

Summary
The genomes of kinetoplastids are organized into polycistronic gene clusters that are flanked by the modified DNA base J. Previous work has established a role of base J in promoting RNA polymerase II termination in Leishmania spp. where the loss of J leads to termination defects and transcription into adjacent gene clusters. It remains unclear whether these termination defects affect gene expression and whether read through transcription is detrimental to cell growth, thus explaining the essential nature of J. We now demonstrate that reduction of base J at specific sites within polycistronic gene clusters in L. major leads to read through transcription and increased expression of downstream genes in the cluster. Interestingly, subsequent transcription into the opposing polycistronic gene cluster does not lead to downregulation of sense mRNAs. These findings indicate a conserved role for J regulating transcription termination and expression of genes within polycistronic gene clusters in trypanosomatids. In contrast to the expectations often attributed to opposing transcription, the essential nature of J in Leishmania spp. is related to its role in gene repression rather than preventing transcriptional interference resulting from read through and dual strand transcription.
Introduction
Kinetoplastids are a group of early-diverged eukaryotes collectively responsible for multiple diseases including African sleeping sickness, Chagas disease and leishmaniasis. Kinetoplastid parasites, which include Trypanosoma brucei, Trypanosoma cruzi and Leishmania major, progress through life stages by cycling between an insect vector and a mammalian host. The parasites must therefore adapt to changes in their environment, such as changes in pH, temperature, oxygen concentrations and host immune responses, among others. Unlike most eukaryotes, the entire genome of kinetoplastids is arranged into gene clusters (Berriman et al., 2005; El-Sayed et al., 2005; Ivens et al., 2005) where RNA Polymerase (RNAP) II initiates transcription at sites called divergent strand switch regions (dSSRs), transcribes polycistronically (Johnson et al., 1987; Martinez-Calvillo et al., 2003; Mottram et al., 1989) through tens to hundreds of genes, and terminates at convergent strand switch regions (cSSRs) (Martinez-Calvillo et al., 2004). Gene clusters are also adjacently arranged on the same DNA strand in what are called a head-tail (HT) arrangement, such that transcription of an upstream cluster terminates and initiation of an independent gene cluster occurs downstream (Kolev et al., 2010; Siegel et al., 2009; Thomas et al., 2009). Pre-mRNAs are processed through trans-splicing with the addition of a 39 nucleotide spliced leader (SL) sequence to the 5′ end of all mRNAs (Agabian, 1990; Boothroyd and Cross, 1982; De Lange et al., 1983; Nelson et al., 1983; Sutton and Boothroyd, 1986; Van der Ploeg et al., 1982) [reviewed in (Borst, 1986)], which is coupled to the 3′ polyadenylation of the upstream transcript (LeBowitz et al., 1993).
Relatively little is known about transcriptional regulation in kinetoplastids, in large part because the polycistronic nature of the genome has led to the belief that gene expression regulation does not occur at the transcriptional level and instead predominately occurs post-transcriptionally through mechanisms such as mRNA processing, mRNA stability and translation efficiency (Campbell et al., 2003; Clayton, 2002). Evidence has begun to emerge however that has challenged the notion that kinetoplastids lack transcriptional regulation. Multiple chromatin modifications are enriched at sites of RNAP II initiation and termination, including histone methylation, acetylation, histone variants and the DNA modification base J that could be utilized by the parasites to regulate transcription (Cliffe et al., 2010; Respuela et al., 2008; Siegel et al., 2009; Thomas et al., 2009). Of these chromatin modifications, base J has been the most studied and demonstrated to play a functional role in the regulation of transcription in all kinetoplastids examined thus far (Ekanayake and Sabatini, 2011; Ekanayake et al., 2011; Reynolds et al., 2014, 2016; van Luenen et al., 2012). J consists of a glucosylated thymidine (Gommers-Ampt et al., 1993) and has been found only in the nuclear DNA of kinetoplastids, Diplonema and Euglena (Dooijes et al., 2000; van Leeuwen et al., 1998). The DNA modification is synthesized through the hydroxylation of thymidine by either JBP1 or JBP2 (Cliffe et al., 2012), forming hydroxymethyluridine, followed by the transfer of a glucose by the glucosyltransferase enzyme JGT (Bullard et al., 2014; Sekar et al., 2014) (reviewed in [Borst and Sabatini, 2008; Reynolds et al., 2015]). Analogous to the TET proteins in mammals (Iyer et al., 2009; Tahiliani et al., 2009), the JBPs utilize 2-oxoglutarate, oxygen and Fe2+ in the hydroxylation reaction (Cliffe et al., 2012). The central roles of JBP1/2 and JGT in synthesizing J have been established in T. brucei, the only kinetoplastid species tested thus far in which genetic deletions of JBP1/2 and JGT are viable (Bullard et al., 2014; Cliffe et al., 2009). Attempts to generate T. cruzi and Leishmania spp. JBP1/2 KO cells have been unsuccessful, suggesting an essential role of J in these kinetoplastids. Addition of the 2-oxoglutarate structural analog dimethyloxalylglycine (DMOG) to the growth medium or limiting oxygen concentrations inhibit hydroxylase activity and thus enable J reduction in cells without genetic modification (Cliffe et al., 2012).
Investigations in T. cruzi have revealed a function of J in the repression of RNAP II initiation, such that J loss increases active chromatin marks and transcription initiation, resulting in global gene expression changes (Ekanayake and Sabatini, 2011; Ekanayake et al., 2011). No defect in RNAP II termination was identified upon J loss in T. cruzi, however in T. brucei and Leishmania spp. J has been found to promote RNAP II termination (Reynolds et al., 2014, 2016; van Luenen et al., 2012). In T. brucei, J located prior to the end of gene clusters promotes termination, repressing downstream genes in the same cluster (Reynolds et al., 2014, 2016). Loss of J from within gene clusters results in read through and expression of the downstream genes, and we therefore refer to these sites as gene cluster internal termination sites. H3.V co-localizes with J at RNAP II termination sites in T. brucei (Cliffe et al., 2010) and has recently been shown to play a similar role in regulating termination (Reynolds et al., 2016; Schulz et al., 2016). J and H3.V independently function to promote termination within gene clusters, such that the combined loss of J and H3.V results in a synergistic increase in read through transcription at gene cluster internal termination sites and derepression of downstream genes (Reynolds et al., 2016). Genes regulated by this process include many that are developmentally regulated in T. brucei and code for proteins involved in optimal growth and immune evasion during infection of the mammalian host (the specific trypanosome life stage where J is synthesized) (Reynolds et al., 2016; Schulz et al., 2016).
While J and H3.V also co-localize at termination sites at the end of gene clusters (i.e. cSSRs) in T. brucei, they are not required for terminating transcription and preventing elongation of RNAP II into the opposing gene cluster (Reynolds et al., 2016). However, in L. major and L. tarentolae J does function to prevent read through transcription at cSSRs and the formation of antisense RNAs (Reynolds et al., 2014; van Luenen et al., 2012). For example, reduction of base J levels in L. major following DMOG treatment results in transcription of the antisense strand of the adjacent gene cluster genome-wide (Reynolds et al., 2014). It has been proposed that dual strand transcription resulting from read through at cSSRs disrupts expression of sense mRNAs via transcriptional interference and is therefore detrimental to Leishmania spp. cell growth (van Luenen et al., 2012). Whether J loss and subsequent read through at cSSRs impact mRNA transcript abundance in Leishmania spp. is not yet clear however. It is also not known if J functions to promote gene cluster internal termination in Leishmania spp., and if so what role this process has in parasite growth and explaining the apparent essential nature of J. The role of H3.V is also unclear. Removal of H3.V in L. major did not reveal defects in RNAP II termination (Anderson et al., 2013), however it is not known if H3.V localizes to termination sites in L. major, as is observed in T. brucei.
The recently identified role of H3.V and base J in promoting RNAP II termination within gene clusters in T. brucei led us to further investigate the function of these epigenetic marks in L. major. We found that like T. brucei, J is localized within several gene clusters in L. major where the acute J loss induced by the J synthesis inhibitor DMOG results in defects of RNAP II termination within the cluster and increased expression of downstream genes. We also demonstrate here that, similar to T. brucei, H3.V co-localizes with base J at termination sites in L. major. Surprisingly, however, H3.V apparently regulates J synthesis in L. major as the loss of H3.V reduces the level of J at termination sites with no effects on RNAP II termination and minimal gene expression changes. Further reduction of J at termination sites in the H3.V knockout (KO) using DMOG revealed greater termination defects, more significant gene expression changes, and greatly reduced cell growth, compared with wild type (WT) cells treated with DMOG. Whilst read through defects in L. major include the extension of RNAP II onto the adjacent opposing gene cluster and dual strand transcription, we saw no evidence of transcription interference resulting in significant downregulation of mRNAs on the opposing gene cluster in either WT or H3.V KO cells treated with DMOG. These results indicate a conserved role for J regulating RNAP II transcription termination and expression of genes within polycistronic gene clusters in trypanosomatids and suggest that the essential nature of J in Leishmania spp. is related to its role in repressing specific genes at the end of gene clusters and not the prevention of dual strand transcription.
Results
L. major H3.V co-localizes with base J at termination sites and regulates J synthesis
We have found that in T. brucei, H3.V and base J co-localize at RNAP II termination sites and both epigenetic marks work together to promote RNAP II termination (Reynolds et al., 2016; Schulz et al., 2016). However, while base J has clearly been shown to regulate RNAP II termination in L. major (Reynolds et al., 2014) the deletion of H3.V from this parasite did not indicate defects in termination (Anderson et al., 2013). To take a closer look, we first determined whether H3.V localizes with base J at termination sites in L. major. Using chromatin immunoprecipitation-qPCR (ChIP-qPCR), and primers that spanned a cSSR (Fig. 1A), we found that while histone H3 is present at similar levels across the cSSR, H3.V is enriched at the RNAP II termination site (Fig. 1B and Supporting Information Fig. S1A). In contrast, we did not detect enrichment of H3.V at dSSRs, where RNAP II initiation takes place (Fig. 1D and E, Supporting Information Fig. S1B). Lack of H3.V ChIP-qPCR signal in H3.V KO cells confirms the specificity of the L. major H3.V antibody (Supporting Information Fig. S1D). Consistent with previous findings (Reynolds et al., 2014), by J IP-qPCR we observed a peak enrichment of J at the RNAP II termination site, confirming that J and H3.V co-localize at RNAP II termination sites in L. major (Fig. 1B and C). As expected, we also detect a peak of J within dSSRs (Fig. 1F).
Fig. 1.
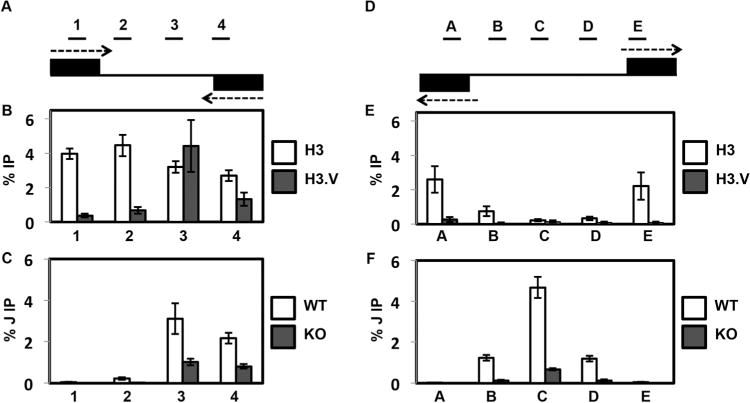
H3.V co-localizes with base J at cSSRs and regulates J synthesis.
A. Map of cSSR 12.1 (12.1 indicates the first termination site on chromosome 12, following nomenclature established by van Luenen et al. (van Luenen et al., 2012). Genomic coordinates for all L. major cSSRs are listed in Reynolds et al. [Reynolds et al., 2014]). Black boxes represent the final gene in the opposing gene clusters. Arrows indicate the direction of transcription. Lines above the map indicate PCR amplified regions 1–4 utilized in the ChIP-qPCR analyses. Not to scale.
B. Localization of H3 and H3.V across cSSR 12.1 in WT cells determined by ChIP-qPCR using H3 antisera and H3.V antisera. The average of three independent IPs is plotted as the percent IP relative to the total input material. All IPs were background subtracted using a no antibody control. White bars, H3 ChIP; dark grey bars, H3.V ChIP. Error bars represent the standard deviation.
C. J localization across a cSSR in WT and H3.V KO cells. Anti-base J IP-qPCR analysis was performed for regions 1–4 within cSSR 12.1 of the indicated cell lines. The peak of J and the TTS have been shown to be within region 3 (Reynolds et al., 2014; van Luenen et al., 2012). The average of three independent IPs is plotted as the percent IP relative to the total input material. All IPs were background subtracted using a no antibody control. White bars, WT; dark grey bars, H3.V KO. Error bars represent the standard deviation. Reduction of J in the H3.V KO is statistically significant at region 3 and 4 as determined by a two-tailed Student’s t-test at a P-value <0.05.
D. Map of dSSR 6.1 (6.1 indicates the first initiation site on chromosome 6, following nomenclature established by van Luenen et al. [van Luenen et al., 2012]). Black boxes represent the initial gene in the opposing gene clusters. Arrows indicate the direction of transcription. Lines above the map indicate PCR amplified regions A–E utilized in the ChIP-qPCR analyses. Not to scale.
E. Localization of H3 and H3.V across dSSR 6.1 in WT cells. H3 and H3.V ChIP-qPCR were performed as described in B.
F. J localization across a dSSR in WT and H3.V KO cells. Anti-base J IP-qPCR analysis was performed for regions A–E within dSSR as described in C. Reduction of J in the H3.V KO is statistically significant at region B–D as determined by a two-tailed Student’s t-test at a P-value <0.05.
While H3.V and base J co-localize and cooperatively regulate RNAP II termination in T. brucei, H3.V does not influence base J synthesis (Cliffe et al., 2010). In contrast, the deletion of H3.V in L. major led to significantly reduced levels of base J in cSSRs (Fig. 1C and Supporting Information Fig. S1C). This loss of J was not restricted to termination sites however, as we also observed reduced J at a dSSR in the H3.V KO, despite the lack of enrichment of H3.V at these sites (Fig. 1E and F). This suggests that, for at least dSSRs, the loss of H3.V indirectly impacts J synthesis. To determine whether J plays a role in histone/nucleosome localization, we analyzed the levels of H3 and H3.V at cSSRs of cells with reduced J following treatment with the J synthesis inhibitor DMOG. We have previously demonstrated that DMOG treatment of L. major leads to significant reductions in base J levels, and corresponding read through transcription, at cSSRs genome-wide (Reynolds et al., 2014). We find here that reduction of base J did not have a significant effect on the abundance of H3.V or canonical histone H3 within cSSRs (Supporting Information Fig. S2A and B). There was a slight trend towards increased H3.V and H3 following J loss, however the increase was not statistically significant at any of the sites analyzed. Thus, the loss of H3.V results in reduced J levels at both RNAP II termination and initiation sites, presumably through an indirect effect. However, J is not required for H3.V or nucleosome localization at RNAP II termination sites.
To begin to investigate how the loss of H3.V decreases J levels at RNAP II termination sites, we analyzed the effect of H3.V loss on nucleosome abundance by H3 ChIP-qPCR. Using primers located near the termination site we found a trend of reduced H3 in cSSRs of the H3.V KO cell line (Supporting Information Fig. S2C). Total protein levels of H3 were similar in the WT and H3.V KO cell lines (Supporting Information Fig. S3) and the loss of H3.V did not affect transcript levels for any of the histones or enzymes involved in J synthesis (see below). Therefore, loss of H3.V leads to reduced assembly of H3 containing nucleosomes at cSSRS. These findings implicate chromatin structure, specifically nucleosome abundance, in the regulation of J synthesis at termination sites, but it remains unclear how the loss of H3.V negatively affects J synthesis at dSSRs.
L. major H3.V is not required for RNAP II termination
Given that we have previously shown that reduction of J in L. major results in read through transcription at RNAP II termination sites, regardless of how J is reduced, we would expect a defect in RNAP II termination in the H3.V KO cells. Anderson et al. have recently found however, that the loss of H3.V does not lead to a detectable termination defect in L. major, as determined by SL RNA-seq (Anderson et al., 2013). While both SL RNA-seq and small RNA-seq are capable of revealing termination defects following J reduction, small RNA-seq is more sensitive (van Luenen et al., 2012) and thus we used this method to further analyze the H3.V KO for termination defects genome-wide by small RNA-seq. As we have previously demonstrated, the reduction of base J in L. major by treatment with DMOG resulted in the production of antisense small RNAs due to read through transcription at cSSRs and into the opposing gene cluster (Fig. 2A, Supporting Information Figs. S4A and S5) (Reynolds et al., 2014). Read through transcription following J loss is confirmed by strand-specific RT-PCR, where an RNA species spanning the termination site is detected only following J loss (Fig. 2B and Supporting Information Fig. S4B), and in agreement with the SL RNA-seq data of Anderson et al., we did not find evidence of read through transcription in the H3.V KO by small RNA-seq or strand-specific RT-PCR (Fig. 2A and B, Supporting Information Figs. S4A and B and S5). Only upon further loss of J in the H3.V KO following DMOG treatment is read through transcription detected (Fig. 2, Supporting Information Figs. S4 and S5). Interestingly, DMOG treatment of the H3.V KO cells results in a greater decrease in J and greater read through transcription than the similar DMOG treatment of WT cells (Fig. 2 and Supporting Information Fig. S4). The increased read through transcription in the H3.V KO+DMOG is observed by small RNA-seq (several cSSRs are shown in Fig. 2A, Supporting Information Figs. S4A and S5 and quantitation across all cSSRs is shown in Supporting Information Fig. S6), and also confirmed by semi-quantitative strand-specific RT-PCR and quantitative strand-specific RT-qPCR analysis of several termination sites (Fig. 2B and C, Supporting Information Fig. S4B). This nicely reflects the direct relationship of J levels and degree of read through transcription previously characterized in L. major (Reynolds et al., 2014). These data support the conclusion that, as opposed to T. brucei, H3.V does not directly regulate RNAP II termination in L. major.
Fig. 2.
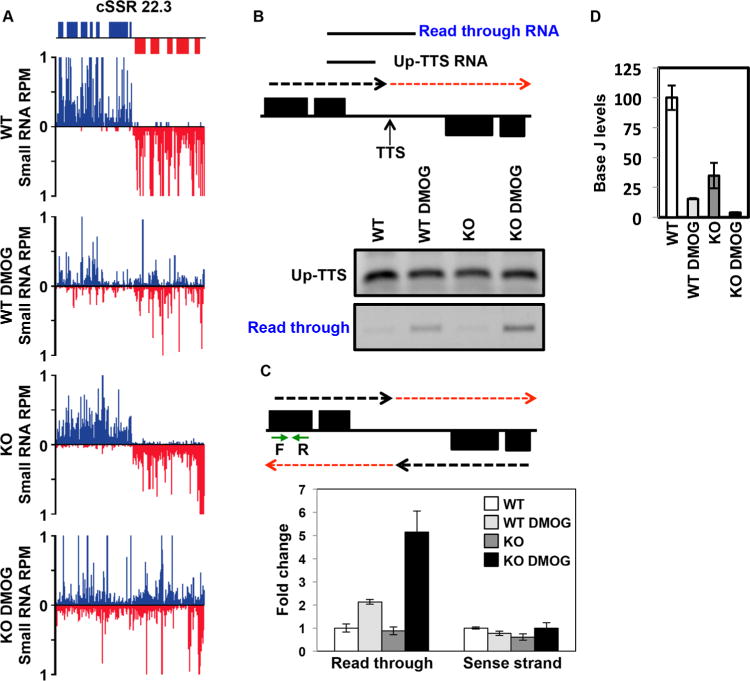
H3.V does not promote transcription termination in L. major.
A. Small RNA-seq reads for a representative cSSR (22.3) are shown where J loss leads to read through transcription, but loss of H3.V does not. Small RNA reads are plotted as reads per million reads mapped (RPM). ORFs are shown above the graphs. The genomic location shown includes position 488–528 kb on chromosome 22. WT: wild type; WT DMOG; WT+DMOG; KO: H3.V KO; KO DMOG: H3.V KO+DMOG. Blue: top strand; red: bottom strand.
B. Strand-specific RT-PCR analysis. Above, schematic representation (not to scale) of cSSR 22.3 illustrating the nascent RNA species assayed by RT-PCR. The dashed red arrow indicates read through transcription past the transcription termination site (TTS). Up-TTS: RNA species upstream of the TTS; Read through RNA: RNA species resulting from read through transcription. Below, cSSR 22.3 was analyzed in WT and H3.V KO cells in the absence and presence of 5mM DMOG.
C. Strand-specific RT-qPCR analysis. Above, schematic representation of cSSR 22.3 as described in panel B. Green arrows indicate the location of primers utilized for strand-specific RT-qPCR analysis. Below, strand-specific RT-qPCR. Read through transcription on the bottom strand was quantitated by performing site-specific cDNA synthesis using primer F illustrated in the diagram above, followed by qPCR using primers F and R. Abundance was normalized using beta tubulin (a gene specific primer against beta tubulin was added to the same cDNA synthesis reaction with primer F, followed by qPCR using beta tubulin primers). White bars: WT; light grey bars: WT+DMOG; dark grey bars: H3.V KO; black bars: H3.V KO+DMOG. Data are plotted as fold change, with WT set to one. Strand-specific RT-qPCR was performed in triplicate, with error bars indicating standard deviation. RNA from the sense strand was similarly quantitated by generating cDNA using primer R, followed by qPCR with primers F and R. Analysis of the sense strand provides a negative control for the strand-specific nature of the assay as well as indicating no change in sense RNA levels following readthrough transcription on the opposing strand.
Increased read through in WT+DMOG and H3.V KO+DMOG relative to WT is statistically significant as determined by a two-tailed Student’s t-test at a P-value <0.05.
D. The amount of read through transcription correlates with the extent of J loss. The levels of J at the TTS in cSSR 22.3 was analyzed by anti-J IP-qPCR for the indicated cell lines as described in Fig. 1C. WT level is set to 100%. Reduction of J is statistically significant in the WT+DMOG, H3.V KO, and H3.V KO+DMOG conditions relative to WT. Increased J reduction in H3.V KO+DMOG compared to WT+DMOG cells is also statistically significant. All statistical significance was determined using two-tailed Student’s t-test and a P-value <0.05.
The fact that reduction of base J in the H3.V KO does not result in termination defects, unless J is further reduced by DMOG, suggests there is a threshold of J required for RNAP II termination.
To address this, we performed a DMOG titration in WT cells to follow the effect of varying levels of J on termination at two cSSRs (Fig. 3). We found that read through transcription does not occur until approximately 75% of the initial J is lost at the termination site. In the DMOG titration we see that ~40% (Fig. 3A) and ~55% (Fig. 3B) reduction of J in WT cells is not sufficient for a termination defect. ~75% (or greater) reduction of J in WT cells, with increasing DMOG concentration, leads to read through transcription. Deletion of H3.V resulted in ~60% reduction in base J, thus explaining the lack of a detectable defect in termination. However, subsequent treatment of the H3.V KO with DMOG decreased J by 90–95% relative to WT cells, and resulted in a termination defect (Fig. 3). As described above, similar treatment of WT and H3.V KO with DMOG resulted in significantly more J loss in the H3.V KO and a stronger defect in termination. In some cases the loss of J in the WT+DMOG (or H3.V KO) is not enough for significant read through transcription at the cSSR and only when J is reduced further in the H3.V KO+DMOG is there strong termination defect (Supporting Information Figs. S4 and S5, cSSR 7.2). We conclude that a threshold of J between 25 and 40% of WT levels is sufficient to prevent read through transcription, thus explaining the lack of read through in the H3.V KO cells. Once below this threshold the degree of termination defect is directly related to the decrease in J, explaining why we detect greater read through in the H3.V KO+DMOG cell line than WT+DMOG. Taken together these data support the conclusion that regulation of termination in L. major is all about base J. H3.V does not regulate termination, but does regulate J levels. However, the loss of H3.V does not decrease J levels below the threshold required for termination.
Fig. 3.
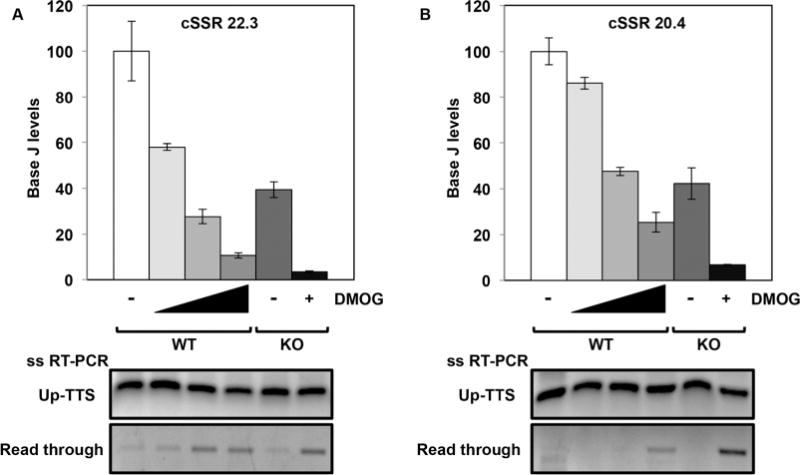
Levels of J remaining in the H3.V KO are sufficient for terminating transcription.
A and B. The levels of J at the TTS of two representative cSSRs were determined by J IP-qPCR analysis in WT cells with increasing DMOG concentrations (0, 1, 2.5 and 5mM) and in H3.V KO cells in the absence and presence of 5 mM DMOG. At cSSR 22.3, reduction of J is statistically significant in each condition relative to WT. At cSSR 20.4, reduction of J is statistically significant in each condition relative to WT except WT+1mM DMOG. All statistical significance was determined using two-tailed Student’s t-test and a P-value <0.05. Below, strand-specific RT-PCR analysis of each of the cSSRs as described in Fig. 2B.
J promotes termination prior to the end of gene clusters, repressing gene expression
We have shown previously that J reduction and subsequent read through transcription in WT L. major cells treated with DMOG result in only a modest defect in cell growth in vitro (Reynolds et al., 2014). These findings contrast with those by van Luenen et al., who found that further reduction of J in a JBP2 KO cell line by bromodeoxyuridine (BrdU) is lethal in L. tarentolae (van Luenen et al., 2012). We now find that the H3.V KO cell line is hypersensitive to DMOG compared to WT cells (Supporting Information Fig. S7). While the loss of H3.V alone has no effect on cell growth (Anderson et al., 2013), H3.V KO+DMOG cells grow significantly slower than WT+DMOG cells after 48 h and continue to slowly divide thereafter. We conclude that the greater J loss and read through defects in the H3.V KO+DMOG cells are detrimental to cell growth and that base J is essential in Leishmania spp. due to its role in regulating RNAP II termination.
One possibility is that the essential nature of J in Leishmania spp. is indicated by the termination defects at cSSRs resulting in RNAP II transcription into opposing gene clusters genome-wide that occur following the loss of J (Reynolds et al., 2014; van Luenen et al., 2012). For example, read through transcription at convergent gene clusters may result in transcriptional interference where transcription of the opposing strand of the adjacent gene cluster exerts a direct negative impact on the transcription of the sense strand. In this collision model, active antisense transcription suppresses sense RNA transcription resulting in downregulation of mRNAs for (essential) genes located at the end of convergent gene clusters. To test this model we performed mRNA-seq in parasites treated with and without DMOG. For each condition (WT, WT+DMOG, H3.V KO and H3.V KO+DMOG) two independent mRNA-seq libraries were sequenced. Replicates were strongly correlated with each other (with R2 values >0.95 for all replicates). We focused on genes that were up or downregulated by twofold or more with an RPKM of at least one in each replicate and a p-value of 0.05 or less. Using these cutoffs we identified very limited gene expression changes in WT L. major cells incubated with DMOG (Supporting Information Table S1). Even fewer gene expression changes were observed in H3.V KO cells, consistent with previous SL RNA-seq (Anderson et al., 2013) (Supporting Information Table S1). mRNA-seq indicated only 17 transcripts were changed more than twofold following the decrease of J in WT cells, the majority of which (16 transcripts) had increased expression. We confirm many of these changes by RT-qPCR (Supporting Information Fig. S8). The limited number of affected genes indicates read through transcription does not lead to significant downregulation of mRNAs. For example, at two cSSRs that are known to have some of the highest levels of read through transcription following loss of J with DMOG (10.2 and 8.1) (Reynolds et al., 2014), none of the mRNAs for genes within 10kb of the termination site of the four gene clusters are significantly downregulated in either WT+DMOG or H3.V KO+DMOG (Fig. 4). In fact, none of the genes on these chromosomes are significantly downregulated in any of the conditions examined here. Consistent with small RNA-seq analysis, read through transcription through the cSSR into the opposing gene cluster following the loss of J is apparent in the mRNA-seq data (Fig. 4A and B). Therefore, in agreement with van Luenen et al., some fraction of read through (antisense) RNA is processed similarly to mRNAs (van Luenen et al., 2012). However, as seen previously, steady-state mRNA does not necessarily reflect the extent of read through transcription compared to small RNA-seq or analysis of nascent RNA by strand-specific RT-PCR. Nevertheless, consistent with small RNA-seq, plotting the mRNA-seq data supports the conclusion that transcriptional read through is highest in H3.V KO+DMOG. Interestingly, while none of genes localized at the end of gene clusters are significantly downregulated, even in the highest degree of read through transcription in the H3.V KO+DMOG, we do detect a slight trend of decreasing mRNA levels with increasing degree of read through (Fig. 4C and D). Therefore, while there may be some transcriptional interference resulting from RNAP II extension into the adjacent convergent gene cluster, this does not result in significant repression of mRNA levels.
Fig. 4.
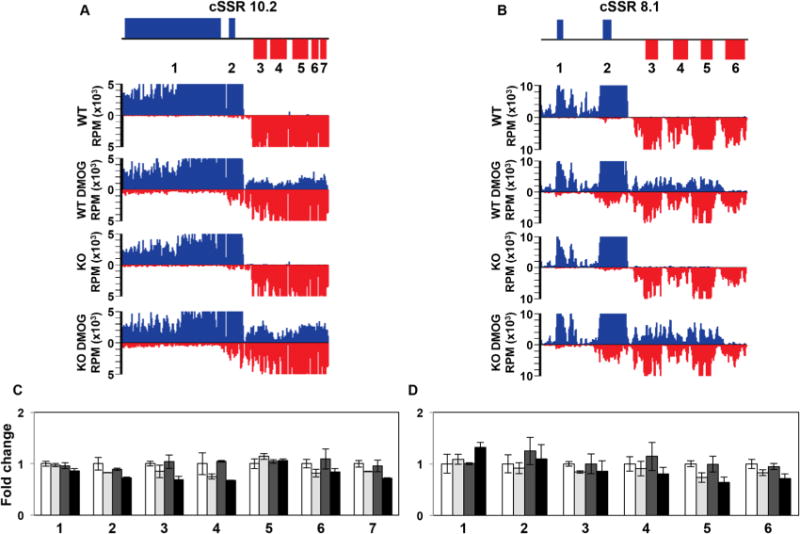
Read through transcription does not lead to transcriptional interference.
A and B. Strand-specific mRNA-seq reads for two representative cSSRs where loss of J leads to high degree of read through transcription into the adjacent gene cluster are shown. The genomic region shown is from 243 to 287 kb on chromosome 10 in (A) and from 384 to 406 kb on chromosome 8 in (B).
C and D. Plot of the mRNA-seq data for the genes indicated (numbered) in the ORF map for the corresponding regions above. The average RPKM of mRNA-seq replicate libraries was used to determine the fold changes, with WT set to one. Error bars indicate the standard deviation between mRNA-seq replicates. White bars: Wild type; grey bars: Wild type+DMOG; dark grey bars: H3.V KO; black bars: H3.V KO+DMOG. None of the genes shown are significantly differentially expressed relative to WT except for gene 5 (LmjF.08.0890) in D in the H3.V KO+DMOG condition, as determined by Cuffdiff (P-value of 0.03), but gene 5 is only downregulated by 1.6 fold, which does not meet our twofold cutoff.
In contrast, we find many more genes upregulated upon stimulation of read through transcription (Supporting Information Table S1). Similar to findings we previously described in T. brucei (Reynolds et al., 2014, 2016), all 16 of the upregulated genes in WT+DMOG L. major cells are located at the end of a gene cluster immediately downstream or within a J peak, where J regulation of transcription termination within a gene cluster attenuates transcription of downstream genes. For example, on chromosome 9 only one gene is upregulated in WT+DMOG, and represents the last gene of a gene cluster at cSSR 9.1 (Fig. 5A). The peak of base J extends from the cSSR into the open reading frame (ORF) of the last gene in the gene cluster that is lowly expressed in the presence of J (WT), but increases upon the loss of J following DMOG treatment (Fig. 5B–E). mRNA levels are further increased upon the loss of J in the H3.V KO+DMOG, presumably due to increased read through transcription. As discussed above, no read through and minimal gene expression changes are detected in the H3.V KO. cSSR 9.1 (Fig. 5) is an example where attenuation of transcription within an ORF is sufficient for gene repression. Termination of transcription prior to the 3′ UTR would prevent the production of full-length precursor transcript and therefore, the production of processed steady-state mRNA. At this gene cluster internal termination site, decreased levels of base J leads to read through transcription on the top strand that results in the complete transcription of the final gene, which is processed to mature mRNA. Read through following the loss of J extends down to an RNAP III-transcribed gene in the cSSR, which are known to terminate RNAP II transcription independent of J in Leishmania spp. (Reynolds et al., 2014; van Luenen et al., 2012). The presence of RNAP III-transcribed genes (a 5S rRNA gene on the bottom strand and tRNA genes on the top strand) at this site terminates transcription on both strands in the absence of base J (Reynolds et al., 2014; van Luenen et al., 2012). Thus, while our previous genome-wide studies quantitating the production of antisense RNAs failed to detect a defect in RNAP II termination at this cSSR due to J-independent mechanisms preventing transcription into the opposing gene clusters, we now illustrate that base J, in fact, regulates termination on the sense strand of a gene cluster prior to the cSSR. No expression change is detected for the genes immediately upstream or the final two genes of the adjacent convergent gene cluster in either WT or H3.V KO cells treated with DMOG (Fig. 5E).
Fig. 5.
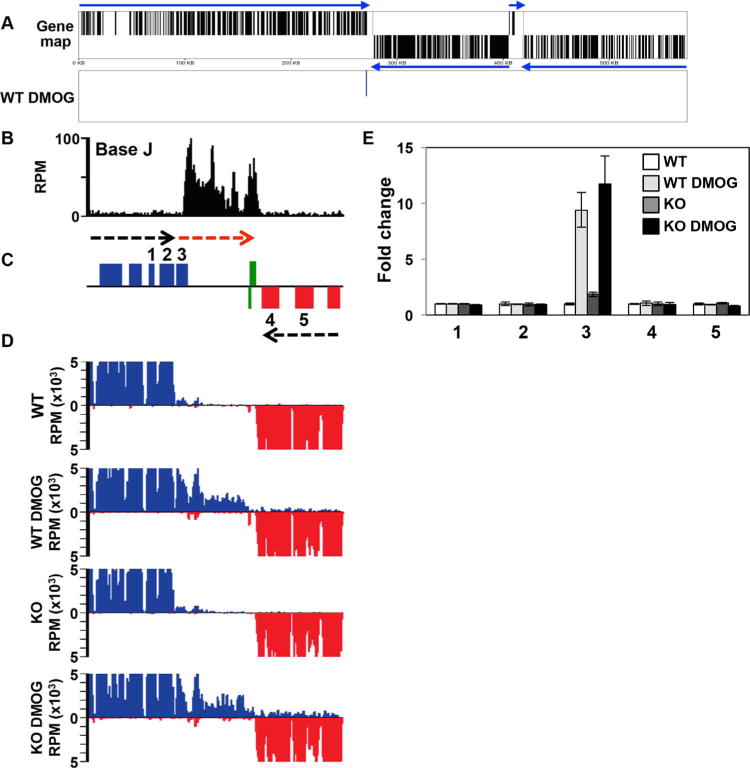
Decreased efficiency of RNAP II termination and increased gene expression following the loss of J.
A. Gene map of chromosome 9 is shown. mRNA coding genes on the top strand are indicated by black lines in the top half of the panel, bottom strand by a line in the bottom half. Genes on the top strand are transcribed from left to right and those on the bottom strand are transcribed from right to left, indicated by blue arrows. Panel below (WT DMOG) indicates the location of the single mRNA found upregulated by at least twofold or more in WT cells treated with DMOG relative to WT. No other expression changes (up or downregulated) were detected.
B–D. A region on chromosome 9 from 263 to 285kb where J regulates transcription termination and gene expression at a cSSR is shown. (B) Base J localizes at the site of RNAP II termination within the gene cluster (prior to the last gene before the cSSR). Base J IP-seq reads are plotted as reads per million (RPM), as previously described (Reynolds et al., 2016). (C) ORFs are shown with the top strand in blue and the bottom strand in red. The red arrow indicates read through transcription following the loss of J. Green boxes indicate RNAP III transcribed genes (tRNA and 5S rRNA). (D) mRNA-seq reads from the indicated cell lines are mapped as described in Fig. 4A and B.
E. Plot of the mRNA-seq data for the genes numbered in the ORF map in panel B, as described in Fig. 4C and D. The upregulated gene, 3, is LmjF.09.0690.
Additional examples of J regulation of gene expression via termination include H-T regions 26.5 (Fig. 6) and 23.1 (Supporting Information Fig. S9A), and cSSR 32.2 (Supporting Information Fig. S9B) where J is found upstream of the last gene within the gene cluster. The downstream (and final) gene is lowly expressed in the presence of J (WT), but increases upon the loss of J following DMOG treatment and further increases in the H3.V KO+DMOG. Adjacent genes, upstream of J or within the neighbouring gene cluster, are not affected. Further evidence for read through transcription at a gene cluster internal termination site following J loss is provided by strand-specific RT-PCR analysis (Fig. 6F). Following the decrease in base J in WT cells we detect an increase in a nascent transcript that extends downstream of the proposed termination site. The level of this RNA species increases along with decreasing levels of J in the H3.V KO+DMOG, directly linking the degree of read through with mRNA abundance of the downstream gene.
Fig. 6.
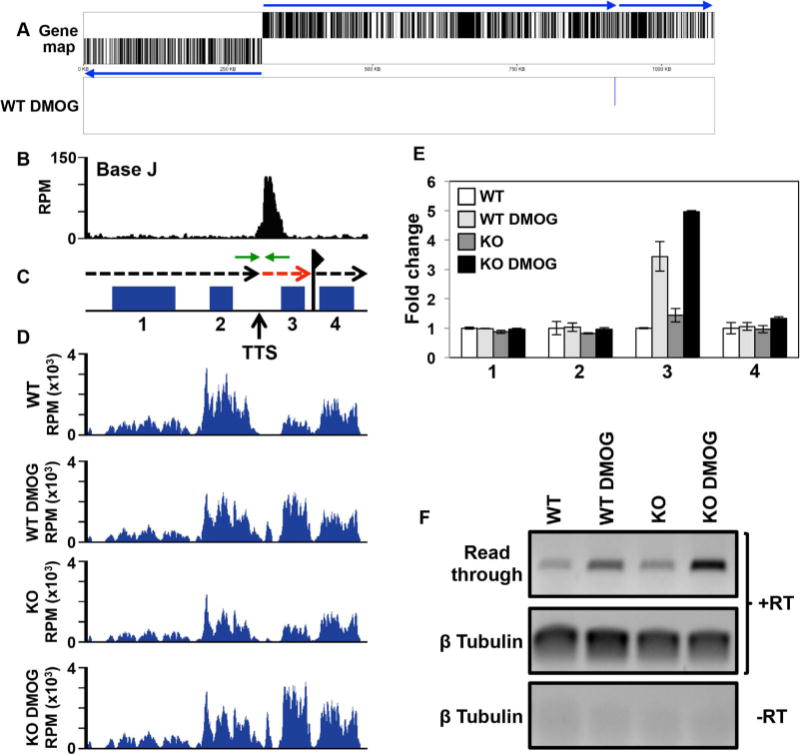
Base J regulates RNAP II termination and gene expression at head-tail regions within gene clusters.
A. Gene map of chromosome 26 is shown where loss of J leads to upregulation of a single mRNA at the end of a gene cluster at a head-tail region. Labeling is as described in Fig. 5A.
B–D. Base J IP-seq reads, ORFs, and mRNA-seq reads are plotted for the head-tail region on chromosome 26 from 912 to 922 kb, as described in Fig. 5. (B) Base J localizes at the transcription termination site (TTS). The vertical arrow indicates the proposed TTS as described in the text (Reynolds et al., 2014, 2016). The black dashed arrow above the map indicates the direction of transcription and the dashed red arrow indicates read through transcription past the TTS. The flag indicates the transcription start site for the downstream gene cluster as indicated by H3 acetylation localization (Thomas et al., 2009). Green arrows flanking the TTS represent the PCR oligos utilized in strand-specific RT-PCR.
E. Plot of the mRNA-seq data for the genes numbered in the ORF map in panel B, as described in Fig. 4C and D. The upregulated gene, 3, is LmjF.26.2280.
F. Strand-specific RT-PCR analysis of read through transcription at the TTS analyzed in B–E. Above the gene map in panel C is a schematic representation (not to scale) of primer location and direction at the TTS. cDNA was synthesized using the reverse primer (relative to transcription) and RNA from WT and H3.V KO cells treated with and without DMOG. Read through was detected by PCR using the same reverse primer used to make the cDNA plus the forward primer, as indicated. Tubulin provides a positive control and a minus RT (-RT) negative control is shown.
Twenty-one of the 33 (64%) upregulated genes in DMOG treated parasites (WT and/or H3.V KO cells) fit a model where derepression occurs as a result of deregulated transcription elongation/termination within a gene cluster following the loss of base J (Supporting Information Table S1 and Fig. 7). These 21 upregulated genes are located in 18 independent gene clusters, out of approximately 184 gene clusters in the L. major genome. In this way, J represses gene expression genome-wide in L. major, often repressing the expression of a single gene, usually the last gene, within a gene cluster. Chromosome maps indicating the genomic location of upregulated genes upon the loss of J are shown in Supporting Information Fig. S10. Thirteen of the 21 genes that fit this model have been identified as developmentally regulated in L. major (Dillon et al., 2015) (see Discussion). These findings overall suggest a conserved role of J in the promotion of RNAP II termination prior to the end of gene clusters in kinetoplastids as a mechanism for regulated gene expression.
Fig. 7.
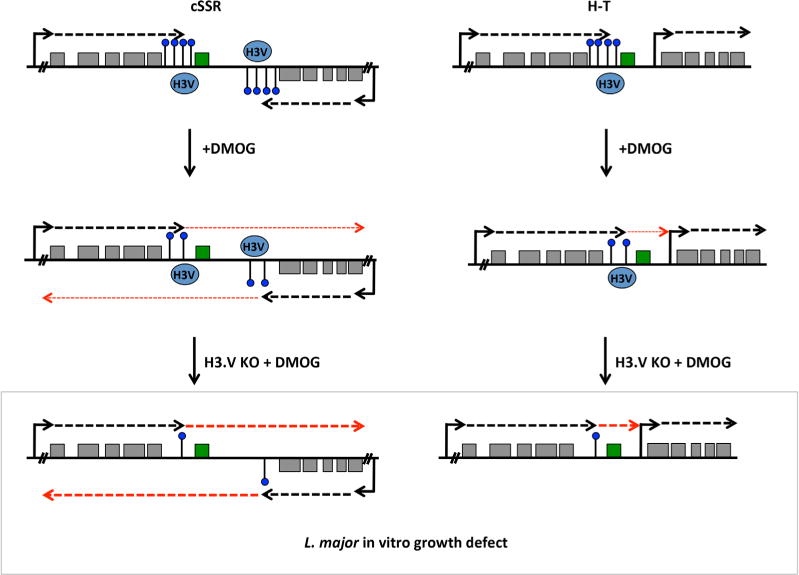
Model of J regulation of RNAP II transcription termination and mRNA expression within gene clusters in L. major.
A cSSR and head-tail region (H-T) is illustrated with polycistronic transcription of genes (grey boxes) indicated by dashed black arrows. For several polycistronic gene clusters RNAP II transcription terminates prior to the final gene in the cluster (highlighted in green), where base J, shown as a black line and a blue dot, is enriched. J is also enriched within the cSSR. H3.V co-localizes with base J at RNAP II termination sites. The transcription start site of the downstream gene cluster at H–T regions is marked by acetylated H3 (not shown). Reduction of base J by DMOG treatment results in read through transcription (thin dashed red arrows), which increases the expression of the last gene on the top strand, but does not significantly affect the expression of other genes. At cSSRs, read through includes transcription into the adjacent opposing gene cluster that does not interfere with production of sense mRNAs. Loss of H3.V leads to some J reduction, but not enough to result in read through transcription (not shown). Loss of H3V in cells treated with DMOG leads to further decreases in J and corresponding increases in read through transcription (thick dashed red arrows) and mRNA levels of the downstream genes. The increased expression of silenced genes in H3.V KO+DMOG correlates with a strong defect in cell growth, suggesting that the essential nature of J in L. major is due to repression of specific genes via regulation of RNAP II termination.
Discussion
The arrangement of functionally unrelated genes into co-transcribed gene clusters and the apparent lack of sequence specific DNA transcription factors has made it difficult to envision regulated gene expression at the level of transcription in kinetoplastids. Evidence has begun to emerge however, that chromatin modifications, including the DNA modification base J and H3.V, can provide some measure of transcriptional control. In T. brucei both J and H3.V promote RNAP II termination prior to the end of gene clusters, thereby repressing downstream genes in the same cluster (Reynolds et al., 2016; Schulz et al., 2016). We demonstrate here that this function of J is conserved between T. brucei and L. major. Whereas H3.V does not have a direct role, base J promotes RNAP II termination and regulation of genes at the end of gene clusters in L. major, including several that are developmentally regulated. We have therefore identified a conserved epigenetic mechanism of regulated gene expression via control of RNAP II transcription termination in kinetoplastids. Consistent with previous findings in L. tarentolae (van Luenen et al., 2012), we also show that a large decrease of base J is detrimental to L. major growth. The absence of major gene expression changes at sites where J loss leads to read through transcription into the adjacent gene cluster and the production of antisense RNAs suggests that defects in cell growth are not due to transcriptional interference, and instead implicates the conserved function of J in the repression genes at the ends of gene clusters in the maintenance of cell viability.
Function of H3.V in termination
In T. brucei, J and H3.V co-localize at RNAP II termination sites genome-wide and loss of either mark at specific sites within gene clusters leads to read through transcription and increases expression of downstream genes (Reynolds et al., 2014, 2016). These studies indicate that base J and H3.V synergistically regulate transcription termination and gene expression. Importantly, the effect of H3.V is not simply due to base J since the levels of the modified base are not reduced in the T. brucei H3.V KO (Cliffe et al., 2010). Rather, H3.V appears to regulate transcription in T. brucei independent of base J. Regions of overlapping transcription in cSSRs of WT T. brucei lead to the production of siRNAs (Tschudi et al., 2012; Zheng et al., 2013). Loss of H3.V, but not base J, leads to increased transcription within these cSSRs and increased production of siRNAs. Variant surface glycoprotein genes within the silent RNAP I transcribed telomeric gene clusters are also derepressed in the H3.V KO. These results indicate a specific role of H3.V, independent of base J, in telomeric gene repression and non-coding RNA expression in T. brucei. Here, we now demonstrate that H3.V and base J co-localize at termination sites in L. major, and confirm that loss of H3.V does not lead to significant effects on RNAP II termination. Therefore, in contrast to its function in T. brucei, H3.V does not directly regulate termination in L. major. Rather, the loss of H3.V in L. major decreases levels of base J by an unknown mechanism, possibly due to subtle chromatin structural changes (Supporting Information Fig. S2). When analyzing read through transcription via antisense RNA production by small RNA-seq (Fig. 2A, Supporting Information Figs. S4A and S5) or SL RNA-seq (Anderson et al., 2013), no evidence of a termination defect was identified in the H3.V KO. Through strand-specific mRNA-seq we did detect a few gene expression changes in the H3.V KO due to read through at internal termination sites (a total of four genes at three different termination sites, see Supporting Information Table S1). Two of these genes are lowly expressed in both WT and H3.V KO cells, potentially explaining why these genes were previously missed. Therefore, at a majority of termination sites (181 out of ~184) enough J remains in the H3.V KO to efficiently terminate transcription, but at three sites J is apparently decreased enough to result in a small degree of read through and subsequently increased steady-state mRNA for the downstream gene(s). Evidence that J regulates these genes is provided by the response to reducing J in WT and the H3.V KO. At each of the three internal termination sites, the genes are upregulated in WT cells where J is reduced via DMOG. Consistent with greater J loss and read through at termination sites upon DMOG treatment compared with deletion of H3.V, the upregulation is higher in WT+DMOG than H3.V KO. Additional loss of J in the H3.V KO, via DMOG, leads to further read through and corresponding increases in the expression of these four genes as well as genes downstream of J in other gene clusters (discussed below). Thus, although the co-localization of J and H3.V at termination sites is conserved between T. brucei and L. major, in contrast to T. brucei, H3.V does not function to promote termination in L. major, but it is involved (potentially indirectly) in the maintenance of J levels.
Essential nature of base J in Leishmania spp
Whereas T. brucei can survive without base J (Cliffe et al., 2009), Leishmania spp. appear to require J because JBP1 KO cells are not viable (Genest et al., 2005). JBP2 KO Leishmania spp. cells are viable, retaining approximately 30% of WT J levels (Vainio et al., 2009; van Luenen et al., 2012). When L. tarentolae JBP2 KO cells are grown in BrdU, they lose more J and die (Vainio et al., 2009). As an explanation for this J-less death, the Borst lab proposed that the resulting massive read through transcription into the adjacent gene cluster interferes with sense RNA synthesis (van Luenen et al., 2012). To test this hypothesis and avoid the toxicity and indirect effects of BrdU, we utilized DMOG to inhibit thymine hydroxylase activity of the JBP enzymes and reduce J synthesis in vivo (Cliffe et al., 2012). DMOG treatment of WT L. major leads to significant reduction of J and read through transcription with minimal effect on parasite growth (Reynolds et al., 2014). Although the deletion of H3.V results in a significant loss of J, the loss is not sufficient for read through transcription or any measurable defect in cell growth. Compared to WT cells, DMOG treatment of the H3.V KO cells leads to the greatest reduction of J (Figs. 2D, 3 and Supporting Information Fig. S4C), more severe read through (Figs. 2A–C, 3, 6F, Supporting Information Figs. S4A and B, S5 and S6), and a more severe growth phenotype (Supporting Information Fig. S7). Therefore, independent approaches to reduce J (BrdU treatment of a JBP2 KO and DMOG treatment of an H3.V KO) have led to the observation that the growth defect following J loss in Leishmania spp. is inversely correlated with J levels and directly correlated with the degree of read through, strongly suggesting that the essential nature of J is due to its role in preventing read through transcription.
To address the essential nature of J, we therefore took advantage of the H3.V KO+DMOG cells to study the consequence of increased read through on gene expression. In contrast to the hypothesis that read through transcription disrupts sense mRNAs, our mRNA-seq analysis indicates only 20 genes are downregulated more than twofold in the H3.V KO+DMOG. Of the 20 downregulated genes, none were reduced by more than 2.4 fold and only four represent the last gene of a convergent gene cluster, where the greatest amount of read through from the opposing gene cluster would be expected. In fact, of the 76 convergent gene clusters in the L. major genome, only six have a significantly downregulated gene within 10kb of the termination site in the H3.V KO+DMOG. Clearly, the expression of convergently arranged genes in L. major is not significantly negatively correlated with the production of antisense RNAs by RNAP II read through transcription. Examination of the genome-wide transcriptome data from WT and the H3.V KO cells with reduced J does reveal slightly downregulated genes near several cSSRs with high read through, suggesting that a transcriptional interference/collision model could explain some of these modest reductions in gene expression. While the changes are much less than our twofold significance cutoff, the downregulation correlates with levels of base J and degree of read through transcription. The lack of significantly downregulated genes at the ends of the majority of gene clusters genome-wide indicates that the process of read through transcription at cSSRs and subsequent dual strand transcription does not negatively affect gene expression at the mRNA level to any significant extent.
Rather than observing downregulation caused by J loss and transcriptional interference, we instead found that the majority of genes affected by J loss were upregulated. Sixteen of the 17 genes affected in WT+DMOG cells were upregulated. The expression of 53 genes was altered in H3.V KO+DMOG cells, which include four highly similar, but distinct genes encoding amastin-like surface proteins. Similar to WT+DMOG cells, 33 out of the 53 genes affected in H3.V KO+DMOG cells were upregulated. 21 of the genes upregulated following J loss are located within at least 10 kb of base J, most of which are immediately downstream of J or overlap regions of J enrichment (Figs. 5, 6 and Supporting Information Table S1), and therefore fit a model in which J promotes termination prior to the gene and represses its expression. Upon J loss, RNAP II elongation continues and gives rise to stabilized processed mRNA. Consistent with this model, of the 16 genes upregulated in WT cells treated with DMOG, 14 are further upregulated in the H3.V KO+DMOG, thus, expression is inversely correlating with J levels. By strand-specific RT-PCR we are also able to detect nascent RNA spanning the termination site and (antisense) RNA downstream of the termination site that is similarly inversely correlated with J levels and directly correlated with increased mRNA levels of the downstream gene. These findings reveal a conserved function of base J among kinetoplastids in the promotion of RNAP II termination prior to the end of gene clusters (Reynolds et al., 2014, 2016; Schulz et al., 2016). In L. major this represents 18 sites of J within gene clusters genome-wide. The location of base J and the genes that are regulated is summarized in Supporting Information Table S1 and genome-wide chromosome maps in Supporting Information Fig. S10.
Regardless of how J loss directly impacts gene expression, 53 genes are differentially expressed after DMOG treating H3.V KO cells, and it is possible that both decreases and increases in transcript abundance contribute to defects in cell growth following J loss. We also cannot exclude the possibility that antisense RNAs generated by transcriptional read through are somehow detrimental to cell viability. These RNAs appear to be processed similarly to sense mRNAs, and although they do not significantly decrease sense mRNA abundance, it is possible that other stages of gene expression are deregulated by antisense RNAs, for example mRNA nuclear export and translation. Future studies are underway to clarify the essential nature of J in L. major. However, given that the major impact of J loss on transcript levels (in terms of the number of genes affected and degree of expression change) is the increased expression of genes at the end of gene clusters, the increasing growth defect with decreased J in L. major promastigotes is more likely linked to the de-repression of genes as opposed to the downregulation of mRNAs or uncharacterized transcriptional interference processes. While the majority of the upregulated genes encode for products of unknown function, approximately 57% of the genes are known to be developmentally regulated in L. major (Dillon et al., 2015). Included are putative amastin-like surface proteins that are upregulated upon differentiation to metacyclic promastigotes. De-repression of these genes following the loss of J may lead to growth defects in our in vitro L. major promastigote cultures. These findings raise the possibility that regulation of J levels during different life stages or in response to environmental signals allows parasites to regulate gene expression at the transcriptional level. Developmental regulation of global levels of base J in kinetoplastids is consistent with this possibility (Ekanayake et al., 2007; van Leeuwen et al., 1998), though high resolution mapping of J across life stages will be necessary to more thoroughly test this hypothesis. Further investigation of the proteins involved in regulating J synthesis, including JBP associated proteins, will also provide insight to the biological significance of J regulation of gene expression in kinetoplastids.
This study has thus identified a conserved function of base J in kinetoplastids in the promotion of RNAP II termination prior to the end of gene clusters and strongly implicates gene repression as the essential role of J in maintaining L. major parasite viability.
Experimental procedures
Enzymes and chemicals
Enhanced chemiluminescence and Hybond-N+ were purchased from Amersham. Goat anti-rabbit HRP (horseradish peroxidase) and goat anti-mouse HRP antibodies were purchased from Southern Biotec Inc. DMOG was purchased from Frontier Scientific, Inc. All other chemicals were purchased from Sigma-Aldrich.
Parasite cell culture
L. major Friedlin strain promastigotes, wild type and the H3.V KO (Anderson et al., 2013), were grown at 26°C in M199 media supplemented with 10% FBS as described (Kapler et al., 1990). DMOG treatment of cells was performed by supplementing media with 5mM DMOG for 2–10 days. Control cells were treated with equal amounts of vehicle (dimethyl sulfoxide).
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR)
To determine histone H3 and H3.V enrichment in the L. major genome, ChIP-qPCR was performed using 2 × 108 cells resuspended in 20 mL of phosphate buffered saline. Cells were cross-linked in 1% formaldehyde for 20 min. Subsequent ChIP steps were performed as previously described (Ekanayake and Sabatini, 2011). L. major histone H3 and H3.V specific antibodies used in this study were previously described (Anderson et al., 2013). Input DNA was used as a positive control for qPCR (10% of the IP). Quantification of selected regions of the genome was performed on an iCycler with an iQ5 multicolor real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA). All primers used in this study were ordered from Integrated DNA Technologies, Inc. or Invitrogen. Primer sequences used in the analysis are listed in Supporting Information Table S4. The reaction mixture contained 5 pmol forward and reverse primer, 2x iQ SYBR green super mix (Bio-Rad Laboratories, Hercules, CA), and 2 μl of template DNA. Standard curves were prepared for each gene using fivefold dilutions of known quantity (100 ng μl−1) of WT DNA. The quantities were calculated using iQ5 optical detection system software.
Determination of the genomic level of J
To quantify the levels of base J at specific regions of the L. major genome, genomic DNA was sonicated and anti J immunoprecipitation was performed as described (Cliffe et al., 2010). Immunoprecipitated J containing DNA was used for qPCR analysis, as described above for ChIP-qPCR analysis.
Western blots
Proteins from 107 cell equivalents were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS page 8% gel), transferred to nitrocellulose and probed with anti-H3 and H3.V as described (Anderson et al., 2013). Anti-alpha tubulin (Sigma-Aldrich) was used as a loading control. Bound antibodies were detected by goat anti-rabbit or goat anti-mouse secondary antibodies conjugated to HRP and visualized by enhanced chemiluminescence. Equal loading was also assessed by Coomassie Brilliant Blue staining.
Strand-specific RNA-seq library construction and data analysis
Small RNAs were isolated from L. major using a Qiagen miRNeasy kit according to the manufacturer’s instructions. 5 × 107 cells were used per sample, and isolated at the log phase of parasite growth. Two small RNA-seq libraries (L. major H3.V KO and H3.V KO+DMOG) were prepared by Vertis Biotechnology AG, Germany, as previously described (Reynolds et al., 2016). All WT and WT+DMOG small RNA-seq data shown here are from our previous study (Reynolds et al., 2014). Libraries were sequenced using Illumina’s HiSeq2000. Reads were mapped to the L. major Friedlin strain genome version 4.2 as previously described (Reynolds et al., 2014). Information about all sequencing data generated in this study is listed in Supporting Information Table S2.
For strand-specific mRNA-seq, total RNA was isolated using Trizol from log phase L. major WT and H3.V KO cells (5 × 107 cells) treated with and without 5mM DMOG for two days. Eight mRNA-seq libraries were constructed (two replicates each; WT, WT+DMOG, H3.V KO and H3.V KO+DMOG) using Illumina’s TruSeq Stranded RNA LT Kit, sequenced by the Georgia Genomics Facility using Illumina’s NextSeq500, and resulting reads were processed and mapped to the L. major Friedlin strain genome version 9.0 as previously described (Reynolds et al., 2016). Determination of transcript abundances was performed as previously described (Reynolds et al., 2016) using the Cufflinks suite version 2.2.1 (Trapnell et al., 2013). To estimate gene expression levels for a condition, replicates were analyzed by Cuffdiff with the L. major Friedlin strain genome version 9.0 annotation. Default parameters were used except specifying library type fr-firststrand. All p-values reported here, determined by Cuffdiff, reflect the FDR-adjusted p-value of 0.05. Correlation coefficients for mRNA-seq replicates of WT, WT+DMOG, H3.V KO and H3.V KO +DMOG were all greater than 0.95. All J IP-seq data shown here are from previously published work (van Luenen et al., 2012). These sequence data have been submitted to NCBI’s Gene Expression Omnibus under GEO Series accession numbers GSE77713 and GSE77632.
Strand-specific reverse transcription-PCR (RT-PCR) analysis of read through transcription
Total RNA was isolated using the hot phenol method, as described previously (Roditi et al., 1989). To ensure complete removal of contaminating genomic DNA, purified RNA was treated with Turbo DNaseI (Invitrogen), followed by phenol:chloroform extraction. Strand-specific RT-PCR was performed as previously described (Reynolds et al., 2014, 2016). Briefly, ThermoScript Reverse Transcriptase from Life Technologies was used for cDNA synthesis at 60–65°C. 1–2 μg of RNA were used to make cDNA using a reverse primer as described in the Figure legends. PCR was performed using GoTaq DNA Polymerase from Promega. A minus-RT control was used to ensure no contaminating genomic DNA was amplified. Primer sequences used in the analysis are listed in Supporting Information Table S4.
Reverse transcription-quantitative PCR (RT-qPCR). Total RNA was obtained using Qiagen RNeasy kits according to manufacturers instructions. First-strand cDNA was synthesized from 1 μg of total RNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) per the manufacturer’s instructions. qPCR was performed using 1–2 μl of template cDNA, and as described above.
Supplementary Material
Acknowledgments
We are grateful for critical reading of the manuscript by Rudo Kieft, Jessica Lopes da Rosa-Spiegler, Whitney Bullard, and Piet Borst. We also thank Nick Rohr for preparing mRNA-seq libraries.
FUNDING
Research reported in this publication was supported by the National Institutes of Health (grant number R01AI109108) to RS; funding from the Office of the Vice President for Research to RJS; funding by the Human Frontier Science Program to TNS; the National Institutes of Health (grant number R0129646) to SMB; a T32 GM007067 fellowship and the Washington University Berg-Morse and Schlesinger Graduate Fellowships to BAA; and the American Heart Association grant 15PRE23240002 to DLR. Funding for open access charge: National Institute of Health R01AI109108.
Footnotes
The authors declare no conflict of interest.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Agabian N. Trans splicing of nuclear pre-mRNAs. Cell. 1990;61:1157–1160. doi: 10.1016/0092-8674(90)90674-4. [DOI] [PubMed] [Google Scholar]
- Anderson BA, Wong IL, Baugh L, Ramasamy G, Myler PJ, Beverley SM. Kinetoplastid-specific histone variant functions are conserved in Leishmania major. Mol Biochem Parasitol. 2013;191:53–57. doi: 10.1016/j.molbiopara.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Boothroyd JC, Cross GA. Transcripts coding for variant surface glycoproteins of Trypanosoma brucei have a short, identical exon at their 5′ end. Gene. 1982;20:281–289. doi: 10.1016/0378-1119(82)90046-4. [DOI] [PubMed] [Google Scholar]
- Borst P. Discontinuous transcription and antigenic variation in trypanosomes. Annu Rev Biochem. 1986;55:701–732. doi: 10.1146/annurev.bi.55.070186.003413. [DOI] [PubMed] [Google Scholar]
- Borst P, Sabatini R. Base J: discovery, biosynthesis, and possible functions. Annu Rev Microbiol. 2008;62:235–251. doi: 10.1146/annurev.micro.62.081307.162750. [DOI] [PubMed] [Google Scholar]
- Bullard W, Lopes da Rosa-Spiegler J, Liu S, Wang Y, Sabatini R. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J Biol Chem. 2014;289:20273–20282. doi: 10.1074/jbc.M114.579821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DA, Thomas S, Sturm NR. Transcription in kinetoplastid protozoa: why be normal? Microbes Infect. 2003;5:1231–1240. doi: 10.1016/j.micinf.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Clayton CE. Life without transcriptional control? From fly to man and back again. EMBO J. 2002;21:1881–1888. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe LJ, Hirsch G, Wang J, Ekanayake D, Bullard W, Hu M, Wang Y, Sabatini R. JBP1 and JBP2 proteins are Fe2+/2-oxoglutarate-dependent dioxygenases regulating hydroxylation of thymidine residues in trypanosome DNA. J Biol Chem. 2012;287:19886–19895. doi: 10.1074/jbc.M112.341974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe LJ, Kieft R, Southern T, Birkeland SR, Marshall M, Sweeney K, Sabatini R. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 2009;37:1452–1462. doi: 10.1093/nar/gkn1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe LJ, Siegel TN, Marshall M, Cross GA, Sabatini R. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 2010;38:3923–3935. doi: 10.1093/nar/gkq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lange T, Liu AY, Van der Ploeg LH, Borst P, Tromp MC, Van Boom JH. Tandem repetition of the 5′ mini-exon of variant surface glycoprotein genes: a multiple promoter for VSG gene transcription? Cell. 1983;34:891–900. doi: 10.1016/0092-8674(83)90546-9. [DOI] [PubMed] [Google Scholar]
- Dillon Laura AL, Okrah K, Hughitt VK, Suresh R, Li Y, Fernandes MC, Belew AT, Corrada Bravo H, Mosser DM, El-Sayed NM. Transcriptomic profiling of gene expression and RNA processing during Leishmania major differentiation. Nucleic Acids Res. 2015;43:6799–6813. doi: 10.1093/nar/gkv656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooijes D, Chaves I, Kieft R, Dirks-Mulder A, Martin W, Borst P. Base J originally found in kinetoplastid is also a minor constituent of nuclear DNA of Euglena gracilis. Nucleic Acids Res. 2000;28:3017–3021. doi: 10.1093/nar/28.16.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekanayake D, Sabatini R. Epigenetic regulation of Pol II transcription initiation in Trypanosoma cruzi: Modulation of nucleosome abundance, histone modification and polymerase occupancy by O-linked thymine DNA glucosylation. Eukaryotic Cell. 2011;10:1465–1472. doi: 10.1128/EC.05185-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekanayake DK, Cipriano MJ, Sabatini R. Telomeric co-localization of the modified base J and contingency genes in the protozoan parasite Trypanosoma cruzi. Nucleic Acids Res. 2007;35:6367–6377. doi: 10.1093/nar/gkm693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekanayake DK, Minning T, Weatherly B, Gunasekera K, Nilsson D, Tarleton R, Ochsenreiter T, Sabatini R. Epigenetic regulation of transcription and virulence in Trypanosoma cruzi by O-linked thymine glucosylation of DNA. Mol Cell Biol. 2011;31:1690–1700. doi: 10.1128/MCB.01277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- Genest PA, ter Riet B, Dumas C, Papadopoulou B, van Luenen HG, Borst P. Formation of linear inverted repeat amplicons following targeting of an essential gene in Leishmania. Nucleic Acids Res. 2005;33:1699–1709. doi: 10.1093/nar/gki304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommers-Ampt JH, Van Leeuwen F, de Beer AL, Vliegenthart JF, Dizdaroglu M, Kowalak JA, Crain PF, Borst P. beta-D-glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell. 1993;75:1129–1136. doi: 10.1016/0092-8674(93)90322-h. [DOI] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PJ, Kooter JM, Borst P. Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell. 1987;51:273–281. doi: 10.1016/0092-8674(87)90154-1. [DOI] [PubMed] [Google Scholar]
- Kapler GM, Coburn CM, Beverley SM. Stable transfection of the human parasite Leishmania major delineates a 30-kilobase region sufficient for extrachromosomal replication and expression. Mol Cell Biol. 1990;10:1084–1094. doi: 10.1128/mcb.10.3.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6:e1001090. doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz JH, Smith HQ, Rusche L, Beverley SM. Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev. 1993;7:996–1007. doi: 10.1101/gad.7.6.996. [DOI] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Nguyen D, Stuart K, Myler PJ. Transcription initiation and termination on Leishmania major chromosome 3. Eukaryotic Cell. 2004;3:506–517. doi: 10.1128/EC.3.2.506-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol Cell. 2003;11:1291–1299. doi: 10.1016/s1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Murphy WJ, Agabian N. A transcriptional analysis of the Trypanosoma brucei hsp83 gene cluster. Mol Biochem Parasitol. 1989;37:115–127. doi: 10.1016/0166-6851(89)90108-4. [DOI] [PubMed] [Google Scholar]
- Nelson RG, Parsons M, Barr PJ, Stuart K, Selkirk M, Agabian N. Sequences homologous to the variant antigen mRNA spliced leader are located in tandem repeats and variable orphons in Trypanosoma brucei. Cell. 1983;34:901–909. doi: 10.1016/0092-8674(83)90547-0. [DOI] [PubMed] [Google Scholar]
- Respuela P, Ferella M, Rada-Iglesias A, Aslund L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J Biol Chem. 2008;283:15884–15892. doi: 10.1074/jbc.M802081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds D, Cliffe L, Forstner KU, Hon CC, Siegel TN, Sabatini R. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014;42:9717–9729. doi: 10.1093/nar/gku714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds D, Cliffe L, Sabatini R. 2-Oxoglutarate-dependent hydroxylases involved in DNA base J (α-D-Glucopyranosyloxymethyluracil) synthesis. In: Schofield CJ, Hausinger RP, editors. 2-Oxoglutarate-Dependent Oxygenases. Cambridge, U.K.: Royal Society of Chemistry; 2015. [Google Scholar]
- Reynolds D, Hofmeister BT, Cliffe L, Alabady M, Siegel TN, Schmitz RJ, Sabatini R. Histone H3 variant regulates RNA polymerase II transcription termination and dual strand transcription of siRNA loci in Trypanosoma brucei. PLoS Genet. 2016;12:e1005758. doi: 10.1371/journal.pgen.1005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roditi I, Schwarz H, Pearson TW, Beecroft RP, Liu MK, Richardson JP, Bühring HJ, Pleiss J, Bülow R, Williams RO. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J Cell Biol. 1989;108:737–746. doi: 10.1083/jcb.108.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Zaringhalam M, Papavasiliou FN, Kim HS. Base J and H3.V Regulate transcriptional termination in Trypanosoma brucei. PLoS Genet. 2016;12:e1005762. doi: 10.1371/journal.pgen.1005762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Merritt C, Baugh L, Stuart K, Myler PJ. Tb927.10.6900 encodes the glucosyltransferase involved in synthesis of base J in Trypanosoma brucei. Mol Biochem Parasitol. 2014;196:9–11. doi: 10.1016/j.molbiopara.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Kemp LE, Figueiredo LM, Lowell JE, Fenyo D, Wang X, Dewell S, Cross GA. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009;23:1063–1076. doi: 10.1101/gad.1790409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RE, Boothroyd JC. Evidence for Trans splicing in trypanosomes. Cell. 1986;47:527–535. doi: 10.1016/0092-8674(86)90617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Green A, Sturm NR, Campbell DA, Myler PJ. Histone acetylations mark origins of polycistronic transcription in Leishmania major. BMC Genomics. 2009;10:152. doi: 10.1186/1471-2164-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschudi C, Fau SH, Ullu E. Small interfering RNA-producing loci in the ancient parasitic eukaryote Trypanosoma brucei. BMC Genomics. 2012;13:427. doi: 10.1186/1471-2164-13-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio S, Genest PA, ter Riet B, van Luenen H, Borst P. Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base J. Mol Biochem Parasitol. 2009;164:157–161. doi: 10.1016/j.molbiopara.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Van der Ploeg LH, Liu AY, Michels PA, De Lange TD, Borst P, Majumder HK, Weber H, Veeneman GH, Van Boom J. RNA splicing is required to make the messenger RNA for a variant surface antigen in trypanosomes. Nucleic Acids Res. 1982;10:3591–3604. doi: 10.1093/nar/10.12.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Taylor MC, Mondragon A, Moreau H, Gibson W, Kieft R, Borst P. beta-D-glucosyl-hydroxymethyluracil is a conserved DNA modification in kinetoplastid protozoans and is abundant in their telomeres. Proc Natl Acad Sci USA. 1998;95:2366–2371. doi: 10.1073/pnas.95.5.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luenen HG, Farris C, Jan S, Genest PA, Tripathi P, Velds A, Kerkhoven RM, Nieuwland M, Haydock A, Ramasamy G, et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional read-through in Leishmania. Cell. 2012;150:909–921. doi: 10.1016/j.cell.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LL, Wen YZ, Yang JH, Liao JY, Shao P, Xu H, Zhou H, Wen JZ, Lun ZR, Ayala FJ, et al. Comparative transcriptome analysis of small non-coding RNAs in different stages of Trypanosoma brucei. RNA. 2013;19:863–875. doi: 10.1261/rna.035683.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.