

Abstract
The Ras/mitogen-activated protein kinase (MAPK) signaling pathway is tightly controlled by negative feedback regulators, such as the tumor suppressor SPRED1. The SPRED1 gene also carries loss-of-function mutations in the RASopathy Legius syndrome. Growth factor stimulation translocates SPRED1 to the plasma membrane, triggering its inhibitory activity. However, it remains unclear whether SPRED1 there acts at the level of Ras or Raf. We show that pharmacological or galectin-1 (Gal-1)-mediated induction of B- and C-Raf-containing dimers translocates SPRED1 to the plasma membrane. This is facilitated in particular by SPRED1 interaction with B-Raf and, via its N terminus, with Gal-1. The physiological significance of these novel interactions is supported by two Legius syndrome-associated mutations that show diminished binding to both Gal-1 and B-Raf. On the plasma membrane, SPRED1 becomes enriched in acidic membrane domains to specifically perturb membrane organization and extracellular signal-regulated kinase (ERK) signaling of active K-ras4B (here, K-ras) but not H-ras. However, SPRED1 also blocks on the nanoscale the positive effects of Gal-1 on H-ras. Therefore, a combinatorial expression of SPRED1 and Gal-1 potentially regulates specific patterns of K-ras- and H-ras-dependent signaling output. More broadly, our results open up the possibility that related SPRED and Sprouty proteins act in a similar Ras and Raf isoform-specific manner.
INTRODUCTION
SPRED (Sprouty-related proteins with an EVH1 domain) proteins are Sprouty-related negative modulators of Ras/mitogen-activated protein kinase (MAPK) signaling, and they have been shown to attenuate extracellular signal-regulated kinase (ERK) activity following various extracellular mitogenic stimuli (e.g., growth factors, cytokines, and chemokines) (1–5). The mammalian SPRED family contains four members, SPRED1, SPRED2, SPRED3, and Eve-3, the last of which is a splice variant of SPRED3 (1, 2, 6). SPRED1 and SPRED2 proteins contain an N-terminal enabled/vasodilator-stimulated phosphoprotein homology-1 (EVH1) domain and a cysteine-rich C-terminal Sprouty-related (SPR) domain, separated by a small c-Kit-binding domain (KBD) (2, 7). The SPR domain is necessary for membrane anchoring following growth factor stimulation (5), and both EVH1 and SPR domains have been implicated in ERK suppression (5, 8, 9). SPRED3 lacks a functional KBD and shows lower inhibitory activity than SPRED1 and -2, suggesting that the KBD also participates in inhibiting ERK activity (1–5).
The precise mechanism of action of how SPRED proteins block MAPK signaling remains unclear. Overexpression of SPRED1 was suggested to increase the recruitment of the Ras effector Raf to the plasma membrane, where its association with Ras does not, however, lead to Raf activation (1, 2, 6). Instead, SPRED1 was reported to prolong Ras-Raf complexation, thus withdrawing Raf from activation by phosphorylation (2, 7). Another study showed that overexpression of SPRED1 inhibits Ras activation, as evidenced by decreased levels of GTP-bound Ras (5, 8). Consistent with this last model, SPRED1 was reported to reduce GTP-Ras levels and MAPK signaling by corecruiting neurofibromin (NF1), a Ras-inactivating and GTPase-activating protein (GAP), to the plasma membrane (5, 8–10).
Despite these mechanistic uncertainties, the important role of SPRED proteins in developmental processes is well appreciated. In vivo studies of SPRED1- or SPRED2-deficient mice have demonstrated that lack of a single SPRED protein causes dysfunctions in bone development, hematopoietic processes, and allergen-induced airway eosinophilia, without affecting viability and fertility of the mice (3, 11, 12). In contrast, the double knockout of SPRED1 and SPRED2 is lethal (13), suggesting a significant role of these proteins during development. In humans, germ line loss-of-function mutations in SPRED1 were discovered in patients with Legius syndrome. This syndrome belongs to a class of developmental disorders called RASopathies, as they are caused by mild overactivation of the Ras/MAPK pathway (14–16). Loss-of-function mutations in this syndrome typically lead to a C-terminally truncated protein (14), underscoring the significance of the C-terminal domain for the biological activity of these proteins.
The Ras/MAPK signaling pathway is an essential regulator of cellular differentiation and survival. Its aberrant regulation is typically implicated in malignant transformation (17, 18). Around 30% of all human tumors carry a mutation in HRAS, NRAS, or KRAS genes. KRAS is the most frequently mutated isoform, with oncogenic mutations detected in 22% of all screened tumors, while the rates for mutations in HRAS and NRAS are lower (8% and 3%, respectively) (19, 20). KRAS is expressed as two splice variants (K-ras4A and K-ras4B) that are both expressed in cancer (21); however, K-ras4B (here, K-ras) is the predominant splice variant and has gained most attention in cancer studies. Ras proteins require membrane association in order to be biologically active (22). GTP-loaded Ras recruits downstream effectors to the plasma membrane (23). Three Raf effector isoforms, A-Raf, B-Raf, and C-Raf (often referred to as Raf-1), can be distinguished in mammals. They function as the initiating activators of the three-tiered MAPK signaling (Raf-MEK-ERK) cascade. All three Ras isoforms can promiscuously activate Raf, with the most potent activator being K-ras (24, 25). Raf binds to the effector region of GTP-Ras, which is the first signal to recruit cytosolic Raf to the plasma membrane. As a consequence, 14-3-3 protein is released from the N-terminal region of Raf, which liberates its autoinhibitory closed conformation (26, 27). Moreover, homo- or heterodimerization of the Raf kinases is a crucial step in Raf activation. Dimerization allows for Raf transactivation; i.e., an “activator” Raf kinase allosterically induces cis-autophosphorylation and thus activation of a “receiver” Raf kinase (28). In this model, phosphorylation of an N-terminal acidic (NtA) motif is required for a Raf kinase to become an activator. As this motif is constitutively phosphorylated in B-Raf, it is the primary activator. On the other hand, C-Raf requires phosphorylation of the NtA in a MEK-dependent manner, explaining the positive feedback loop between MEK and C-Raf. Moreover, B-Raf/C-Raf heterodimers are more potent MEK activators than the respective homodimers (29–31). In recent years, it has become clear that Raf inhibitors can paradoxically activate the MAPK pathway by promoting homo- and/or heterodimerization of the Raf isoforms (32–34).
Galectin-1 (Gal-1) belongs to a family of β-galactoside binding proteins with conserved carbohydrate recognition domains (35). Galectins are multifunctional proteins, which regulate both intra- and extracellularly several functions, including cell-cell adhesion, migration, and protein trafficking (36, 37). Galectins also have a role in tumor progression and metastasis, and their expression is often associated with a poor prognosis (38). We have recently shown that dimeric Gal-1 binds to the Ras binding domain (RBD) of effectors, and both the intact Gal-1 dimer interface and that to the RBD are required for Gal-1 to positively regulate the nanoscale clustering of GTP–H-ras and negatively regulate that of GTP–K-ras (39). These results therefore imply the striking possibility that Gal-1 is an endogenous Raf dimer stabilizer.
While SPRED1 is a well-established negative regulator of the Ras/MAPK signaling pathway, it is not known exactly how it is translocated to the plasma membrane and which Ras or Raf isoform it targets. Here, we show that either epidermal growth factor (EGF) stimulation or Gal-1 expression leads to SPRED1 plasma membrane translocation in a B-Raf- and C-Raf-dependent manner. Gal-1 appears to directly interact with SPRED1, while SPRED1 most prominently binds to B-Raf. Legius syndrome-associated SPRED1 mutants are defective in Gal-1 and B-Raf binding. Gal-1-mediated SPRED1 plasma membrane translocation then specifically disturbs K-ras but not H-ras membrane anchorage with a subsequent loss of ERK signaling. Our results reveal a complex mechanism of how SPRED1 specifically attenuates K-ras-associated MAPK signaling.
MATERIALS AND METHODS
Plasmids and molecular cloning.
Mouse wild-type pcDNA3-SPRED1 and a pcDNA3-ΔC-SPRED1 deletion mutant (26-amino-acid deletion in the C terminus) have been described previously (2, 5). To generate plasmids pmGFP-SPRED1 (where mGFP is monomeric green fluorescent protein) and pmGFP-ΔC-SPRED1, SPRED1 sequences from pcDNA3-SPRED1 and pcDNA3-ΔC-SPRED1, respectively, were subcloned into the XbaI and HindIII restriction sites of the plasmid pmGFP-H-rasG12V. To generate the construct mCherry-SPRED1, the SPRED1 sequence from pcDNA3-SPRED1 was subcloned into the EcoRI and XbaI restriction sites of the plasmid pmCherry-C1 (Clontech Laboratories, Inc., Mountain View, CA). In order to generate the N-terminal EVH1 deletion of SPRED1 (124-amino-acid deletion; ΔN-SPRED1), the sequence of SPRED1 was first amplified from plasmid mGFP-SPRED1 by PCR using the forward primer 5′-CAGGATCCGGGTGCCCAGCGTC-3′ (Clontech) and the reverse primer 5′-TCTAGATCCGGTGGATCCCGGGCC-3′ (Clontech). The amplified PCR product was purified and subcloned into pCR II-Blunt-TOPO (Invitrogen, Carlsbad, CA). Then, ΔN-SPRED1 (SPRED1125–444) was subcloned into XbaI and BamHI restriction sites of mGFP-SPRED1. Plasmid mGFP-SPRED1 served as a template to which mutations were introduced by site-directed mutagenesis (GenScript USA, Inc., Piscataway, NJ) in order to generate plasmids pmGFP-SPRED1-T102R and pmGFP-SPRED1-P415A. Plasmids for mCherry–H-rasG12V, mCherry–K-rasG12V, and mGFP–K-rasG12V were described elsewhere (40). Plasmids for mRFP–Gal-1 (where mRFP is monomeric red fluorescent protein) and mGFP–H-rasG12V were described in Abankwa et al. (41, 42), and pcDNA3-Gal-1 was described in Paz et al. (43). Plasmids containing the full C-terminal hypervariable region of K-ras4B or H-ras (pmGFP-CTK or pmGFP-CTH, respectively) were described elsewhere (42, 44, 45). Plasmids pmRFP-CTK, pmRFP-CTH, and pEGFP-B-Raf (where EGFP is enhanced GFP) were generous gifts from John F. Hancock. Plasmids pEGFP-LactC2 (46) and GFP-ARNO-PH (where PH is pleckstrin homology) (47) were described previously. GFP-C1-PLCδ-PH (where PLCδ is phospholipase Cδ) was from Addgene (21179; Cambridge, MA). pEGFP-A-Raf was from Angela Baljuls, and pEGFP-C-Raf was from Krishnaraj Rajalingam.
Cell culture and transfections.
Human embryonic kidney 293 (HEK293)-EBNA cells were described in Meissner et al. (48), and MDCK cells stably expressing mGFP-K-rasG12V or mGFP-H-rasG12V were described in Cho et al. (49). Baby hamster kidney 21 (BHK21) cells were obtained from the ATCC repository. All cell lines were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were passaged at 80 to 90% confluence every 3 to 4 days.
JetPRIME transfection reagent (Polyplus Transfection, Inc., Illkirch, France) was used for transfections of subconfluent HEK293-EBNA or MDCK cells according to the manufacturer's instructions. The cells were used for experiments after an expression time of 24 h.
Growth factor and inhibitor treatments.
Cells were first transfected for 24 h with mGFP-SPRED1 alone or together with mCherry-SPRED1 using JetPRIME according to the manufacturer's protocol. For EGF stimulation, the cells were washed twice with starvation medium (DMEM without 10% FCS) and starved for 5 h at 37°C. The cells were then stimulated with 100 ng/ml EGF (Sigma-Aldrich) for 10 min at 37°C and fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). For inhibitor treatments, the cells were treated with 50 μM sorafenib (S1040; Selleckchem, Munich, Germany) for 2 h, with 10 μM PLX4720 (S1152; Selleckchem) for 1 h, with 10 μM PLX7904 (HY-18997; MedChem Express, Princeton, NJ), or with 10 μM PLX8394 (HY-18972; MedChem Express) for 1 h at 37°C and fixed with 4% PFA. For treatment with inhibitor plus EGF, the cells were first starved as above and then treated with sorafenib (50 μM; 2 h at 37°C), PLX7904 (10 μM; 1 h at 37°C), or PLX8394 (10 μM; 1 h at 37°C) and finally stimulated with EGF (100 ng/ml; 10 min at 37°C) and fixed with PFA. Control samples for sorafenib were treated with 0.5% dimethyl sulfoxide (DMSO; diluted in the growth medium) for 2 h at 37°C, and control samples for PLX4720, PLX7904, and PLX8394 were treated with 0.1% DMSO for 1 h at 37°C.
Western blot analysis.
Samples were separated in 10% SDS-polyacrylamide gels and electroblotted onto polyvinylidene difluoride membrane (Immobilon-P transfer membrane; Merck Millipore, Darmstadt, Germany) or onto nitrocellulose membrane (Amersham Hybond-ECL; GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). The following antibodies were used: phospho-p44/42 MAPK (9101) and p44/42 MAPK (9102) (both from Cell Signaling Technology, Beverly, MA); ARAF (sc408), BRAF (sc166), CRAF (sc133), and SPRED1 (sc98290) (all from Santa Cruz, Inc., CA); galectin-1 (500-P210; Peprotech); GFP (3999-100; BioVision); and β-actin (A-1978; Sigma-Aldrich). Horseradish peroxidase (HRP)-conjugated secondary antibodies (chicken anti-mouse IgG–HRP [sc2954] and goat anti-rabbit IgG-HRP [sc2004]; Santa Cruz) were used as secondary antibodies. The bands were visualized by enhanced chemiluminescence (ECL; Bio-Rad, Hercules, CA) using a Chemidoc MP system (Bio-Rad). Band intensities were normalized to the β-actin or ERK1/2 level using Image Lab software (Bio-Rad). Averages from two to three biological repeats were calculated.
Coimmunoprecipitation assay (GFP-Trap).
EGFP-tagged full-length A-Raf, B-Raf, or C-Raf was cotransfected in HEK cells with untagged SPRED1 for 24 h. Cells were harvested and washed with ice-cold phosphate-buffered saline (PBS) and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, 137 mM NaCl, 1% glycerin, 1 mM sodium orthovanadate, 0.5 mM EDTA). Immunoprecipitation was then performed using GFP-Trap A (agarose) beads according to the manufacturer's protocol (ChromoTek). Samples were separated in a 10% SDS-polyacrylamide gel and electroblotted onto polyvinylidene difluoride membrane (Merck Millipore).
siRNA experiments.
The following small inhibitory RNAs (siRNAs) were used: human RAF-1 (L-003601-0005), human BRAF (L-003460-0005), human ARAF (L-003563-0005), and, as a control, a nontargeting negative-control siRNA (D-001810-10-05) (all On-Target Plus SMART Pool siRNAs; Dharmacon RNA Technologies, Lafayette, CO). First, cells were transfected with siRNAs (25 nM each) using JetPRIME according to the manufacturer's protocol for siRNA transfections. After 24 h, the cells were transfected with mGFP-SPRED1 alone or cotransfected with mCherry-SPRED1 or mRFP–Gal-1 using JetPRIME according to the manufacturer's instructions, and the incubations were continued for another 24 h. For EGF stimulations, the cells were first transfected with siRNAs, as described above, for 24 h. The cells were then serum starved for 5 h, which was followed by transfection with mGFP-SPRED1 for another 24 h under serum-free conditions; alternatively, after siRNA treatments, the cells were cotransfected with mGFP-SPRED1 and mCherry-SPRED1 for 24 h, followed by starvation for 5 h. The knockdown efficiency of the siRNAs was shown by SDS-PAGE and Western blot analysis of the respective proteins. Band intensities were normalized to the level of β-actin using Image Lab software (Bio-Rad).
In situ PLA.
BHK21 cells were cultured on coverslips and fixed with ice-cold methanol for 10 min. Samples were incubated with Santa Cruz mouse monoclonal antibodies against A-Raf (sc-135820), B-Raf (sc-5284), or C-Raf (sc-376142) and rabbit polyclonal anti-SPRED1-antibody (PA5-20617; Thermo Fisher) for 1 h. A proximity ligation assay (PLA) was carried out with a Duolink II in situ PLA kit (Sigma-Aldrich) according to the instructions of the manufacturer (Olink Biosciences, Sweden). Coverslips were mounted on glass slides using Duolink in situ mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma-Aldrich).
Immunofluorescence (IF) labeling and confocal microscopy.
MDCK cells were fixed with ice-cold methanol (10 min) and ice-cold acetone (5 min). Cells were then blocked with PBS (Sigma-Aldrich) containing 3% bovine serum albumin (BSA) for 1 h at RT and treated with antibodies diluted in 3% BSA-PBS. Monoclonal anti-c-myc antibody (clone 9E10; Sigma-Aldrich), which specifically recognizes the c-myc tag, was used to detect myc-tagged wild-type SPRED1. Highly pre-cross-absorbed goat anti-mouse antibody–Alexa Fluor 647 (A-21235; Thermo Fisher Scientific, Waltham, MA) was used as a secondary antibody against mouse primary antibody. The cells were mounted in Mowiol 4-88 (81381; Sigma-Aldrich).
HEK cells were fixed with 4% PFA for 20 min at RT and blocked with blocking solution (3% BSA and 0.3% Triton X-100 in PBS) for 1 h at RT. The cells were then treated with myc-tagged mouse monoclonal antibody labeled with Alexa Fluor 647 conjugate (2233; Cell Signaling), which was diluted in antibody blocking solution (1% BSA and 0.3% Triton X-100 in PBS). After overnight incubation at +4°C, the cells were washed with PBS and mounted in Mowiol 4-88.
Both IF-labeled proteins and fluorescently tagged plasmids were examined with a Zeiss LSM780 confocal microscope (Zeiss, Jena, Germany) (63× water immersion objective; numerical aperture [NA], 1.2; mGFP excitation at 488 nm, mRFP/mCherry excitation at 543 nm, and Alexa Fluor 647-secondary antibody excitation at 633 nm) by using Zen2010 software (Zeiss). For the PLA assay, images were acquired with a 40× water immersion objective (NA, 1.2; DAPI excitation at 360 nm and Duolink in situ detection reagent red excitation at 594 nm). Imaging was performed at the Cell Imaging Core, Turku Center for Biotechnology, University of Turku and Åbo Akademi University. All images were prepared and PLA signals were analyzed with Fiji, an image processing platform based on ImageJ (50). Approximately 30 cells from two to three independent experiments, with 10 to 15 cells from each experiment, were imaged.
FRET imaging using fluorescence lifetime microscopy (FLIM).
HEK cells were grown on coverslips overnight and transfected with an mGFP-tagged donor construct and mCherry- or mRFP-tagged acceptor construct (1:3 ratio; 0.5 μg:1.5 μg of plasmid DNA) using JetPRIME for 24 h. This realizes comparable expression ratios across all expression levels, as shown previously (41). Cells were then fixed with 4% PFA and mounted with Mowiol 4-88 on microscope slides. Fluorescence lifetimes of the mGFP- or EGFP-tagged donor constructs were measured using a fluorescence lifetime imaging attachment (Lambert Instruments, Leutingwolde, The Netherlands) on an inverted microscope (Zeiss Axio Observer.D1) as previously described (51). Fluorescein (0.01 mM, pH 9) was used as a lifetime reference standard. In addition, it served to calibrate a fixed setting that allows acquisition of data from cells with comparable expression levels. Two or three biological repeats were performed, and the apparent fluorescence resonance energy transfer (FRET) efficiency was calculated from obtained fluorescence lifetimes (51). Our well-established membrane FRET assay has been described elsewhere (45, 52–54).
Statistical analysis.
Statistical differences were determined using analysis of variance (ANOVA) complemented by Tukey's honest significant difference (HSD) test. Prism, version 5, software (GraphPad Software, Inc., La Jolla, CA) was used to perform the analyses. Statistical significance levels are shown in the figure legends.
RESULTS
Raf inhibitor or galectin-1 treatment translocates SPRED1 to the plasma membrane.
The C terminus of SPRED1 is required for its plasma membrane translocation and ability to suppress MAPK signaling (5, 55). Translocation can be triggered by stimulation with growth factors, such as EGF (Fig. 1A), suggesting that it is required for mediating the negative feedback on MAPK signaling. However, it is presently unclear how plasma membrane translocation is enabled.
FIG 1.

Raf inhibitor sorafenib or galectin-1 induces SPRED1 plasma membrane translocation. (A) Confocal images of HEK cells transiently transfected with mGFP-SPRED1 for 24 h, followed by control treatment (0.5% DMSO in growth medium) for 2 h, serum starvation for 5 h and stimulation with EGF (100 ng/ml, 10 min), serum starvation for 5 h and sorafenib treatment (50 μM, 2 h) followed by EGF stimulation (100 ng/ml, 10 min), or sorafenib treatment alone (50 μM, 2 h), as indicated. Scale bars, 10 μm. (B) HEK cells were transiently transfected with mGFP-SPRED1 for 24 h, followed by control treatment (0.1% DMSO in growth medium) for 1 h, serum starvation for 5 h and stimulation with EGF (100 ng/ml, 10 min), serum starvation for 5 h and PLX7904 treatment (10 μM, 1 h) followed by EGF stimulation (100 ng/ml, 10 min), or PLX7904 treatment alone (10 μM, 1 h), as indicated. The cells were imaged by confocal microscopy. Scale bars, 10 μm. (C) A SPRED1 membrane translocation FRET assay was conducted in HEK cells transiently coexpressing mGFP-/mCherry-SPRED1. After 24 h of transfection, the cells were treated as described for panel B and fixed. Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001; ns, nonsignificant. (D) mGFP-SPRED1 and mRFP–Gal-1 were transiently cotransfected in HEK cells for 24 h and imaged by confocal microscopy. Scale bars, 10 μm. (E) A SPRED1 membrane translocation FRET assay was conducted in HEK cells transiently coexpressing mGFP-/mCherry-SPRED1. The cells were coexpressed with untagged Gal-1 or starved for 5 h and stimulated with EGF (100 ng/ml, 10 min) or treated with sorafenib (50 μM, 2 h). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. (F) A SPRED1 membrane translocation FRET assay was conducted in HEK cells transiently coexpressing mGFP-/mCherry-SPRED1. The cells were starved for 5 h (control) or coexpressed with untagged Gal-1 and starved for 5 h, or starved for 5 h and stimulated with EGF (100 ng/ml, 10 min), or coexpressed with untagged Gal-1 followed by starvation for 5 h and stimulation with EGF (100 ng/ml, 10 min). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. **, P < 0.01; ***, P < 0.001. (G) mGFP-ΔC-SPRED1, a C-terminal deletion mutant, or mGFP-ΔN-SPRED1, an N-terminal deletion mutant, was cotransfected together with mRFP–Gal-1 for 24 h in HEK cells and imaged by confocal microscopy, as indicated. The schematic representation of domain structures shows the differences between wild-type (WT) SPRED1 and the deletion mutants. The ΔC-SPRED1 mutant lacks 26 amino acids from the C terminus, and the ΔN-SPRED1 mutant lacks 124 amino acids from the N terminus. EVH1, Ena/vasodilator-stimulated phosphoprotein (VASP) homology-1 domain; KBD, c-Kit-binding domain; SPR, Sprouty-related domain. Scale bars, 10 μm. (H) Interaction between WT mGFP-SPRED1 or mGFP-tagged SPRED1 mutants with mRFP–Gal-1 was detected using FRET in HEK cells transiently expressing the constructs as indicated. Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001.
In order to establish whether signaling downstream of Raf kinase activity is required for this relocalization event, we stimulated HEK293-EBNA (here HEK) cells expressing mGFP-SPRED1 with EGF for 10 min in the presence of the Raf inhibitor sorafenib. Sorafenib was not able to block SPRED1 translocation, suggesting that events downstream of Raf kinase activity are not required (Fig. 1A). Surprisingly, the control sample revealed that sorafenib alone was sufficient to efficiently translocate SPRED1 to the plasma membrane (Fig. 1A). This was also the case with another potent Raf inhibitor, PLX4720, which clearly induced SPRED1 translocation to the plasma membrane (see Fig. S1A and B in the supplemental material). We hypothesized that this sorafenib- and PLX4720-triggered event is associated with their ability to dimerize Raf kinases (33, 34, 56). This was confirmed using the next-generation Raf inhibitor PLX7904 and its optimized analogue PLX8394, which do not induce Raf dimers or paradoxical activation of the MAPK pathway in the presence of mutant Ras (so called “paradox breakers”) (57). Confocal imaging showed that under normal serum conditions, neither of the next-generation Raf inhibitors induced SPRED1 translocation to the plasma membrane (Fig. 1B; see also Fig. S1C). Yet EGF stimulation was able to induce SPRED1 translocation also in the presence of PLX7904 (Fig. 1B) and PLX8394 (see Fig. S1C), demonstrating that Raf phosphorylation is not essential for SPRED1 relocalization.
In order to quantify these SPRED1 translocation events to the plasma membrane, we conducted fluorescence resonance energy transfer (FRET) experiments. In general, FRET reports on molecular proximities due to clustering or interaction events. Typically, a donor and an acceptor fluorophore are attached to the complexation partners allowing the detection of FRET if the fluorophore distance remains approximately below 10 nm in the complex. We employed fluorescence lifetime imaging microscopy (FLIM) for fast, robust, and precise quantification of FRET (58). Due to the limited resolution capabilities of our microscopy setup, whole-cell FLIM-FRET values were determined; however, confocal imaging data of the FRET donor readily provided information on the subcellular structures from which FRET emerged.
Specifically, we coexpressed mGFP- and mCherry-tagged SPRED1, expecting that increased concentration of SPRED1 on the plasma membrane would lead to an increase of FRET (SPRED1 membrane translocation FRET assay), as observed for similar biosensors (52, 53). Indeed, compared to the normal serum level control, EGF stimulation significantly increased the FRET (Fig. 1C; see also Fig. S1D in the supplemental material), in line with the increased plasma membrane localization (Fig. 1A; see also Fig. S1C). Conversely, neither of the paradox breaker inhibitors, PLX7904 or PLX8394, blocked the EGF-induced increase in FRET (Fig. 1C; see also Fig. S1D), as expected based on confocal imaging data showing that SPRED1 readily translocates to the plasma membrane (Fig. 1B; see also Fig. S1C).
Moreover, these FRET experiments could quantify the effect of Raf inhibitors on SPRED1 plasma membrane translocation under normal serum levels. While both sorafenib and PLX4720 increased the FRET (Fig. 1E; see also Fig. S1B), this was not the case with the paradox breakers (Fig. 1C; see also Fig. S1D).
Therefore, these Raf inhibitor experiments suggest that Raf dimer induction by first-generation Raf inhibitors is sufficient to translocate SPRED1 to the plasma membrane under normal serum levels. Moreover, EGF-induced translocation of SPRED1 is insensitive to Raf kinase inhibition.
We have recently described the small, dimeric protein galectin-1 (Gal-1) as a candidate Raf dimer stabilizer (39). We therefore explored whether expression of Gal-1 in HEK cells is sufficient to induce SPRED1 translocation. Intriguingly, expression of mRFP-tagged Gal-1 in HEK cells under normal serum levels led to a clear relocalization of mGFP-tagged SPRED1 from the cell cytosol to the plasma membrane (Fig. 1D), which was indistinguishable from what we observed after acute EGF stimulation (Fig. 1A). This was supported by quantification with our SPRED1 membrane translocation FRET assay (Fig. 1E).
However, when EGF stimulation and Gal-1 expression are combined in HEK cells that are naturally almost devoid of Gal-1, their joint effect exceeded that of the individual treatments (Fig. 1F). This suggests that the EGF-induced increased membrane concentration of SPRED1 is significantly enhanced by Gal-1.
SPRED1 requires its C-terminal cysteine-rich Sprouty-related domain for plasma membrane localization (2, 55). On the other hand, the N-terminal EVH1 domains of SPRED proteins also appear to participate in ERK inhibition (2, 9, 59). We therefore studied whether N- or C-terminal SPRED1 deletion mutants still translocated after Gal-1 expression. Confocal imaging showed that Gal-1-induced SPRED1 translocation required an intact C terminus of SPRED1, while an N-terminal deletion mutant (lacking the EVH1 domain) still exhibited some degree of plasma membrane localization (Fig. 1G).
In order to establish whether this effect was mediated by the interaction of Gal-1 and SPRED1, we conducted FRET experiments on HEK cells. We observed significant FRET between SPRED1 and Gal-1 (Fig. 1H), consistent with a direct interaction or close complexation of these proteins. Interestingly, the C-terminal truncation mutant of SPRED1, which localized to the cytoplasm (Fig. 1G), also showed FRET levels similar to those of the full-length protein (Fig. 1H), suggesting that Gal-1 and SPRED1 already exist in a complex in the cell cytoplasm. For the N-terminal truncation mutant, we observed a significant loss of FRET compared to the level in the full-length SPRED1, which may suggest that this part of the protein is involved in mediating the SPRED1–Gal-1 interaction (Fig. 1H). This partial loss of interaction also explains the compromised Gal-1-mediated membrane translocation of the N-terminally truncated SPRED1 (Fig. 1G).
In conclusion, Raf dimer induction by first-generation Raf inhibitors or expression of the bona fide Raf dimer stabilizer Gal-1 drives SPRED1 to the plasma membrane under normal serum levels. Similar to the other biological activities of SPRED1 (2), the activity of Gal-1 required the C terminus of SPRED1, while the N terminus appeared to mediate interaction with Gal-1. Gal-1 may therefore be a novel, significant enhancer of SPRED1 activity.
Depletion of B- or C-Raf abolishes SPRED1 membrane translocation.
In order to establish whether Raf homo- or heterodimers mediate the Gal-1-induced SPRED1 relocalization, we knocked down one or two Raf isoforms, thus relatively enriching concentrations of the remaining isoforms. All specific siRNAs against A-Raf, B-Raf, or C-Raf silenced the expression of their target proteins by 80 to 90% (see Fig. S2A in the supplemental material) without affecting the expression of the other Raf isoforms. We first silenced Raf isoforms for 24 h, followed by cotransfection of mGFP-tagged SPRED1 and mRFP-tagged Gal-1 for a further 24 h. Confocal imaging showed that in A-Raf-silenced cells, SPRED1 translocation to the plasma membrane was unaffected and showed plasma membrane localization similar to that in the cells transfected with negative-control siRNA (Fig. 2A and B). However, knockdown of either B-Raf or C-Raf prevented SPRED1 translocation (Fig. 2B), suggesting an important role of these Raf isoforms in Gal-1-induced SPRED1 translocation. Likewise, EGF-induced translocation was evident in cells transfected with a negative-control siRNA (Fig. 2A, right), but it was abolished if B- or C-Raf expression was downmodulated (Fig. 2C; see also Fig. S2B). This was confirmed by quantification with our SPRED1 membrane translocation FRET assay (Fig. 2D; see also Fig. S2C). Therefore, B-/C-Raf specificity is probably not associated with Gal-1 but with SPRED1. These data were supported by Raf double-knockdown experiments that would have shifted the equilibrium to remaining Raf homodimers. Basically all double knockdowns compromised the Gal-1-induced SPRED1 relocalization (see Fig. S2D).
FIG 2.

Silencing of B-Raf or C-Raf abrogates SPRED1 plasma membrane translocation. (A) Gal-1- or EGF-induced SPRED1 localization under nontargeting control siRNA treatment. (B) A-Raf, B-Raf, and C-Raf were first silenced with specific siRNAs (si- prefixes) in HEK cells for 24 h, and then the cells were transfected with mGFP-SPRED1 and with mRFP–Gal-1 for another 24 h. Scale bars, 10 μm. (C) EGF-induced SPRED1 localization after knockdown of individual Raf proteins. The cells were first treated with an siRNA for 24 h and then serum starved for 5 h, followed by transfection of mGFP-SPRED1 for another 24 h under serum-starved conditions. After that, the cells were stimulated with EGF (100 ng/ml) for 10 min and fixed. Scale bars, 10 μm. (D) A SPRED1 membrane translocation FRET assay was conducted in HEK cells transiently coexpressing mGFP-/mCherry-SPRED1. HEK cells were first siRNA treated for 24 h, followed by cotransfections of SPRED1 constructs for another 24 h. The cells were kept under normal serum conditions and fixed (gray bar). Other HEK cells were first treated with siRNA for 24 h, followed by cotransfections of SPRED1 constructs for another 24 h. After that, the cells were starved for 5 h and stimulated with EGF (100 ng/ml) for 10 min and fixed (blue bars). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001; ns, nonsignificant. (E) HEK cells were transfected with an EGFP-tagged A-Raf, B-Raf, or C-Raf construct together with myc-tagged SPRED1 for 24 h, followed by labeling with myc tag antibody (Alexa Fluor 647 conjugate). The cells were imaged by confocal microscopy. Scale bars, 10 μm.
Next, we examined whether overexpression of Raf isoforms alone could trigger SPRED1 translocation. While overexpression of individual Raf isoforms did not change SPRED1 localization (see Fig. S2E in the supplemental material), coexpression of B-Raf together with C-Raf clearly translocated SPRED1 to the plasma membrane, while the other Raf pairs were not sufficient (Fig. 2E).
In summary, these results suggest that B-/C-Raf dimers are required to translocate SPRED1 to the plasma membrane.
B-Raf is the major Raf isoform interacting with SPRED1.
We next examined whether SPRED1 interacts with the different Raf isoforms. Coimmunoprecipitation analysis of mGFP-tagged SPRED1 found it in a complex with B-Raf but not A-Raf or C-Raf (Fig. 3A). Interestingly, the SPRED1/B-Raf complex was also found in serum-starved cells (Fig. 3B), indicating that complex formation is constitutive. To further analyze the interaction of SPRED1 with Raf isoforms in intact cells, we conducted FLIM-FRET experiments in HEK cells. High FRET was detected between SPRED1 and B-Raf but not with the other Raf isoforms (Fig. 3C). Finally, to examine the association of endogenously expressed SPRED1 and Raf isoforms in situ, we performed proximity ligation assays (PLAs) (Fig. 3D). These experiments confirmed a specific interaction of endogenous SPRED1 with B-Raf.
FIG 3.

SPRED1 specifically interacts with B-Raf. (A) A coimmunoprecipitation assay (GFP-Trap IP) was performed in HEK cells by transfecting the cells with EGFP-tagged A-Raf, B-Raf, or C-Raf together with untagged wild-type SPRED1 for 24 h. Immunoblots were probed with the indicated antibodies. I, input; FT, flowthrough; B, bound fraction. (B) HEK cells were serum starved for 6 h, followed by transfections as described for panel A but under serum-free conditions. (C) Interaction between wild-type mCherry-tagged SPRED1 and EGFP-tagged full-length Raf proteins was detected using FRET in HEK cells transiently expressing the constructs as indicated. Numbers inside or next to the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. (D) A proximity ligation assay (PLA) was performed in BHK21 cells using the indicated combinations of A-Raf, B-Raf, C-Raf, and SPRED1 antibodies. Quantification shows the PLA signal per cell in each case (the total numbers of PLA signal foci were counted and normalized to the number of cells). Sample images are shown below the graph. An endogenous SPRED1-Raf interaction was visualized as red dots by PLA and detected by confocal microscopy. Cell nuclei were stained with DAPI. Numbers inside or above the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. Scale bars, 10 μm.
SPRED1 selectively disturbs membrane anchorage of K-ras.
Sorafenib-induced B-/C-Raf heterodimers have been linked to specific increases in the nanoscale clustering of oncogenic K-ras4B (here, K-ras) (60, 61), which resides in acidic lipid nanodomains (62). While evidence for the localization of SPRED1 to acidic lipid domains exists (55), details on the composition are not known.
We therefore used FRET analysis to map in detail the nanoscale colocalization of SPRED1 and specific lipid probes. Confocal imaging revealed that under normal serum levels, lipid probes EGFP-ARNO {a probe for phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3]} (47), EGFP-LactC2 (phosphatidylserine [PS]) (46), and EGFP-PLCδ [(PI(4,5)P2] (63) localized primarily on the plasma membrane, irrespective of whether SPRED1 is expressed (Fig. 4A; see also Fig. S3 in the supplemental material), whereas under serum-free conditions they localized more to the cell cytoplasm (see Fig. S3).
FIG 4.

SPRED1 disturbs membrane anchorage of K-ras but not H-ras. (A) Confocal images of HEK cells cotransfected with mCherry-SPRED1 together with EGFP-ARNO, EGFP-LactC2, or EGFP-PLCδ for 24 h under normal serum conditions. Scale bars, 10 μm. (B) Nanoscale colocalization of mCherry-SPRED1 and EGFP-tagged lipid probes in HEK cells was detected using FRET. The lipid recognition specificity of the indicated lipid probes is given in italics (right). The cells were transfected with mCherry-SPRED1 and EGFP-tagged lipid probes for 24 h, followed by starvation for 5 h, or kept under normal serum conditions, or starved for 5 h, followed by EGF stimulation (100 ng/ml, 10 min). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. (C) Schematic representation of full-length Ras, and the K- and H-ras hypervariable region (HVR)-derived extended membrane anchors CTK and CTH, respectively. The impact of SPRED1 on nanoscale clustering and membrane anchorage-dependent FRET of HEK cells expressing mGFP/mRFP-CTK or mGFP/mRFP-CTH is illustrated. Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001; ns, nonsignificant. (D) Interaction between mCherry-SPRED1 and mGFP-tagged K-rasG12V or H-rasG12V was detected using FRET in HEK cells transiently expressing the constructs as indicated. After transfection for 24 h, the cells were starved for 5 h, or kept under normal serum conditions, or starved for 5 h followed by EGF stimulation (100 ng/ml, 10 min). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. (E) MDCK cells stably expressing mGFP–K-rasG12V or mGFP–H-rasG12V were transfected with myc-tagged SPRED1. Cells immunolabeled with anti-c-myc antibody followed by labeling with Alexa Fluor 647-secondary antibody were imaged for the indicated constructs using confocal microscopy. Scale bars, 10 μm.
In agreement with the distribution, increased FRET was found between mCherry-SPRED1 and the PS-specific probe EGFP-LactC2 and the PI(4,5)P2-specific probe EGFP-PLCδ under normal serum conditions compared to levels in serum-starved cells (Fig. 4B). In contrast, the PI(3,4,5)P3-specific probe EGFP-ARNO showed very low FRET with SPRED1 under both conditions (Fig. 4B). EGF stimulation significantly increased the FRET of SPRED1 with the PI(3,4,5)P3 probe and with the PS probe (Fig. 4B) but not with the PI(4,5)P2 probe, which already showed very high FRET under normal serum levels (Fig. 4B).
Therefore, these results suggest that upon EGF stimulation and therefore SPRED1 plasma membrane translocation, SPRED1 becomes relatively enriched in PS- and PI(3,4,5)P3-containing domains.
In agreement with the localization in acidic membrane domains that are also populated by K-ras (62, 64), SPRED1 expression perturbed nanoscale clustering-associated FRET of a K-ras-derived but not H-ras-derived FRET biosensor (52), suggesting an effect on nano- or microscale membrane organization (Fig. 4C). SPRED1 specificity toward K-ras was further confirmed by FRET experiments between mGFP-tagged RasG12V and mCherry-SPRED1, which after stimulation with EGF showed a significant increase in FRET with K-rasG12V but not H-rasG12V (Fig. 4D). This confirms that acute EGF stimulation induces SPRED1 translocation to such acidic lipid-enriched plasma membrane domains, where K-ras also resides (62).
Finally, confocal image analysis of the full-length oncogenic Ras proteins stably expressed in MDCK cells confirmed that, after 24 h of expression, SPRED1 led to a specific loss of plasma membrane anchorage of K-rasG12V but not H-rasG12V (Fig. 4E).
Therefore, we conclude that SPRED1 becomes specifically enriched in PS-containing membrane domains to interfere with K-ras membrane anchorage.
Galectin-1 augments the negative activity of SPRED1 on K-ras but not H-ras signaling.
Given that SPRED1 translocation to the plasma membrane is further enhanced by Gal-1 after acute EGF stimulation (Fig. 1F), we tested whether Gal-1-induced translocation can further increase the displacement of K-ras. To this end, we employed our well-established membrane FRET assay that can quantify membrane anchorage and nanoscale clustering of constitutively active Ras (40, 51). FRET of K-rasG12V significantly decreased when K-rasG12V was coexpressed with Gal-1 (Fig. 5A). This observation is consistent with the negative effect of H-rasG12V on K-rasG12V nanoscale clustering (62), which may be further augmented by increased nanoscale clustering of H-ras by Gal-1 (39). A similar negative effect on K-ras FRET was observed for SPRED1 alone (Fig. 5A), which we attributed to the observed loss of plasma membrane anchorage of K-ras (Fig. 4E). However, additional expression of Gal-1 significantly potentiated this effect, suggesting that full plasma membrane translocation of SPRED1 most efficiently disrupts K-ras (Fig. 5A).
FIG 5.
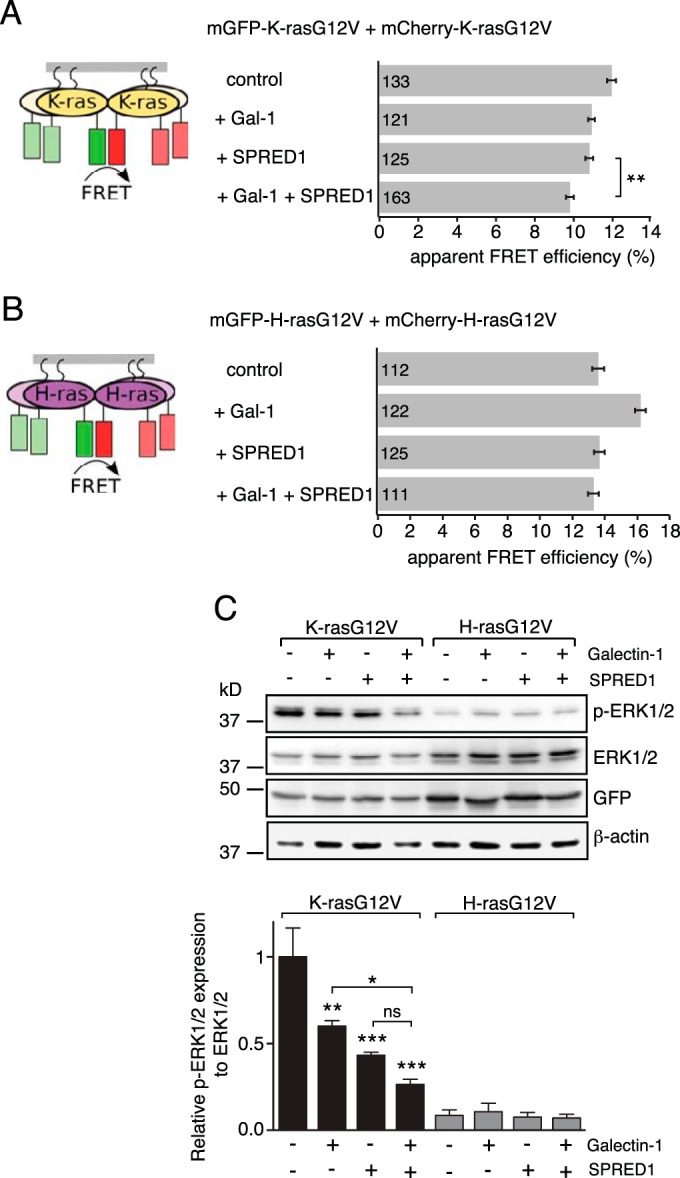
K-ras specific p-ERK suppression by SPRED1 is augmented by Gal-1. (A and B) Membrane FRET of K-rasG12V (A) or H-rasG12V (B) in the presence of SPRED1 and/or Gal-1 in HEK cells is illustrated. Cells were transiently transfected with mGFP–/mCherry–K-rasG12V or mGFP–/mCherry–H-rasG12V and, as indicated, with untagged SPRED1 and/or Gal-1. Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. **, P < 0.01. (C) Representative Western blots of the samples from the experiments shown in panels A and B that were partially processed for immunoblotting. Indicated samples were probed with antibodies as shown on the right. β-Actin was used as a loading control. p-ERK1/2 protein expression levels were normalized to the level of ERK1/2 (three independent biological repeats). Error bars indicate the standard errors of the means. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, nonsignificant.
In contrast, FRET of H-rasG12V remained apparently unaffected (Fig. 5B), except for the well-documented case when Gal-1 was expressed, which increased FRET due to an increase in GTP-H-ras nanoclustering (51, 65, 66). However, coexpression of SPRED1 abrogated this Gal-1 effect, consistent with utilization of Gal-1 in the SPRED1/K-ras context (Fig. 5B).
Importantly, the K-ras-specific effects had a direct impact on ERK signaling (Fig. 5C), as determined on the same samples that were used for the FRET analysis. Quantification of the Western blots showed a significant decrease in phospho-ERK (p-ERK) levels in the K-rasG12V-expressing samples, which correlated with the loss of FRET (Fig. 5A and C). Of note, expression of SPRED1 alone had a significantly stronger negative effect on p-ERK levels than that of Gal-1 alone, and the combination of the two showed a stronger effect than SPRED1 alone. In contrast, cells expressing H-rasG12V had low overall p-ERK levels, which did not show significant differences under the treatment conditions (Fig. 5C).
In summary, our results show that SPRED1 specifically blocks K-ras signaling by disturbing membrane anchorage of K-rasG12V. Gal-1 can significantly augment this negative effect of SPRED1 probably by facilitating plasma membrane localization of SPRED1.
SPRED1 mutations found in Legius syndrome show decreased interaction with Gal-1 and B-Raf.
Germ line loss-of-function mutations in SPRED1 cause Legius syndrome, a rare developmental disorder that shares clinical features with another RASopathy, neurofibromatosis type 1 (14). Our data suggest that SPRED1 translocation depends on B-Raf-containing Raf dimers. Accordingly, we observed FRET between B-Raf (Fig. 3C) and the putative Raf dimer scaffold Gal-1 (Fig. 1H). We therefore asked whether pathogenic missense mutations SPRED1-T102R and SPRED1-P415A, which are found in Legius syndrome (67), would show altered interaction with Gal-1 or B-Raf by FRET.
Both the T102R mutant (harboring the point mutation in the N-terminal EVH1 domain) and the P415A mutant (with a point mutation in the C-terminal SPR domain) showed decreased FRET with Gal-1 compared to the level in the wild-type SPRED1 (Fig. 6A). Furthermore, both mutants showed decreased FRET with B-Raf compared to the level of the wild-type control (Fig. 6B). Thus, our findings propose a substantive role for correct SPRED1/Gal-1/B-Raf complexation in normal SPRED1 function as both of the Legius syndrome mutants showed defective complex formation.
FIG 6.

SPRED1 mutants found in Legius syndrome show decreased interactions with Gal-1 and B-Raf. (A and B) Interaction between mGFP-tagged SPRED1 mutants and mRFP–Gal-1 or mCherry–B-Raf was detected using FRET in HEK cells transiently expressing the constructs as indicated. Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001. (C) SPRED1 membrane translocation FRET assay was conducted in HEK cells transiently coexpressing an mGFP-tagged SPRED1 mutant and mCherry-tagged wild-type SPRED1. The cells were treated with 0.5% DMSO (control cells), or starved for 5 h and stimulated with EGF (100 ng/ml, 10 min), or treated with sorafenib (50 μM, 2 h). Numbers inside the bars correspond to the total number of cells studied in each case. Error bars indicate the standard errors of the means. ***, P < 0.001; ns, nonsignificant.
As the mutants showed decreased complexation with both Gal-1 and B-Raf, we next tested whether plasma membrane translocation of SPRED1 mutants is affected when cells are stimulated with EGF or sorafenib, two treatments which clearly translocated wild-type SPRED1 to the plasma membrane (Fig. 1A and E). We here modified our SPRED1 membrane translocation FRET assay, using mGFP-tagged mutant SPRED1 and mCherry-tagged wild-type SPRED1 as a FRET pair. While the T102R mutant FRET pair showed an EGF- and sorafenib-induced increase of FRET (Fig. 6C) similar to that of the wild-type SPRED1 FRET pair (Fig. 1E), the P415A mutant FRET pair failed to increase FRET, indicating that it cannot translocate to the plasma membrane (Fig. 6C). These results are in agreement with previous observations by Stowe et al. showing that the T102R mutant is able to localize to the plasma membrane, whereas the P415A mutant is mainly in the cytoplasm (9).
In conclusion, two Legius syndrome-associated SPRED1 mutations were defective in B-Raf and Gal-1 complexation, supporting the significance of this newly described interaction also in a pathological setting.
DISCUSSION
SPRED proteins are important tumor suppressors in human hepatocellular carcinoma, where SPRED1 and SPRED2 proteins are downregulated in 84% of cases (68). SPRED1 is also a disease gene in the RASopathy Legius syndrome (14, 15, 69). Likewise, the role of SPRED and Sprouty proteins during development is well established (7, 70). Considering the significance of these proteins in health and disease, it is surprising that their exact mechanism of action is still rather poorly understood.
Here, we present new insight into how SPRED1 negatively regulates Ras/MAPK signaling. Our data suggest the following model (Fig. 7A). In the cell cytoplasm, SPRED1 associates constitutively with B-Raf. SPRED1 translocation to the plasma membrane occurs in conjunction with B-Raf/C-Raf dimerization, which can be stimulated constitutively by Gal-1 and serum or acutely by EGF. Interestingly, this Raf dimer specificity also matches the specificities of sorafenib and PLX4720 (33, 56, 61), which also translocated SPRED1 (Fig. 1A; see also Fig. S1A in the supplemental material). SPRED1 localizes to PS- and PIP2-enriched membrane domains. Colocalization with PS is consistent with the colocalization of SPRED proteins and Rab11 (71). Rab11 is an essential Rab protein in the recycling endosome, a compartment that is enriched in PS (72, 73). SPRED1 localization in PS- and PIP2-enriched domains of the plasma membrane specifically leads to a loss of plasma membrane localization of K-ras but not of H-ras. Another interesting feature of SPRED1-regulated ERK signaling inhibition is our observation that high Gal-1 and SPRED1 expression levels switch off Gal-1-induced H-ras nanoclustering enhancement, which is evident from our FRET data (Fig. 5B). Thus, with high Gal-1 and SPRED1 expression levels, both K-ras- and H-ras-mediated signaling is relatively compromised. Although the exact details of how SPRED1 disrupts K-ras membrane anchorage are still elusive, we conclude that SPRED1 translocates via B-Raf/C-Raf dimers to the plasma membrane to negatively affect K-ras signaling.
FIG 7.

Predicted model for SPRED1-regulated K-ras signaling inhibition. (A) Without Gal-1 expression, SPRED1 localizes mainly to the cell cytoplasm and interacts with B-Raf even in the absence of serum (1). When Gal-1 is expressed at high enough levels in the cells, Gal-1 dimerizes and binds to Raf (2). Gal-1 may thus stabilize Raf dimers on the plasma membrane (that originate from serum stimulation), thus translocating B-Raf-bound SPRED1 to the plasma membrane. SPRED1 specifically requires B-Raf/C-Raf heterodimers for this, which can also be induced by EGF and sorafenib. In the complex, SPRED1 may also interact with C-Raf through dimerization. On the plasma membrane, SPRED1 disrupts specifically K-ras membrane anchorage (3), leading to K-ras mislocalization from the plasma membrane into the cell and subsequent ERK signaling inhibition (4). (B) When Gal-1 levels are low and SPRED1 levels are high, SPRED1 attenuates K-ras-mediated ERK signaling (left). An increase in Gal-1 expression further augments the negative activity of SPRED1 (right). (C) At low Gal-1 levels, SPRED1 does not affect H-ras-mediated ERK signaling (left). If only Gal-1 is present, H-ras-mediated signaling is enhanced (right). Concurrent expression of SPRED1 turns off Gal-1-induced H-ras enhancement. Arrows indicate the strength of the Ras isoform-specific membrane FRET and signaling according to our results.
This mechanistic setting may explain why it has been difficult to pinpoint whether SPRED1 acts at the level of Ras or Raf on MAPK signaling (2, 8). Our new mechanism does not exclude the possibility that the GTPase-activating protein (GAP) NF1 is in addition recruited to the membrane together with SPRED1, as suggested before (9, 10). NF1 recruitment may then in addition deactivate Ras. However, it is questionable whether the NF1-mediated mechanism alone could account for the RasG12V isoform specificity observed here as these oncogenic variants are GAP insensitive.
Synthesizing existing data on the role of Gal-1 in Ras signaling and our new results, the following picture emerges. Gal-1 stabilizes GTP–H-ras signaling by increasing nanoscale clustering (termed nanoclustering) of H-ras, thus increasing effector recruitment (51, 74). GTP–H-ras by itself negatively regulates K-ras by redistributing PS on the nanoscale in the plasma membrane (62). Gal-1 can increase this negative effect on K-ras nanoclustering and signaling, probably via its clustering effect on GTP–H-ras (Fig. 5A and C) (39). We also recently described dimeric Gal-1 as a candidate Raf dimer stabilizer, which binds to the Ras binding domain (RBD) of Raf proteins (39). Our data suggest that by stabilizing B-/C-Raf dimers, Gal-1 facilitates plasma membrane localization of SPRED1, where the latter may then act on the membrane organization of K-ras, thus increasing the already Gal-1-associated inhibition of K-ras (Fig. 1 and 5). We show that this obvious redundancy goes together with changes in potency of inhibition and also with a loss of an increase in Gal-1-mediated nanoclustering of GTP–H-ras if SPRED1 is present (Fig. 7B and C).
Thus, the following scheme for differential Ras signaling output becomes apparent. If only Gal-1 is present, then H-ras signaling is relatively enhanced compared to the level of K-ras signaling. Concomitant suppression of K-ras signaling can be significantly increased if SPRED1 is expressed in addition. In the latter scenario, utilization of Gal-1 by SPRED1 would remove the stimulatory effect of Gal-1 from H-ras signaling. This is evident from our FRET data (Fig. 5B). In summary, depending on the combination of Gal-1 and SPRED1 expression, these proteins could critically regulate differential signaling output from H- and K-ras.
In which biological context could this be relevant? It was recently suggested that K-ras is the major driver of stem cell or progenitor cell proliferation, while H-ras drives differentiation (75–77). Therefore, SPRED1's well-documented role in regulating differentiation (2) may integrate in the following way, according to our differential Ras regulation scenario outlined above. If Gal-1 is high, then H-ras is enhanced, and K-ras is suppressed. Subsequent induction of SPRED1 would turn off H-ras enhancement and further suppress K-ras. This hypothetical sequence would allow a stepwise down-modulation of Ras isoform-specific activities.
Compared to SPRED2 which is strongly expressed throughout development and adulthood, SPRED1 is mainly expressed in the embryo, with high levels in brain and lung and also in other tissues (1, 71). According to gene expression databases, Gal-1 does not seem to be much expressed in the developing brain or lung (78). However, these data would have to be further validated. Interestingly, in human adult tissue, highly related SPRED2 can be found in the liver and small intestine (71), where Gal-1 is also expressed (79). It is thus possible that the observations that we have made here also apply to the functioning of SPRED2.
Given that Gal-1 could be such a strong potentiator of SPRED activity, it is interesting that typically higher Gal-1 levels are associated with a worse outcome in cancer (80, 81). This would put the apparently small loss in K-ras inhibition, and also the increase in H-ras signaling due to SPRED loss-of-function mutations, in the spotlight of cancer progression. However, the effects of Gal-1 expression in cancer are quite diverse (38). It is therefore hard to predict what the combined outcome of a gain- and loss-of-function genetic event of Gal-1 and SPRED1, respectively, would lead to in a tumor.
Finally, our results suggest that pathogenic SPRED1 point mutations found in Legius syndrome are not able to form correct complexes with Gal-1 and B-Raf. As these mutants might be able to translocate to the plasma membrane but are still defective in inhibiting ERK signaling, our results further propose a significant role for both Gal-1 and B-Raf in completing the inhibitory activity of SPRED1. Furthermore, these results point out that it is crucial to understand the highly complex plasma membrane interactions to fully understand the molecular mechanism of SPRED1.
While our new mechanistic data suggest an important Raf and Ras isoform-specific function of SPRED1, there are still several open mechanistic questions. More broadly, our results may provide important new clues regarding the functioning of other SPRED and Sprouty proteins and of their role during development in conjunction with specific Ras and Raf isoforms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Akihiko Yoshimura (Keio University) for providing SPRED1 plasmid constructs and John F. Hancock (The University of Texas Medical School at Houston, TX) for providing plasmid constructs and MDCK cell line.
We declare that we have no conflict of interests.
D.A. conceived the project, E.S. conducted the experiments, and E.S. and D.A. designed the experiments and wrote the manuscript.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00191-16.
REFERENCES
- 1.Kato R, Nonami A, Taketomi T, Wakioka T, Kuroiwa A, Matsuda Y, Yoshimura A. 2003. Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk activation. Biochem Biophys Res Commun 302:767–772. doi: 10.1016/S0006-291X(03)00259-6. [DOI] [PubMed] [Google Scholar]
- 2.Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K, Tsuneoka M, Komiya S, Baron R, Yoshimura A. 2001. Spred is a Sprouty-related suppressor of Ras signalling. Nature 412:647–651. doi: 10.1038/35088082. [DOI] [PubMed] [Google Scholar]
- 3.Inoue H, Kato R, Fukuyama S, Nonami A, Taniguchi K, Matsumoto K, Nakano T, Tsuda M, Matsumura M, Kubo M, Ishikawa F, Moon B-G, Takatsu K, Nakanishi Y, Yoshimura A. 2005. Spred-1 negatively regulates allergen-induced airway eosinophilia and hyperresponsiveness. J Exp Med 201:73–82. doi: 10.1084/jem.20040616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyoshi K, Wakioka T, Nishinakamura H, Kamio M, Yang L, Inoue M, Hasegawa M, Yonemitsu Y, Komiya S, Yoshimura A. 2004. The Sprouty-related protein, Spred, inhibits cell motility, metastasis, and Rho-mediated actin reorganization. Oncogene 23:5567–5576. doi: 10.1038/sj.onc.1207759. [DOI] [PubMed] [Google Scholar]
- 5.Nonami A, Taketomi T, Kimura A, Saeki K, Takaki H, Sanada T, Taniguchi K, Harada M, Kato R, Yoshimura A. 2005. The Sprouty-related protein, Spred-1, localizes in a lipid raft/caveola and inhibits ERK activation in collaboration with caveolin-1. Genes Cells 10:887–895. doi: 10.1111/j.1365-2443.2005.00886.x. [DOI] [PubMed] [Google Scholar]
- 6.King JAJ, Corcoran NM, D'Abaco GM, Straffon AF, Smith CT, Poon CLC, Buchert M, I S, Hall NE, Lock P, Hovens CM. 2006. Eve-3: a liver enriched suppressor of Ras/MAPK signaling. J Hepatol 44:758–767. doi: 10.1016/j.jhep.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 7.Bundschu K, Walter U, Schuh K. 2007. Getting a first clue about SPRED functions. Bioessays 29:897–907. doi: 10.1002/bies.20632. [DOI] [PubMed] [Google Scholar]
- 8.King JAJ, Straffon AFL, D'Abaco GM, Poon CLC, I ST, Smith CM, Buchert M, Corcoran NM, Hall NE, Callus BA, Sarcevic B, Martin D, Lock P, Hovens CM. 2005. Distinct requirements for the Sprouty domain for functional activity of Spred proteins. Biochem J 388:445–454. doi: 10.1042/BJ20041284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, Burlingame AL, McCormick F. 2012. A shared molecular mechanism underlies the human rasopathies Legius syndrome and neurofibromatosis-1. Genes Dev 26:1421–1426. doi: 10.1101/gad.190876.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirata Y, Brems H, Suzuki M, Kanamori M, Okada M, Morita R, Llano-Rivas I, Ose T, Messiaen L, Legius E, Yoshimura A. 2016. Interaction between a domain of the negative regulator of the Ras-ERK Pathway, SPRED1 protein, and the GTPase-activating protein-related domain of neurofibromin is implicated in legius syndrome and neurofibromatosis type 1. J Biol Chem 291:3124–3134. doi: 10.1074/jbc.M115.703710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bundschu K, Knobeloch K-P, Ullrich M, Schinke T, Amling M, Engelhardt CM, Renné T, Walter U, Schuh K. 2005. Gene disruption of Spred-2 causes dwarfism. J Biol Chem 280:28572–28580. doi: 10.1074/jbc.M503640200. [DOI] [PubMed] [Google Scholar]
- 12.Nobuhisa I, Kato R, Inoue H, Takizawa M, Okita K, Yoshimura A, Taga T. 2004. Spred-2 suppresses aorta-gonad-mesonephros hematopoiesis by inhibiting MAP kinase activation. J Exp Med 199:737–742. doi: 10.1084/jem.20030830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi K, Kohno R-I, Ayada T, Kato R, Ichiyama K, Morisada T, Oike Y, Yonemitsu Y, Maehara Y, Yoshimura A. 2007. Spreds are essential for embryonic lymphangiogenesis by regulating vascular endothelial growth factor receptor 3 signaling. Mol Cell Biol 27:4541–4550. doi: 10.1128/MCB.01600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns J-P, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. 2007. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet 39:1120–1126. doi: 10.1038/ng2113. [DOI] [PubMed] [Google Scholar]
- 15.Pasmant E, Sabbagh A, Hanna N, Masliah-Planchon J, Jolly E, Goussard P, Ballerini P, Cartault F, Barbarot S, Landman-Parker J, Soufir N, Parfait B, Vidaud M, Wolkenstein P, Vidaud D, France RNF. 2009. SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. J Med Genet 46:425–430. doi: 10.1136/jmg.2008.065243. [DOI] [PubMed] [Google Scholar]
- 16.Spurlock G, Bennett E, Chuzhanova N, Thomas N, Jim H-P, Side L, Davies S, Haan E, Kerr B, Huson SM, Upadhyaya M. 2009. SPRED1 mutations (Legius syndrome): another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J Med Genet 46:431–437. doi: 10.1136/jmg.2008.065474. [DOI] [PubMed] [Google Scholar]
- 17.Dhillon AS, Hagan S, Rath O, Kolch W. 2007. MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 18.Fernández-Medarde A, Santos E. 2011. Ras in cancer and developmental diseases. Genes Cancer 2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. 2011. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res 72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR. 2015. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A 112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hancock JF. 2003. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 23.Mott HR, Owen D. 2015. Structures of Ras superfamily effector complexes: what have we learnt in two decades? Crit Rev Biochem Mol Biol 50:85–133. doi: 10.3109/10409238.2014.999191. [DOI] [PubMed] [Google Scholar]
- 24.Voice JK, Klemke RL, Le A, Jackson JH. 1999. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J Biol Chem 274:17164–17170. doi: 10.1074/jbc.274.24.17164. [DOI] [PubMed] [Google Scholar]
- 25.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. 1998. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem 273:24052–24056. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- 26.Baljuls A, Kholodenko BN, Kolch W. 2013. It takes two to tango—signalling by dimeric Raf kinases. Mol Biosyst 9:551–558. doi: 10.1039/C2MB25393C. [DOI] [PubMed] [Google Scholar]
- 27.Udell CM, Rajakulendran T, Sicheri F, Therrien M. 2011. Mechanistic principles of RAF kinase signaling. Cell Mol Life Sci 68:553–565. doi: 10.1007/s00018-010-0520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, Shaw AS. 2013. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freeman AK, Ritt DA, Morrison DK. 2013. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 49:751–758. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber CK, Slupsky JR, Kalmes HA, Rapp UR. 2001. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res 61:3595–3598. [PubMed] [Google Scholar]
- 31.Rushworth LK, Hindley AD, O'Neill E, Kolch W. 2006. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol 26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carnahan J, Beltran PJ, Babij C, Le Q, Rose MJ, Vonderfecht S, Kim JL, Smith AL, Nagapudi K, Broome MA, Fernando M, Kha H, Belmontes B, Radinsky R, Kendall R, Burgess TL. 2010. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther 9:2399–2410. doi: 10.1158/1535-7163.MCT-10-0181. [DOI] [PubMed] [Google Scholar]
- 33.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 34.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. 2010. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K. 1994. Galectins: a family of animal beta-galactoside-binding lectins. Cell 76:597–598. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- 36.Delacour D, Koch A, Jacob R. 2009. The role of galectins in protein trafficking. Traffic 10:1405–1413. doi: 10.1111/j.1600-0854.2009.00960.x. [DOI] [PubMed] [Google Scholar]
- 37.Elola MT, Wolfenstein-Todel C, Troncoso MF, Vasta GR, Rabinovich GA. 2007. Galectins: matricellular glycan-binding proteins linking cell adhesion, migration, and survival. Cell Mol Life Sci 64:1679–1700. doi: 10.1007/s00018-007-7044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thijssen VL, Heusschen R, Caers J, Griffioen AW. 2015. Galectin expression in cancer diagnosis and prognosis: a systematic review. Biochim Biophys Acta 1855:235–247. doi: 10.1016/j.bbcan.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Blaževitš O, Mideksa YG, Šolman M, Ligabue A, Ariotti N, Nakhaeizadeh H, Fansa EK, Papageorgiou AC, Wittinghofer A, Ahmadian MR, Abankwa D. 2016. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci Rep 6:24165. doi: 10.1038/srep24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Šolman M, Ligabue A, Blaževitš O, Jaiswal A, Zhou Y, Liang H, Lectez B, Kopra K, Guzmán C, Härmä H, Hancock JF, Aittokallio T, Abankwa D. 2015. Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. eLife 4:e08905. doi: 10.7554/eLife.08905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abankwa D, Gorfe AA, Inder K, Hancock JF. 2010. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci U S A 107:1130–1135. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abankwa D, Hanzal-Bayer M, Ariotti N, Plowman SJ, Gorfe AA, Parton RG, McCammon JA, Hancock JF. 2008. A novel switch region regulates H-ras membrane orientation and signal output. EMBO J 27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. 2001. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 20:7486–7493. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 44.Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF. 2001. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol 3:368–375. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 45.Abankwa D, Vogel H. 2007. A FRET map of membrane anchors suggests distinct microdomains of heterotrimeric G proteins. J Cell Sci 120:2953–2962. doi: 10.1242/jcs.001404. [DOI] [PubMed] [Google Scholar]
- 46.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 47.Venkateswarlu K, Oatey PB, Tavaré JM, Cullen PJ. 1998. Insulin-dependent translocation of ARNO to the plasma membrane of adipocytes requires phosphatidylinositol 3-kinase. Curr Biol 8:463–466. doi: 10.1016/S0960-9822(98)70181-2. [DOI] [PubMed] [Google Scholar]
- 48.Meissner P, Pick H, Kulangara A, Chatellard P, Friedrich K, Wurm FM. 2001. Transient gene expression: recombinant protein production with suspension-adapted HEK293-EBNA cells. Biotechnol Bioeng 75:197–203. doi: 10.1002/bit.1179. [DOI] [PubMed] [Google Scholar]
- 49.Cho K-J, Park J-H, Piggott AM, Salim AA, Gorfe AA, Parton RG, Capon RJ, Lacey E, Hancock JF. 2012. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem 287:43573–43584. doi: 10.1074/jbc.M112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guzmán C, Šolman M, Ligabue A, Blaževitš O, Andrade DM, Reymond L, Eggeling C, Abankwa D. 2014. The efficacy of Raf kinase recruitment to the GTPase H-ras depends on H-ras membrane conformer-specific nanoclustering. J Biol Chem 289:9519–9533. doi: 10.1074/jbc.M113.537001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köhnke M, Schmitt S, Ariotti N, Piggott AM, Parton RG, Lacey E, Capon RJ, Alexandrov K, Abankwa D. 2012. Design and application of in vivo FRET biosensors to identify protein prenylation and nanoclustering inhibitors. Chem Biol 19:866–874. doi: 10.1016/j.chembiol.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 53.Najumudeen AK, Köhnke M, Šolman M, Alexandrov K, Abankwa D. 2013. Cellular FRET-biosensors to detect membrane targeting inhibitors of N-myristoylated proteins. PLoS One 8:e66425. doi: 10.1371/journal.pone.0066425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sykes AM, Palstra N, Abankwa D, Hill JM, Skeldal S, Matusica D, Venkatraman P, Hancock JF, Coulson EJ. 2012. The effects of transmembrane sequence and dimerization on cleavage of the p75 neurotrophin receptor by γ-secretase. J Biol Chem 287:43810–43824. doi: 10.1074/jbc.M112.382903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim J, Yusoff P, Wong ESM, Chandramouli S, Lao DH, Fong CW, Guy GR. 2002. The cysteine-rich Sprouty translocation domain targets mitogen-activated protein kinase inhibitory proteins to phosphatidylinositol 4,5-bisphosphate in plasma membranes. Mol Cell Biol 22:7953–7966. doi: 10.1128/MCB.22.22.7953-7966.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. 2010. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, Tsang G, Marimuthu A, Cho H, Wu G, Wang W, Fong D, Nguyen H, Shi S, Womack P, Nespi M, Shellooe R, Carias H, Powell B, Light E, Sanftner L, Walters J, Tsai J, West BL, Visor G, Rezaei H, Lin PS, Nolop K, Ibrahim PN, Hirth P, Bollag G. 2015. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 58.Guzmán C, Oetken-Lindholm C, Abankwa D. 2016. Automated high-throughput fluorescence lifetime imaging microscopy to detect protein-protein interactions. J Lab Autom 21:238–245. doi: 10.1177/2211068215606048. [DOI] [PubMed] [Google Scholar]
- 59.Nonami A, Kato R, Taniguchi K, Yoshiga D, Taketomi T, Fukuyama S, Harada M, Sasaki A, Yoshimura A. 2004. Spred-1 negatively regulates interleukin-3-mediated ERK/mitogen-activated protein (MAP) kinase activation in hematopoietic cells. J Biol Chem 279:52543–52551. doi: 10.1074/jbc.M405189200. [DOI] [PubMed] [Google Scholar]
- 60.Cho K-J, Kasai RS, Park J-H, Chigurupati S, Heidorn SJ, van der Hoeven D, Plowman SJ, Kusumi A, Marais R, Hancock JF. 2012. Raf inhibitors target Ras spatiotemporal dynamics. Curr Biol 22:945–955. doi: 10.1016/j.cub.2012.03.067. [DOI] [PubMed] [Google Scholar]
- 61.Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DYL, Bouvier M, Sicheri F, Therrien M. 2013. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol 9:428–436. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG, Hancock JF. 2014. Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol Cell Biol 34:862–876. doi: 10.1128/MCB.01227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Várnai P, Lin X, Lee SB, Tuymetova G, Bondeva T, Spät A, Rhee SG, Hajnóczky G, Balla T. 2002. Inositol lipid binding and membrane localization of isolated pleckstrin homology (PH) domains. Studies on the PH domains of phospholipase C δ1 and p130. J Biol Chem 277:27412–27422. [DOI] [PubMed] [Google Scholar]
- 64.Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. 2006. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. 2008. Galectin-1 is a novel structural component and a major regulator of H-Ras nanoclusters. Mol Biol Cell 19:1404–1414. doi: 10.1091/mbc.E07-10-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prior IA, Muncke C, Parton RG, Hancock JF. 2003. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol 160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brems H, Legius E. 2013. Legius syndrome, an update. Molecular pathology of mutations in SPRED1. Keio J Med 62:107–112. [DOI] [PubMed] [Google Scholar]
- 68.Yoshida T, Hisamoto T, Akiba J, Koga H, Nakamura K, Tokunaga Y, Hanada S, Kumemura H, Maeyama M, Harada M, Ogata H, Yano H, Kojiro M, Ueno T, Yoshimura A, Sata M. 2006. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 25:6056–6066. doi: 10.1038/sj.onc.1209635. [DOI] [PubMed] [Google Scholar]
- 69.Ma X-N, Liu X-Y, Yang Y-F, Xiao F-J, Li Q-F, Yan J, Zhang Q-W, Wang L-S, Li X-Y, Wang H. 2011. Regulation of human hepatocellular carcinoma cells by Spred2 and correlative studies on its mechanism. Biochem Biophys Res Commun 410:803–808. doi: 10.1016/j.bbrc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 70.Masoumi-Moghaddam S, Amini A, Morris DL. 2014. The developing story of Sprouty and cancer. Cancer Metastasis Rev 33:695–720. doi: 10.1007/s10555-014-9497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Engelhardt CM, Bundschu K, Messerschmitt M, Renné T, Walter U, Reinhard M, Schuh K. 2004. Expression and subcellular localization of Spred proteins in mouse and human tissues. Histochem Cell Biol 122:527–538. doi: 10.1007/s00418-004-0725-6. [DOI] [PubMed] [Google Scholar]
- 72.Baetz NW, Goldenring JR. 2014. Distinct patterns of phosphatidylserine localization within the Rab11a-containing recycling system. Cell Logist 4:e28680. doi: 10.4161/cl.28680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M, Uchiyama Y, Iemura S-I, Natsume T, Kuwahara R, Nakagawa T, Nishikawa K, Mukai K, Miyoshi E, Taniguchi N, Sheff D, Lencer WI, Taguchi T, Arai H. 2011. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci U S A 108:15846–15851. doi: 10.1073/pnas.1109101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rotblat B, Belanis L, Liang H, Haklai R, Elad-Zefadia G, Hancock JF, Kloog Y, Plowman SJ. 2010. H-Ras nanocluster stability regulates the magnitude of MAPK signal output. PLoS One 5:e11991. doi: 10.1371/journal.pone.0011991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quinlan MP, Quatela SE, Philips MR, Settleman J. 2008. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol 28:2659–2674. doi: 10.1128/MCB.01661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Najumudeen AK, Jaiswal A, Lectez B, Oetken-Lindholm C, Guzmán C, Siljamäki E, Posada IMD, Lacey E, Aittokallio T, Abankwa D. 14 March 2016. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene doi: 10.1038/onc.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang M-T, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, McCormick F. 2015. K-Ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell 163:1237–1251. doi: 10.1016/j.cell.2015.10.041. [DOI] [PubMed] [Google Scholar]
- 78.Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I, Lin-Marq N, Koch M, Bilio M, Cantiello I, Verde R, De Masi C, Bianchi SA, Cicchini J, Perroud E, Mehmeti S, Dagand E, Schrinner S, Nürnberger A, Schmidt K, Metz K, Zwingmann C, Brieske N, Springer C, Hernandez AM, Herzog S, Grabbe F, Sieverding C, Fischer B, Schrader K, Brockmeyer M, Dettmer S, Helbig C, Alunni V, Battaini M-A, Mura C, Henrichsen CN, Garcia-Lopez R, Echevarria D, Puelles E, Garcia-Calero E, Kruse S, Uhr M, Kauck C, Feng G, Milyaev N, Ong CK, Kumar L, Lam M, Semple CA, Gyenesei A, Mundlos S, Radelof U, Lehrach H, Sarmientos P, Reymond A, Davidson DR, Dollé P, Antonarakis SE, Yaspo M-L, Martinez S, Baldock RA, Eichele G, Ballabio A. 2011. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9:e1000582. doi: 10.1371/journal.pbio.1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dias-Baruffi M, Stowell SR, Song S-C, Arthur CM, Cho M, Rodrigues LC, Montes MAB, Rossi MA, James JA, McEver RP, Cummings RD. 2010. Differential expression of immunomodulatory galectin-1 in peripheral leukocytes and adult tissues and its cytosolic organization in striated muscle. Glycobiology 20:507–520. doi: 10.1093/glycob/cwp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang P, Zhang P, Shi B, Zhou M, Jiang H, Zhang H, Pan X, Gao H, Sun H, Li Z. 2014. Galectin-1 overexpression promotes progression and chemoresistance to cisplatin in epithelial ovarian cancer. Cell Death Dis 5:e991. doi: 10.1038/cddis.2013.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, Giaccia AJ, Koong AC, Le Q-T. 2011. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res 71:4423–4431. doi: 10.1158/0008-5472.CAN-10-4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.