests that SAA administration does not completely recapitulate the effect on the intestinal mucosal that was observed during SFB administration. Alteration of the microbiota during SFB administration may also contribute to protection, and this possibility will be examined in future studies. Recent studies have also shown that neutrophilia can occur independently of IL-17A induction in a fungal infection model (45). Therefore, while IL-17A may be important in neutrophil infiltration to mucosal sites during infection with E. histolytica (46), it is not necessarily the sole factor governing it.
In conclusion, these data reveal key details of host-pathogen interaction. Alteration of the microbiota via introduction of SFB can have a persistent effect on hematopoiesis and GM-CSF signaling. GM-CSF signaling is also needed for microbiota-mediated expansion of myeloid precursor cells and protection from ameba infection. These data further demonstrate that a host DAMP (SAA) can, independently of SFB colonization, have epigenetic mediator (Jmjd3)-dependent and GM-CSF-associated effects on myelopoiesis and thereby provide protection from amebiasis. To our knowledge, this is the first demonstration that a systemic, yet transient, increase in a host-derived DAMP can alter bone marrow hematopoiesis, GM-CSF signaling, and susceptibility to parasitic infection. SAA and GM-CSF may therefore provide useful targets to explore in the treatment of intestinal parasitic infection. Future studies will determine if commensal clostridia found in populations exposed to E. histolytica can alter the progression of amebiasis and will explore which time points are key for protection. The ameba burden may significantly influence the long-term sequelae of amebic colitis and the progression from asymptomatic (47) to fulminant colitis in patients. Increased E. histolytica burden has also been shown to be associated with diarrhea in children (48). Therefore, a decreased ameba burden caused by a component of the microbiota may have a significant influence on amebic colitis in patients.
These observations may have broader implications for the field of immunology and infectious disease. SAA is upregulated in the serum during many infectious and inflammatory diseases (49). We suspect that this effect is generalizable and that increased serum SAA via many mechanisms might lead to more robust mucosal immune responses to subsequent infectious or inflammatory challenges. Therefore, mechanisms observed in the amebiasis model might help to better explain the newly emerging field of innate trained immunity (4), as well as the poorly understood, but widespread, observation of postinfectious chronic gut dysfunction (50).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH grant 5R01 AI-026649.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00316-16.
REFERENCES
- 1.Netea MG, Quintin J, van der Meer JWM. 2011. Trained immunity: a memory for innate host defense. Cell Host Microbe 9:355–361. doi: 10.1016/j.chom.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg B-J, Wijmenga C, Joosten LAB, Xavier RJ, van der Meer JWM, Stunnenberg HG, Netea MG. 2012. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. 2014. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 15:374–381. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, ONeill LAJ, Xavier RJ. 22 April 2016. Trained immunity: a program of innate immune memory in health and disease. Science doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, Cheng S-C, Ratter J, Berentsen K, van der Ent MA, Sharifi N, Janssen-Megens EM, Ter Huurne M, Mandoli A, van Schaik T, Ng A, Burden F, Downes K, Frontini M, Kumar V, Giamarellos-Bourboulis EJ, Ouwehand WH, van der Meer JWM, Joosten LAB, Wijmenga C, Martens JHA, Xavier RJ, Logie C, Netea MG, Stunnenberg HG. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quintin J, Cheng S-C, van der Meer JW, Netea MG. 2014. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol 29C:1–7. doi: 10.1016/j.coi.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Obata Y, Furusawa Y, Hase K. 2015. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol 93:226–232. doi: 10.1038/icb.2014.114. [DOI] [PubMed] [Google Scholar]
- 8.Balmer ML, Schurch CM, Saito Y, Geuking MB, Li H, Cuenca M, Kovtonyuk LV, McCoy KD, Hapfelmeier S, Ochsenbein AF, Manz MG, Slack E, Macpherson AJ. 2014. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol 193:5273–5283. doi: 10.4049/jimmunol.1400762. [DOI] [PubMed] [Google Scholar]
- 9.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. 2006. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu A, Wang Y, Ding Y, Baez I, Payne KJ, Borghesi L. 2015. Hematopoietic stem cell expansion and common lymphoid progenitor depletion require hematopoietic-derived, cell-autonomous TLR4 in a model of chronic endotoxin. J Immunol 195:2524–2528. doi: 10.4049/jimmunol.1501231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgess SL, Buonomo E, Carey M, Cowardin C, Naylor C, Noor Z, Wills-Karp M, Petri WA. 2014. Bone marrow dendritic cells from mice with an altered microbiota provide interleukin 17A-dependent protection against Entamoeba histolytica colitis. mBio 5:e01817-14. doi: 10.1128/mBio.01817-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, Testa G, Sung W-K, Wei C-L, Natoli G. 2009. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J 28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Q, Sun L, Zhu Z, Wang L, Li S, Ye RD. 2014. Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell Signal 26:1783–1791. doi: 10.1016/j.cellsig.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanther M, Tomkovich S, Xiaolun S, Grosser MR, Koo J, Flynn EJ, Jobin C, Rawls JF. 2014. Commensal microbiota stimulate systemic neutrophil migration through induction of serum amyloid A. Cell Microbiol 16:1053–1067. doi: 10.1111/cmi.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton JA. 2008. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 17.Hamano S, Asgharpour A, Stroup SE, Wynn TA, Leiter EH, Houpt E. 2006. Resistance of C57BL/6 mice to amoebiasis is mediated by nonhemopoietic cells but requires hemopoietic IL-10 production. J Immunol 177:1208–1213. doi: 10.4049/jimmunol.177.2.1208. [DOI] [PubMed] [Google Scholar]
- 18.Swamydas M, Lionakis MS. 2013. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp 2013:e50586. doi: 10.3791/50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman PS, Marshall AM, Olekhnovich I, Warren CA, Burgess SL, Hontecillas R, Viladomiu M, Bassaganya-Riera J, Guerrant RL, Macdonald TL. 2014. Preclinical studies of amixicile: a systemic therapeutic developed for treatment of Clostridium difficile infections also shows efficacy against Helicobacter pylori. Antimicrob Agents Chemother 58:4703–4712. doi: 10.1128/AAC.03112-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller PY, Janovjak H, Miserez AR, Dobbie Z. 2002. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32:1372–1374, 1376, 1378–1379. [PubMed] [Google Scholar]
- 21.Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, Budelsky AL, Wills-Karp M. 2010. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 11:928–935. doi: 10.1038/ni.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy S, Kabir M, Mondal D, Ali IKM, Petri WA, Haque R. 2005. Real-time-PCR assay for diagnosis of Entamoeba histolytica infection. J Clin Microbiol 43:2168–2172. doi: 10.1128/JCM.43.5.2168-2172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, Bovio I, Akira S, Kumagai Y, Moldawer LL. 2010. Bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol 184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Álvarez-Errico D, Vento-Tormo Sieweke R, Ballestar ME. 2015. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol 15:7–17. doi: 10.1038/nri3777. [DOI] [PubMed] [Google Scholar]
- 25.Burgess AW, Metcalf D. 1980. The nature and action of granulocyte-macrophage colony stimulating factors. Blood 56:947–958. [PubMed] [Google Scholar]
- 26.Martinez-Moczygemba M, Huston DP. 2003. Biology of common β receptor-signaling cytokines IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol 112:653–665. doi: 10.1016/j.jaci.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 27.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. 2009. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 114:1289–1298. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guthridge MA, Powell JA, Barry EF, Stomski FC, McClure BJ, Ramshaw H, Felquer FA, Dottore M, Thomas DT, To B, Begley CG, Lopez AF. 2006. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. EMBO J 25:479–489. doi: 10.1038/sj.emboj.7600948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosas M, Gordon S, Taylor PR. 2007. Characterisation of the expression and function of the GM-CSF receptor alpha-chain in mice. Eur J Immunol 37:2518–2528. doi: 10.1002/eji.200636892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, Solomon D, Mueller S, Paris PL, Zhang Z, Petritsch C, Gupta N, Waldman TA, James CD. 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20:1394–1396. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruidenier L, Chung C, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. 2012. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Kleer I, Willems F, Lambrecht B, Goriely S. 2014. Ontogeny of myeloid cells. Front Immunol 5:423. doi: 10.3389/fimmu.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang HQ, Bos NA, Cebra JJ. 2001. Timing, localization, and persistence of colonization by segmented filamentous bacteria in the neonatal mouse gut depend on immune status of mothers and pups. Infect Immun 69:3611–3617. doi: 10.1128/IAI.69.6.3611-3617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takizawa H, Boettcher S, Manz MG. 2012. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 35.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. 2010. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 36.Burchfield JS, Li Q, Wang HY, Wang R-F. 2015. JMJD3 as an epigenetic regulator in development and disease. Int J Biochem Cell Biol 67:148–157. doi: 10.1016/j.biocel.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwasaki H, Mizuno S, Mayfield R, Shigematsu H, Arinobu Y, Seed B, Gurish MF, Takatsu K, Akashi K. 2005. Identification of eosinophil lineage-committed progenitors in the murine bone marrow. J Exp Med 201:1891–1897. doi: 10.1084/jem.20050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naylor C, Burgess S, Madan R, Buonomo E, Razzaq K, Ralston K, Petri WA. 2014. Leptin receptor mutation results in defective neutrophil recruitment to the colon during Entamoeba histolytica infection. mBio 5:e02046-14. doi: 10.1128/mBio.02046-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, Boyson JE, Fitzgerald KA, Flavell RA, Eisenbarth SC, Poynter ME. 2011. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol 187:64–73. doi: 10.4049/jimmunol.1100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farkas AM, Panea C, Goto Y, Nakato G, Galan-Diez M, Narushima S, Honda K, Ivanov II. 2015. Induction of Th17 cells by segmented filamentous bacteria in the murine intestine. J Immunol Methods 421:104–111. doi: 10.1016/j.jim.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MGG, Laufer TMM, Ignatowicz L, Ivanov III. 2014. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, Sczesnak A, Liao J-J, Torres VJ, Jenkins MK, Lafaille JJ, Littman DR. 2014. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov II, Sugiyama T, Nuñez G, Camp JG, Hattori M, Umesaki Y, Honda K. 2015. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163:367–380. doi: 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng M, Horne W, McAleer JP, Pociask D, Eddens T, Good M, Gao B, Kolls JK. 2016. Therapeutic role of interleukin 22 in experimental intra-abdominal Klebsiella pneumoniae infection in mice. Infect Immun 84:782–789. doi: 10.1128/IAI.01268-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S. 2015. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol 8:221–231. doi: 10.1038/mi.2014.57. [DOI] [PubMed] [Google Scholar]
- 46.Rubino SJ, Geddes K, Girardin SE. 2012. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol 33:112–118. doi: 10.1016/j.it.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe K, Nagata N, Sekine K, Watanabe K, Igari T, Tanuma J, Kikuchi Y, Oka S, Gatanaga H. 2014. Asymptomatic intestinal amebiasis in Japanese HIV-1-infected individuals. Am J Trop Med Hyg 91:816–820. doi: 10.4269/ajtmh.14-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taniuchi M, Sobuz SU, Begum S, Platts-Mills JA, Liu J, Yang Z, Wang X-Q, Petri WA, Haque R, Houpt ER. 2013. Etiology of diarrhea in Bangladeshi infants in the first year of life analyzed using molecular methods. J Infect Dis 208:1794–1802. doi: 10.1093/infdis/jit507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV. 2014. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. Elife 3:e03206. doi: 10.7554/eLife.03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collins SM, Chang C, Mearin F. 2012. Postinfectious chronic gut dysfunction: from bench to bedside. Am J Gastroenterol Suppl 1:2–8. doi: 10.1038/ajgsup.2012.2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.