

Abstract
Nonalcoholic fatty liver disease (NAFLD) has become the most common liver disease, and decreased fatty acid oxidation is one of the important contributors to NAFLD. Mitochondrial trifunctional protein α-subunit (MTPα) functions as a critical enzyme for fatty acid β-oxidation, but whether dysregulation of MTPα is pathogenically connected to NAFLD is poorly understood. We show that MTPα is acetylated at lysine residues 350, 383, and 406 (MTPα-3K), which promotes its protein stability by antagonizing its ubiquitylation on the same three lysines (MTPα-3K) and blocking its subsequent degradation. Sirtuin 4 (SIRT4) has been identified as the deacetylase, deacetylating and destabilizing MTPα. Replacement of MTPα-3K with either MTPα-3KR or MTPα-3KQ inhibits cellular lipid accumulation both in free fatty acid (FFA)-treated alpha mouse liver 12 (AML12) cells and primary hepatocytes and in the livers of high-fat/high-sucrose (HF/HS) diet-fed mice. Moreover, knockdown of SIRT4 could phenocopy the effects of MTPα-3K mutant expression in mouse livers, and MTPα-3K mutants more efficiently attenuate SIRT4-mediated hepatic steatosis in HF/HS diet-fed mice. Importantly, acetylation of both MTPα and MTPα-3K is decreased while SIRT4 is increased in the livers of mice and humans with NAFLD. Our study reveals a novel mechanism of MTPα regulation by acetylation and ubiquitylation and a direct functional link of this regulation to NAFLD.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) has emerged as a common public health problem that encompasses a spectrum of liver damage ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis, and even hepatocellular carcinoma (HCC) (1, 2). The prevalence of NAFLD has increased to more than 30% of adults in developed countries and is still rising (3, 4). NAFLD is strongly associated with obesity, insulin resistance, hypertension, and dyslipidemia, making it the liver manifestation of metabolic syndrome (MS) (1, 5). Although the “two-hit” hypothesis has been widely accepted to depict the pathogenesis of NAFLD (6, 7), the molecular mechanism underlying NAFLD initiation and progression remains imperfectly understood.
NAFLD is characterized by the increased accumulation of lipid in the liver, which can result from decreased fatty acid oxidation in the liver cells (8, 9). Mitochondrial trifunctional protein α-subunit (MTPα) contains both the long-chain hydratase and 3-hydroxyacyl coenzyme A (3-hydroxyacyl-CoA) dehydrogenase functions that catalyze the second and third steps, respectively, of fatty acid β-oxidation (10). Children with MTPα defects present with hepatic dysfunction associated with predominantly microvesicular hepatic steatosis that resembles Reye syndrome (11). Furthermore, heterozygous mice lacking MTPα have significantly reduced fatty acid oxidation in the liver and develop hepatic steatosis and insulin resistance (12, 13). Thus, MTPα is a critical enzyme in fatty acid β-oxidation. However, it remains unclear to what degree the enzyme is involved in obesity-related derangement of hepatic lipid metabolism and NAFLD.
Lysine acetylation is a conserved protein posttranslational modification in the regulation of a wide range of cellular processes, including cellular metabolism (14, 15). An increasing number of studies have shown that many metabolic enzymes can be acetylated, and the functional importance of their acetylation is being elucidated. For example, the M2 isoform of pyruvate kinase (PKM2) is acetylated at lysine residue 433 (K433), which promotes its nuclear accumulation and protein kinase activity to facilitate cell proliferation and tumorigenesis (16). In another instance, acetylation of aldehyde dehydrogenase (ALDH) inhibits its enzyme activity, thereby suppressing breast cancer stem cells (17). Considering the importance of lysine acetylation in the regulation of metabolism, dissecting the function and regulatory mechanism of the acetylation of metabolic enzymes is instrumental in our fight against metabolic disorders, such as cancer, diabetes, and fatty liver disease.
Since NAFLD is a metabolic disease, dysregulation of lysine acetylation could be involved in the development of NAFLD. However, this field remains poorly investigated. In this study, we demonstrate that MTPα is acetylated on three lysine residues, 350, 383, and 406 (MTPα-3K). Acetylation of MTPα blocks its ubiquitylation on the same three lysine residues (MTPα-3K) and its subsequent degradation, thereby promoting MTPα protein stability. Moreover, the functional importance of the dysregulated acetylation of MTPα in the pathogenesis of NAFLD is examined.
MATERIALS AND METHODS
Cell culture.
HEK293T and HEK293A cells were maintained and propagated in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). Alpha mouse liver 12 (AML12) cells were maintained and propagated in DMEM–F-12 with 10% FBS, ITS Liquid Media Supplement (Sigma), and 0.1 μM dexamethasone. Primary hepatocytes were isolated by the collagenase perfusion method, as previously described (18). The digested liver tissue was filtered through a 70-μm-mesh filter and centrifuged at 520 rpm for 2 min. The cell pellets were resuspended and cultured in DMEM with 10% FBS. The deacetylase inhibitor trichostatin A (TSA) (Sigma) was added to the cells 16 h before harvest at a final concentration of 10 μM, while 10 mM nicotinamide (NAM) (Sigma) was added to the medium 8 h before harvest. The proteasomal inhibitor MG132 (Sigma) was added 4 h before harvest at a final concentration of 10 μM. For fat overloading induction of cells, AML12 cells and primary hepatocytes were exposed to a mixture of free fatty acids (FFAs) (oleate and palmitate from Sigma) at a final ratio of 2:1 and a final concentration of 1 mM for 24 h.
Reagents.
Antibodies against MTP and sirtuin 4 (SIRT4) were purchased from Santa Cruz; β-actin was from Abcam; antibodies against Flag, hemagglutinin (HA), and Myc were from Earthox; antibodies against panacetyllysine were from ImmunoChem Pharmaceuticals. Mitotracker probe was from Invitrogen. Isolation of mitochondrial and cytosol fractions of cells was performed by using a Mitochondria Isolation kit (Thermo Fisher Scientific). Liver and plasma triglyceride and cholesterol levels and plasma β-hydroxybutyrate, alanine aminotransferase (ALT), and aspartate transaminase (AST) levels were measured using kits from Stanbio. Antibodies specifically recognizing acetylation at lysines 350, 383, and 406 of MTPα (MTPα-3K-Ac) were prepared commercially at Shanghai Genomics, Inc., by immunizing rabbits. To generate the anti-MTPα-3K-Ac antibody, the MTPα peptide harboring acetylated K350, acetylated K383, or acetylated K406 was applied to immunize the rabbits to produce antibody against acetylated K350, acetylated K383, or acetylated K406. After confirming the titers and specificity of these three antibodies, they were mixed to make the anti-MTPα-3K-Ac antibody.
Animals.
Male C57BL/6J mice, ob/ob mice, db/db mice, and their control littermates were purchased from the Model Animal Research Center of Nanjing University. The mice were maintained on a normal diet. To produce diet-induced obesity, 8-week-old C57BL/6J mice were fed a high-fat/high-sucrose (HF/HS) diet (research diet) for 2 months, with normal-chow diet mice as controls. Then, adenovirus injection was performed in HF/HS diet-fed mice. All the studies involving animal experimentation were approved by the Animal Care and Use Committee of the Fudan University Shanghai Medical College and followed the National Institutes of Health guidelines on the care and use of animals.
Western blotting.
Cells and tissues were harvested and homogenized with lysis buffer containing 2% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 6.8), 10 mM dithiothreitol (DTT), 10% glycerol, 0.002% bromphenol blue, protease inhibitor mixture (Roche), 50 mM NAM, and 20 μM TSA. For protein stability assays, cells were treated with cycloheximide (CHX) (Sigma) before harvest. Equal amounts of protein were resolved on an SDS-polyacrylamide gel for electrophoresis, transferred to a polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA), immunoblotted with primary antibodies, and visualized with horseradish peroxidase-coupled secondary antibodies.
RNA extraction and reverse transcription (RT)-qPCR.
Total RNA from cells and tissues was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from total RNA with PrimeScript reverse transcriptase and random primers (TaKaRa Bio, Otsu, Japan). cDNAs were amplified with Power SYBR green PCR master mix (Applied Biosystems, Carlsbad, CA) and a Prism 7500 instrument (Applied Biosystems), with 18S rRNA as an endogenous control. The quantitative PCR (qPCR) was done in triplicate and repeated at least 3 times. The primers for qPCR are available upon request.
Confocal microscopic analyses.
Cells were fixed in 4% formaldehyde for 10 min at room temperature prior to cell permeabilization with 0.1% Triton X-100 (4°C; 10 min). The cells were blocked with phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA) for 1 h at room temperature and processed for immunostaining. In some cases, cells were stained with 200 nM MitoTracker for 20 min before fixation. Fluorescence images were taken and analyzed with a Leica confocal microscope (TCS SP5; Leica, Germany).
Ubiquitylation assay.
Ubiquitylation assays were performed following a protocol as previously described (19). Briefly, cells were lysed in 1% SDS buffer (Tris [pH 7.5], 0.5 mM EDTA, 1 mM dithiothreitol) and boiled for 10 min. For immunoprecipitation, the lysates were diluted 10-fold in Tris-HCl buffer. Analyses of ubiquitination were performed by anti-HA blotting.
Peptide deacetylation assay.
The peptide deacetylation assay was performed based on a previously described method (20). Acetylated peptides used in deacetylation experiments were as follows: K350-Ac peptide, GQVLCK(Ac)KNKFG; K383-Ac peptide, IAQVSVDK(Ac)GLKTL; K406-Ac peptide, GQQQVFK(Ac)GLNDKV; and PDH-Ac peptide, KADQLYK(Ac)QKFIRG. Briefly, the peptide deacetylation reaction mixture contained 20 mM Tris-HCl, pH 7.8, 150 mM NaCl, 1 mM NAD+, 2 mM DTT, 0.5 mM substrate peptide, and 20 μg SIRT4. Reaction mixtures were incubated at 37°C for 30 min and then mixed with the same volume of matrix (α-cyano-4-hydroxycinnamic acid [CHCA] in 30% acetonitrile [ACN] and 0.1% trifluoroacetic acid [TFA]) before spotting on the target plate. Peptide detection was performed on an AB Sciex matrix-assisted laser desorption ionization–time of flight (MALDI-TOF)-TOF 5800 analyzer (AB Sciex, Foster City, CA) equipped with a neodymium-yttrium-aluminum-garnet laser (laser wavelength, 349 nm). The TOF-TOF calibration mixtures (AB Sciex) were used to calibrate the spectrum to a mass tolerance within 100 ppm. Peptide mass maps were acquired in positive reflection mode, and a 700 to 5,000 m/z mass range was used with 1,000 laser shots per spectrum.
RNA interference (RNAi).
Short hairpin RNAs (shRNAs) were constructed in the adenovirus vectors (Invitrogen) for the infection of mice. The sequences of small interfering RNAs (siRNAs) and shRNAs were as follows: siMTPα, GCCAATACAGAATAGCAACAA; shMTPα, GCTGACCAGAACCCATATTAA; shSIRT4, TGGAGAGTTGCTGCCTTTAAT; shNC, AATTTAACCGCCAGTCAGGCT. To make shRNA-resistant MTPα, a mouse MTPα construct altered in codon usage by synonymous mutation was designed to generate base pair mismatches with shMTPα. The synonymous mutation was as follows (mutated residues are underlined): ACCCATATT (coding sequence positions 115 to 123) was mutated to ACACACATC.
Oil Red O staining and H&E staining.
Cells were washed with cold PBS and fixed for 10 min with 3.7% formaldehyde. Oil Red O (Sigma; 0.5% in isopropanol) was diluted with water (3:2), filtered through a 0.45-μm filter, and incubated with the fixed cells for 4 h at room temperature. The cells were washed with water, and stained fat droplets in the cells were examined by light microscopy and photographed. Oil Red O staining and hematoxylin and eosin (H&E) staining of mouse liver tissues were conducted as described previously (21).
GTT and ITT.
Levels of blood glucose were measured using a glucometer monitor (Roche). For the glucose tolerance test (GTT), mice were injected intraperitoneally with d-glucose (2 mg/g body weight) after overnight fasting, and tail blood glucose levels were monitored every 0.5 h. For the insulin tolerance test (ITT), mice were injected intraperitoneally with human insulin (Eli Lilly; 0.75 mU/g body weight) after 4 h of fasting, and tail blood glucose levels were monitored every 0.5 h.
Cellular oxygen consumption rate.
The cellular oxygen consumption rate was measured by using an OxygenMeter (Strathkelvin Instruments) with a Mitocell (MT200) mixing chamber. Cells (2 × 104) were suspended in 400 μl culture medium, and the oxygen concentration was recorded for 5 min. The oxygen consumption rate was calculated using software (782 Oxygen System version 4.0) and normalized to the protein content.
Knocking down and restoring stable AML12 cell lines.
The mouse shRNA-resistant wild-type (WT) MTPα or 3K mutants of MTPα were cloned into the pBABE retroviral vector and cotransfected into HEK293T cells together with vectors expressing gag and vsvg (vesicular stomatitis virus G) genes. Retroviral supernatants were harvested by filtration (0.45-μm filters) 48 h after the initial plasmid transfection and mixed with Polybrene (8-μg/ml final concentration) to increase the infection efficiency before being applied to AML12 cells. The AML12 cells were infected with the prepared virus for 12 h and screened with hygromycin (250-μg/ml final concentration) for at least 2 weeks. Control shRNA (shNC) or shMTPα was cloned into the pMSCV retroviral vector, and the retroviruses were prepared as described above. Stable AML12 cells expressing WT MTPα or 3K mutants of MTPα were infected with the pMSCV retroviruses harboring the shRNAs. The MTPα knockdown efficiency of the positive clones was determined after 2 weeks of drug selection (puromycin; 5-μg/ml final concentration).
Generation and administration of recombinant adenoviruses.
Recombinant adenoviruses for overexpression and knockdown were generated using the ViraPower adenoviral expression system (Invitrogen) and the Block-iT adenoviral RNAi expression system (Invitrogen), respectively. High-titer stocks of amplified recombinant adenoviruses were purified with Sartorius adenovirus purification kits. Viral titers were determined by the 50% tissue culture infectious dose (TCID50) method using 293A cells. For tail vein injection, 3 × 108 PFU/mouse of adenoviruses harboring shNC or shMTPα was mixed with 2 × 108 PFU/mouse of adenoviruses harboring shRNA-resistant WT MTPα or 3K mutants of MTPα and then diluted to 100 μl with PBS and administered through tail vein injection (once a week). Measurements were performed at 15 days postinfection. To overexpress SIRT4 and/or MTPα in mouse livers, 2 × 108 PFU/mouse of adenoviruses harboring SIRT4 and/or 2 × 108 PFU/mouse of adenoviruses harboring MTPα (WT or mutants) was administered through tail vein injection as described above. To knock down SIRT4 in mouse livers, 3 × 108 PFU/mouse of adenoviruses harboring shSIRT4 was administered through tail vein injection as described above. For infection of primary hepatocytes, adenoviruses harboring shNC or shMTPα at a multiplicity of infection (MOI) of 4 were mixed with adenoviruses harboring shRNA-resistant WT MTPα or 3K mutants of MTPα at an MOI of 3 and then added to the cell culture medium. After adding the viruses, the cell culture plates were centrifuged at 3,000 rpm for 30 min at the lowest brake speed. Then, cells were cultured with the viruses for 24 h before replacement with fresh culture medium. Experiments were done 48 h after infection. To knock down SIRT4 in AML12 cells, adenoviruses harboring shNC or shSIRT4 at an MOI of 4 were used to infect AML12 cells as described above.
Human liver samples and immunohistochemistry.
Human liver samples were acquired from Zhongshan Hospital of Fudan University. A physician obtained informed consent from the patients. The procedures related to human subjects were approved by the Ethics Committee of the School of Basic Medical Sciences, Fudan University. All of the NAFLD patients were diagnosed with simple steatosis according to the histological manifestation. Patients were excluded if alcohol consumption was more than 40 g/day for men and more than 20 g/day for women and if other liver diseases were detected by serological testing and imaging studies. Paraffin-embedded liver tissue was cut, deparaffinized, and hydrated, and immunohistochemistry was performed as described previously (22). Images were captured using a charge-coupled device camera and analyzed using Image-Pro Plus software. An average staining score was calculated by dividing the positive areas by the total area.
Statistics.
Results are presented as means and standard deviations (SD) from at least 3 independent experiments. P values were determined by Student's t test, with P values of <0.05 taken as significant.
RESULTS
MTPα is acetylated at lysine residues 350, 383, and 406, leading to its enhanced protein stability.
We conducted mass spectrometry analyses using mouse livers and identified a number of potentially acetylated proteins, including MTPα. In our mass spectrometry analyses, nine putative acetylation sites were identified on MTPα (Table 1). Among the nine sites, five lysine residues, lysine 60 (K60), K350, K383, K406, and K505, are highly conserved among species (Fig. 1A).
TABLE 1.
Identification of acetylated MTPα peptides by mass spectrometry
| MTPα acetylation site | Peptide sequencea | Peptide m/z | Peptide Mr | Peptide scoreb | P value |
|---|---|---|---|---|---|
| K60 | INSPNSK(Ac)VNTLNK | 735.8986 | 1,469.783 | 44.1 | 3.90E−05 |
| K309 | IIDAVK(Ac)AGLEQGSDAGYLAESQK | 802.4161 | 2,404.227 | 55.33 | 2.90E−06 |
| K350 | FGELALTKESKALMGLYNGQVLCK(Ac) | 946.8339 | 2,837.48 | 15.51 | 0.028 |
| K383 | NVQQLAILGAGLMGAGIAQVSVDK(Ac)GLK | 898.5126 | 2,692.516 | 25.47 | 0.0028 |
| K406 | GQQQVFK(Ac)GLNDK | 702.368 | 1,402.721 | 59.23 | 1.20E−06 |
| K436 | DSIFSNLIGQLDYK(Ac)GFEK | 1,058.529 | 2,115.044 | 21.09 | 0.0078 |
| K440 | GFEK(Ac)ADMVIEAVFEDLGVK | 713.6966 | 2,138.068 | 39.24 | 0.00012 |
| K505 | VIGMHYFSPVDK(Ac)MQLLEIITTDK | 907.4805 | 2,719.42 | 23.88 | 0.0041 |
| K728 | FVDLYGAQK(Ac)VVDR | 776.4136 | 1,550.813 | 39.9 | 0.0001 |
(Ac), acetylation site.
Peptide score = −10·log P.
FIG 1.
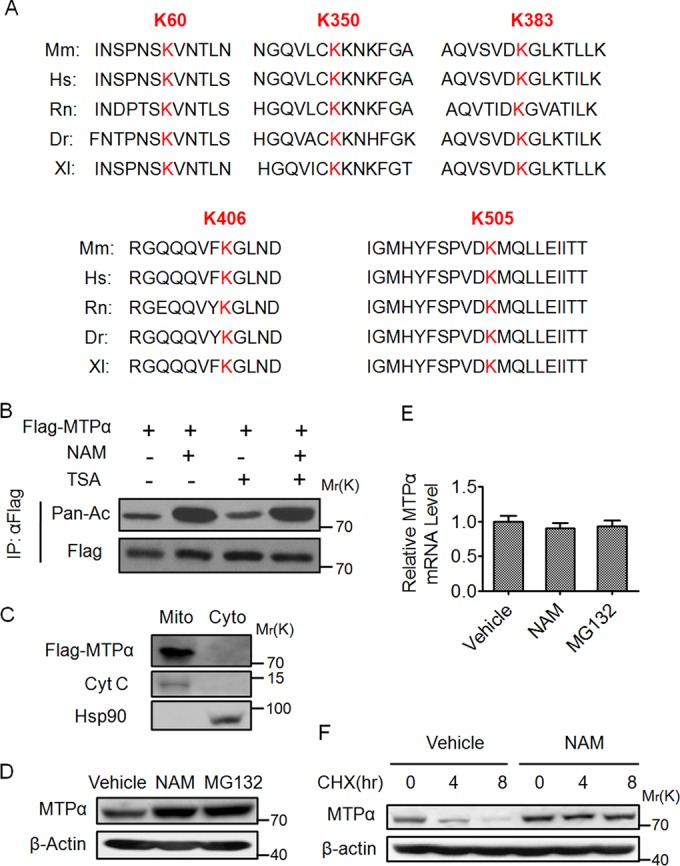
Acetylation of MTPα enhances its protein stability. (A) Multiple alignments of the protein sequences surrounding the potential acetylated sites of MTPα by mass spectrometry from different organisms. Mm, Mus musculus (mouse); Hs, Homo sapiens (human); Rn, Rattus norvegicus (Norway rat); Dr, Danio rerio (zebrafish); Xl, Xenopus laevis (African clawed frog). Letters in red indicate the conserved lysines among species. (B) HEK293T cells were transfected with Flag-tagged MTPα (Flag-MTPα) and left untreated or treated with NAM-TSA. Immunoprecipitated Flag-MTPα was subjected to Western blotting. (C) HEK293T cells transfected with Flag-tagged MTPα were subjected to isolation of mitochondrial (Mito) and cytoplasmic (Cyto) fractions. Equal amounts of protein from each fraction were analyzed by Western blotting. (D and E) AML12 murine hepatocytes were untreated or treated with NAM-MG132. The endogenous protein and mRNA levels of MTPα were determined. The error bars indicate SD. (F) AML12 cells were left untreated or treated with NAM in the presence of CHX for the indicated times and then subjected to Western blotting.
To confirm the acetylation modification of MTPα, we detected the acetylation level of ectopically expressed MTPα, followed by Western blotting with panspecific anti-acetylated-lysine antibody (Pan-Ac). This experiment demonstrated that MTPα was indeed acetylated and that its acetylation was significantly elevated in HEK293T cells after treatment with NAM, an inhibitor of the sirtuin (SIRT) family deacetylases, but not by TSA, an inhibitor of histone deacetylase (HDAC) classes I, II, and IV (Fig. 1B). Because MTPα is a mitochondrial protein, the subcellular localization of ectopically expressed MTPα was examined. HEK293T cells overexpressing Flag-tagged MTPα were subjected to mitochondrial isolation. Western blotting revealed that ectopically expressed MTPα was exclusively localized in the mitochondrial fraction (Fig. 1C), ruling out the possibility that ectopically expressed MTPα is mislocalized in the cell. The purity of the fractions was confirmed by probing for the mitochondrial marker cytochrome c and the cytosolic marker Hsp90 (Fig. 1C).
Interestingly, it was found that MTPα acetylation affected its protein level. We treated murine AML12 hepatocytes with NAM and found an increase in the endogenous MTPα protein level (Fig. 1D), whereas the mRNA level of MTPα was not significantly affected by the treatment (Fig. 1E), indicating that the upregulation of MTPα is mostly achieved at the posttranscriptional level. Meanwhile, treatment of AML12 cells with the proteasomal inhibitor MG132 also increased the MTPα protein level (Fig. 1D), suggesting that MTPα could be degraded by the ubiquitin proteasome pathway. Inhibition of protein synthesis with CHX showed that MTPα is an unstable protein with a half-life of about 4 h, and blocking SIRT deacetylase activity with NAM substantially extended the half-life of MTPα (Fig. 1F). These data confirm the existence of MTPα acetylation and that this modification promotes the protein stability of MTPα.
As described above, five putative acetylation sites with high species conservation were identified by the mass spectrometry analyses (Fig. 1A). Single mutation of each lysine to an arginine (R) was then performed to examine its contribution to the regulation of MTPα protein stability. HEK293T cells were transfected with wild-type MTPα or MTPα mutants, treated with CHX and then subjected to Western blotting. Mutation of K350, K383, and K406 partially increased the protein stability of MTPα, while mutation of K60 and K505 had no effect on its stability (Fig. 2A to C). We then generated triple R and Q mutants of K350, K383, and K406 (3KR and 3KQ) and found that both the 3KR and 3KQ mutants blocked the degradation of MTPα almost completely (Fig. 2D and E). Moreover, treatment with NAM or MG132 could not further increase the protein levels of 3KR and 3KQ mutants (Fig. 2F). These data indicate that K350, K383, and K406 are the critical sites regulating the stability of MTPα. To confirm the acetylation of the three residues, we examined the acetylation of wild-type and 3K mutant MTPα and found that mutation of 3K significantly reduced the acetylation level of MTPα, indicating that the 3K residues are the important acetylation sites of MTPα (Fig. 2G).
FIG 2.

Acetylation at lysine residues 350, 383, and 406 stabilizes MTPα. (A to C) HEK293T cells were transfected with WT or mutant MTPα, treated with CHX for the indicated times, and then subjected to Western blotting and quantified. Quantification of Western blotting was done by using ImageJ. The error bars indicate SD. (D and E) HEK293T cells were transfected with WT or 3K mutant MTPα, treated with CHX for the indicated times, and then subjected to Western blotting and quantified (ImageJ). (F) HEK293T cells were transfected with WT or 3K mutant MTPα, left untreated or treated with NAM-MG132, and then subjected to Western blotting. (G) HEK293T cells were transfected with WT or 3K mutant MTPα. Immunoprecipitated Flag-MTPα was subjected to Western blotting. (H) The specificity of antibody against MTPα-3K-Ac was determined by dot blot assay. A nitrocellulose membrane was spotted with different amounts of MTPα-3K-Ac peptides or unmodified peptides and probed with anti-MTPα-3K-Ac antibody. (I) Endogenous MTPα is acetylated on 3K. AML12 cells were transfected with or without siMTPα and then subjected to Western blotting with the indicated antibodies. (J) Treatment with NAM increases endogenous MTPα acetylation on 3K. AML12 cells were left untreated or treated with NAM and then subjected to Western blotting with the indicated antibodies. (K) Anti-MTPα-3K-Ac antibody has high specificity for MTPα-3K. HEK293T cells were transfected with WT or 3K mutant MTPα. Immunoprecipitated Flag-MTPα was subjected to Western blotting with the indicated antibodies.
To better examine the acetylation of 3K, we generated an antibody specific for acetylated K350, K383, and K406 (anti-MTPα-3K-Ac). The specificity of this antibody was first confirmed by its ability to recognize the acetylated, but not the unacetylated, 3K peptides (Fig. 2H). We then used the anti-MTPα-3K-Ac antibody to determine the acetylation of endogenous MTPα. Acetylation of MTPα in AML12 cells could be readily detected by the antibody. The signal was diminished by MTPα knockdown and was completely blocked by preincubation with the antigen peptides (Fig. 2I). Using this antibody, we found that the acetylation of endogenous MTPα was clearly increased after treatment of AML12 cells with NAM (Fig. 2J). Furthermore, the antibody recognized ectopically expressed wild-type MTPα quite well but not 3KR mutants of MTPα (Fig. 2K). Collectively, these results demonstrate the specificity of our anti-MTPα-3K-Ac antibody in recognizing the 3K acetylation of MTPα.
K350, K383, and K406 acetylation stabilizes MTPα by blocking its ubiquitylation.
We then explored the mechanism underlying the acetylation-regulated MTPα protein stability. As shown in Fig. 1D, the proteasomal inhibitor of MG132 had a role similar to that of NAM in increasing the protein level of MTPα. This prompted us to investigate the potential cross talk between MTPα acetylation and MTPα ubiquitylation. Our data confirmed the existence of MTPα ubiquitylation (Fig. 3A), which is consistent with a previous report showing that MTPα could be ubiquitylated (23). Also, the ubiquitylation level of MTPα was reduced by inhibiting SIRT deacetylases with NAM (Fig. 3A). In order to identify the ubiquitylation sites, we tested the ubiquitylation levels of double mutants, K350/383R, K383/406R, and K350/406R (Fig. 3B). We found that the ubiquitylation of the double mutants K350/383R, K383/406R, and K350/406R was partially decreased, while mutation of K350, K383, and K406 (3KR), which changes all three putative acetylation lysine residues of MTPα to arginine residues, dramatically reduced the MTPα ubiquitylation level (Fig. 3B), indicating that 3K lysines might also be ubiquitylation target residues. Moreover, inhibition of deacetylases by NAM decreased ubiquitylation of the WT but not the 3KQ or 3KR mutant (Fig. 3C). These results imply an antagonizing role of acetylation toward the ubiquitylation of MTPα at these three lysine residues.
FIG 3.

Acetylation of MTPα at lysine residues 350, 383, and 406 blocks its ubiquitylation. (A) Flag-MTPα was cotransfected with HA-tagged ubiquitin (HA-Ub) into HEK293T cells with or without treatment with NAM. Ubiquitylation of immunoprecipitated MTPα was determined by Western blotting. (B) Flag-tagged wild-type or 2KR or 3KR mutant MTPα was cotransfected with HA-Ub into HEK293T cells. Ubiquitylation of immunoprecipitated MTPα was determined by Western blotting. (C) WT MTPα or 3K mutant MTPα was cotransfected with HA-Ub into HEK293T cells with or without treatment with NAM. Ubiquitylation of immunoprecipitated proteins was determined by Western blotting. (D) K350/383R, K383/406R, and K350/406R left with one lysine or K350/383/406R with all three lysines mutated to arginines was cotransfected with Flag-tagged ubiquitin (Flag-Ub) into HEK293T cells in which the endogenous human MTPα had been knocked down by siRNA. The anti-Flag-Ub immunoprecipitates (assumed to be a mixture of all the ubiquitylated substrates) and the remaining supernatants (assumed to be a mixture of nonubiquitylated substrates) were blotted to compare the MTPα 3K acetylation levels.
There are two possible mechanisms by which acetylation may block MTPα ubiquitylation. (i) Acetylation at 3K indirectly antagonizes MTPα ubiquitylation on other lysines (but not 3K) by recruiting a factor promoting MTPα deubiquitylation. (ii) Alternatively, the three lysine residues could also be the sites of ubiquitylation, with their acetylation directly blocking MTPα ubiquitylation, as both modifications are covalently linked to the same epsilon-amino group of lysine. If it were due to the first mechanism, the acetylation-deficient 3KR mutant would block the recruitment of the MTPα deubiquitylation factor, thereby increasing MTPα ubiquitylation. However, the fact is that both the acetylation-deficient 3KR mutant and the acetylation-mimetic 3KQ mutant dramatically reduced the MTPα ubiquitylation level (Fig. 3B and C), ruling out the first mechanism. To verify the second mechanism, where the three lysine residues (3K) could be sites of ubiquitylation with their acetylation directly blocking MTPα ubiquitylation on the same sites, we compared the acetylation levels of the ubiquitylated and nonubiquitylated MTPα 2KR mutants, in which two of the three lysine residues (K350 and K383, K383 and K406, or K350 and K406) were mutated to arginine. We ectopically expressed Flag-tagged ubiquitin, together with MTPα 2KR mutants or the MTPα 3KR mutant, in HEK293T cells lacking endogenous human MTPα. The cell lysates were immunoprecipitated with anti-Flag antibody to enrich ubiquitylated proteins, followed by Western blotting using anti-MTPα-3K-Ac and anti-MTPα antibodies (Fig. 3D). Anti-MTPα-3K-Ac can recognize MTPα as long as one of the 3K lysine residues (K350, K383, or K406) is acetylated. As shown in Fig. 3D (top, lanes 1, 3, and 5), the remaining K406, K350, or K383 in these 2KR mutants can still be acetylated. Also, these 2KR mutants can still be ubiquitylated (Fig. 3D, bottom, lanes 2, 4, and 6), because the 2KR mutation is not sufficient to block MTPα ubiquitylation (as shown in Fig. 3B). As a control, the MTPα 3KR mutant could not be recognized by anti-MTPα-3K-Ac antibody (Fig. 3D, top, lanes 7 and 8) and also could not be efficiently ubiquitylated (Fig. 3D, bottom, lane 8). It was found that the K350, K383, or K406 acetylation of these MTPα 2KR mutants was detected only in the nonubiquitylated (Fig. 3D, top, lanes 1, 3, and 5, which are the supernatants after Flag-ubiquitin IP), but not the ubiquitylated (Fig. 3D, top, lanes 2, 4, and 6, which are the precipitants of Flag-ubiquitin IP), MTPα species. If MTPα were ubiquitylated on the other lysines but not 3K (K350/K383/K406), the acetylation of K350, K383, or K406 in these MTPα 2KR mutants should also exist in the ubiquitylated MTPα species. However, that is not the case, as shown in Fig. 3D (top, lanes 2, 4, and 6). Thus, this result indicates that MTPα acetylation and ubiquitylation are mutually exclusive, supporting the model that K350, K383, and K406 (3K) are the sites of both acetylation and ubiquitylation. Consequently, acetylation of these lysines would block ubiquitylation on the same lysines.
SIRT4 deacetylates MTPα.
Our earlier observation that NAM increases MTPα acetylation (Fig. 1B) led us to investigate a possible involvement of NAD+-dependent sirtuins in MTPα deacetylation. Because MTPα is a mitochondrial enzyme, the three mammalian sirtuins (SIRT3, SIRT4, and SIRT5), which are located in the mitochondria, were selected for further investigation. We performed coimmunoprecipitation experiments and found that MTPα physically bound with SIRT4 but not SIRT3 or SIRT5 (Fig. 4A), indicating a specific interaction between MTPα and SIRT4. We also confirmed the colocalization between the ectopically expressed MTPα and SIRT4 in the HEK293T cells using confocal microscopy (Fig. 4B). In addition, the ectopically expressed MTPα and SIRT4 were well localized in the mitochondria (Fig. 4B). Furthermore, overexpression of wild-type SIRT4 but not the catalytically inactive H162Y mutant led to decreased 3K acetylation of MTPα (Fig. 4C). To further confirm the deacetylase activity of SIRT4 toward MTPα, mass spectrometry-based deacetylation assays using chemically synthesized acetylated peptides and SIRT4 recombinant protein were performed. As shown in Fig. 4D to F, after 30 min of in vitro reaction, SIRT4 dramatically deacetylated the K350-Ac peptide, the K383-Ac peptide, and the K406-Ac peptide. As a negative control, SIRT4 did not deacetylate pyruvate dehydrogenase (PDH) peptide, suggesting that SIRT4 deacetylase activity is substrate specific (Fig. 4G). Importantly, overexpression of wild-type SIRT4 but not the H162Y mutant increased the MTPα ubiquitylation level (Fig. 4H). In addition, knockdown of SIRT4 in AML12 cells increased the protein level but not the mRNA level of MTPα (Fig. 4I). Consistently, knockdown of SIRT4 promoted the protein stability of MTPα in AML12 cells (Fig. 4J). Together, these results demonstrate that SIRT4 deacetylates MTPα at the 3K sites to facilitate its ubiquitylation and degradation.
FIG 4.

SIRT4 deacetylates MTPα and decreases its protein level. (A) Sirtuin-MTPα interactions were assessed by cotransfecting expression vectors for SIRT3, SIRT4, or SIRT5 (HA tagged at the C terminus) with an expression vector for Flag-MTPα in HEK293T cells. Flag-MTPα was immunoprecipitated, and interactions were detected by Western blotting. (B) HEK293T cells were cotransfected with Flag-MTPα and HA-SIRT4 or transfected with Flag-MTPα and HA-SIRT4. The subcellular localization of Flag-MTPα (green) and HA-SIRT4 (red), Flag-MTPα (green) and MitoTracker (red), or HA-SIRT4 (green) and MitoTracker (red) was examined by immunofluorescence (magnification, ×200). (C) SIRT4, but not the catalytically inactive mutant SIRT4-H162Y, deacetylates MTPα. HEK293T cells were cotransfected with the indicated vectors. Flag-MTPα was immunoprecipitated and subjected to Western blotting. Numbers indicate molecular weights in thousands. (D to G) The 3K lysine residues of MTPα are deacetylated by SIRT4 in vitro. Recombinant SIRT4 was incubated for the indicated times with synthesized acetylated peptides of MTPα, and peptide deacetylation was assessed using mass spectrometry. (D to F) Compared with 0 min, the mass peak shifts leftward due to the removal of an acetyl group from the peptide by SIRT4. (G) Acetylated peptide from PDH was included as a negative control. The arrowheads show the positions of acetylated peptides, and the arrows show the positions of deacetylated peptides. (H) Flag-MTPα was cotransfected with Myc-tagged SIRT4 or SIRT4-H162Y into HEK293T cells. Ubiquitylation of purified proteins was detected. (I) AML12 cells were infected with adenovirus expressing shNC or shSIRT4 and then subjected to Western blotting. Numbers indicate molecular weights in thousands. The mRNA levels of SIRT4 and MTPα were determined by RT-qPCR. *, P < 0.05. The error bars indicate SD. (J) AML12 cells were infected with adenovirus as for panel I, treated with CHX for the indicated times, and then subjected to Western blotting.
Acetylation of MTPα ameliorates cellular lipid accumulation in AML12 cells and primary hepatocytes induced by FFA.
Because MTPα is a critical enzyme regulating lipid oxidation, the functional importance of MTPα acetylation in hepatic lipid homeostasis was examined. To this end, stable cell lines were generated in the AML12 mouse hepatocyte line, in which endogenous MTPα was knocked down by shRNA and the shRNA-resistant WT MTPα or 3KR or 3KQ mutant MTPα was stably expressed. At the mRNA level, the endogenous MTPα was effectively knocked down and WT MTPα, MTPα-3KR, or MTPα-3KQ was expressed at a level similar to that of endogenous MTPα (Fig. 5A). At the protein level, a similar result was obtained, except that the protein level of MTPα 3KR or 3KQ was a bit higher than that of WT MTPα (Fig. 5B), which is consistent with the notion that MTPα-3K acetylation promotes MTPα protein stability. To determine the effect of MTPα acetylation on lipid accumulation in hepatocytes, AML12 cells were cultured in complete medium with or without FFA mixtures. Oil Red O staining (Fig. 5C) and cellular triglyceride determination (Fig. 5D) demonstrated that increased lipid stores were present in hepatocytes when the cells were treated with FFAs. The increase was markedly augmented in cells infected with MTPα shRNA, confirming the role of MTPα in lipid combustion (Fig. 5C and D). In addition, the cellular oxygen consumption rate was significantly diminished when MTPα was knocked down, suggesting impaired fatty acid oxidation (Fig. 5E). As expected, reexpression of WT MTPα depleted the marked augmentation of lipid stores caused by MTPα shRNA and restored the cellular oxygen consumption rate. Importantly, reexpression of MTPα-3KR or MTPα-3KQ led to significantly less lipid accumulation than that of WT MTPα (Fig. 5C and D) and a higher cellular oxygen consumption rate (Fig. 5E). The same experiments were also done in primary hepatocytes, which were infected with adenovirus harboring shNC or shMTPα, together with or without adenovirus encoding shRNA-resistant WT MTPα or 3K mutants of MTPα, and similar results were obtained (Fig. 5F to J). Together, these results indicate a functional role of MTPα acetylation in promoting lipid catabolism in hepatic-cell models.
FIG 5.

Acetylation of MTPα ameliorates FFA-induced lipid accumulation in AML12 cells and primary hepatocytes in vitro. (A to C) Stable cell lines were generated in AML12 cells in which endogenous MTPα was knocked down by shRNA and the shRNA-resistant WT MTPα or 3K mutant MTPα was stably expressed. The stable AML12 cells were then treated with FFA for 24 h or left untreated and subjected to RT-qPCR (*, P < 0.05 versus LacZ plus shNC plus FFA) (A), Western blotting (B), and Oil Red O staining (C). (D and E) Cellular triglyceride was determined (D) and the cellular oxygen consumption rate was measured (E) (§, P < 0.05 versus LacZ plus shNC plus vehicle; *, P < 0.05 versus LacZ plus shNC plus FFA; #, P < 0.05 versus LacZ plus shMTPα plus FFA; †, P < 0.05 versus MTPα-WT plus shMTPα plus FFA). (F to J) Primary hepatocytes were infected with adenovirus harboring shNC or shMTPα, together with or without adenovirus encoding shRNA-resistant WT MTPα or 3K mutant MTPα. Then, the same experiments as for stable AML12 cells were done in primary hepatocytes. Symbols are as defined for panels D and E. The error bars indicate SD.
Acetylation of MTPα ameliorates hepatic steatosis in mice challenged by an HF/HS diet.
On the basis of in vitro results, we next examined whether the acetylation of MTPα also plays a role in HF/HS diet-induced hepatic steatosis in mice. We used the recombinant adenovirus to deliver the shRNA and the shRNA-resistant expression constructs to mouse livers through tail vein injection. Mice fed an HF/HS diet were infected once a week with adenoviruses harboring the indicated shRNA and expression constructs via tail vein injection and sacrificed 15 days after injection. The knockdown of MTPα and reexpression of WT MTPα, MTPα-3KR, or MTPα-3KQ in the mouse livers was confirmed by RT-qPCR and Western blotting (Fig. 6A and B, respectively). The reexpression of WT MTPα, MTPα-3KR, or MTPα-3KQ yielded mRNA levels similar to the endogenous MTPα mRNA level (Fig. 6A). However, at the protein level, the MTPα-3KR or MTPα-3KQ protein level was a bit higher than that of WT MTPα protein (Fig. 6B), supporting the notion that MTPα-3K acetylation promotes MTPα protein stability. The plasma β-hydroxybutyrate, an indicator of hepatic fatty acid oxidation, was then measured. Knockdown of MTPα decreased the level of plasma β-hydroxybutyrate and reexpression of MTPα WT restored it, while reexpression of MTPα-3KR or MTPα-3KQ more potently increased the level of plasma β-hydroxybutyrate (Fig. 6C). These data suggest an important role of MTPα-3K acetylation in promoting hepatic fatty acid oxidation. Knockdown of MTPα exacerbated steatotic changes in the livers of HF/HS diet-fed mice and its accompanying pathology, as shown by increased liver weight (Fig. 6D), increased fat accumulation in the liver (Fig. 6E), increased hepatic triglyceride and cholesterol (Fig. 6F), elevated plasma ALT and AST levels (Fig. 6G), enhanced inflammatory gene expression in the liver (Fig. 6H), and impaired insulin sensitivity and glucose tolerance (Fig. 6I). As expected, reexpression of MTPα-3KR or MTPα-3KQ ameliorated hepatic steatosis and its accompanying pathology and metabolic disorders better than that of WT MTPα (Fig. 6D to I). Taken together, these data confirmed the functional importance of MTPα acetylation in hepatic steatosis and its associated pathology and metabolic disorders.
FIG 6.

Acetylation of MTPα ameliorates hepatic steatosis in mice challenged by an HF/HS diet. (A and B) Male C57BL/6J mice fed an HF/HS diet for 2 months were injected once a week with adenovirus harboring shNC or shMTPα, together with or without adenovirus harboring shRNA-resistant WT MTPα or 3K mutant MTPα, through the tail vein. Measurements were performed at 15 days postinfection. The mRNA levels and protein levels of MTPα in the livers were determined by RT-qPCR (A) and Western blotting (B), respectively. (C) Plasma β-hydroxybutyrate levels. (D) Organ weights. (E) Representative images of H&E staining and Oil Red O staining of liver sections (magnification, ×100). (F) Triglyceride and cholesterol levels in livers. (G) Levels of AST and ALT in plasma. (H) Expression levels of inflammatory genes in livers. (I) Results of GTT (left) and ITT (right). *, P < 0.05 versus LacZ plus shNC; #, P < 0.05 versus LacZ plus shMTPα; †, P < 0.05 versus MTPα-WT plus shMTPα. n = 6 to 10/group. The error bars indicate SD.
Knockdown of SIRT4 could phenocopy the effects of MTPα-3K mutant expression in mouse livers.
Our data have demonstrated that Sirt4 is the deacetylase of MTPα, facilitating its ubiquitylation and proteasome degradation. We asked whether knockdown of SIRT4 alone could phenocopy the effects of MTPα-3K mutant expression in the mouse liver. Indeed, adenovirus-mediated knockdown of SIRT4 in the livers of HF/HS diet-fed mice increased both the protein level and the 3K acetylation level of MTPα (Fig. 7A to C); promoted hepatic fatty acid oxidation, as indicated by elevated plasma β-hydroxybutyrate (Fig. 7D); and inhibited hepatic steatosis and its accompanying pathology and metabolic disorders (Fig. 7E to J). Our results showing that knockdown of SIRT4 promotes hepatic lipid catabolism are consistent with previous studies (24, 25).
FIG 7.

Knockdown of SIRT4 alone could phenocopy the effects of MTPα-3K mutant expression in mouse livers. (A and B) Male C57BL/6J mice fed an HF/HS diet for 2 months were injected once a week with adenovirus harboring shNC or shSIRT4. Measurements were performed at 15 days postinfection. The mRNA levels and protein levels of SIRT4 and MTPα in the livers were determined by RT-qPCR (A) and Western blotting (B), respectively. (C) MTPα-3K acetylation levels were compared to MTPα protein levels. (D) Plasma β-hydroxybutyrate levels. (E) Organ weights. (F) Representative images of H&E staining and Oil Red O staining of liver sections (magnification, ×100). (G) Triglyceride and cholesterol levels in livers. (H) Levels of AST and ALT in plasma. (I) Expression levels of inflammatory genes in livers. (J) GTT (left) and ITT (right) results. *, P < 0.05 versus shNC. n = 6 to 10/group. The error bars indicate SD.
Deacetylation of MTPα by SIRT4 contributes to SIRT4-mediated hepatic steatosis in HF/HS diet-fed mice.
To further investigate the link between SIRT4 and MTPα acetylation in NAFLD, SIRT4, with or without MTPα or its mutants, was overexpressed in the livers of HF/HS diet-fed mice. The mRNA levels of overexpressed WT MTPα, MTPα-3KR, and MTPα-3KQ were similar (Fig. 8A). Meanwhile, the mRNA and protein levels of overexpressed SIRT4 were not affected by the overexpression of MTPα or its mutants (Fig. 8A and B). Overexpression of SIRT4 alone decreased the endogenous MTPα protein level (Fig. 8B) but not its mRNA level (Fig. 8A), and it also attenuated the acetylation of endogenous MTPα (Fig. 8C). Consistently, overexpression of SIRT4 together with MTPα WT displayed an MTPα protein level only slightly higher than overexpression of SIRT4 alone, while overexpression of SIRT4 together with MTPα-3KR or MTPα-3KQ exhibited a much higher MTPα protein level than overexpression of SIRT4 alone (Fig. 8B). These data further support the notion that SIRT4 deacetylates MTPα to inhibit its protein stability. Functionally, overexpression of SIRT4 alone inhibited hepatic fatty acid oxidation (Fig. 8D) and promoted hepatic steatosis and its accompanying pathology and metabolic disorders (Fig. 8E to J). Compared to overexpression of WT MTPα, overexpression of MTPα-3KR or MTPα-3KQ more potently inhibited the effect of SIRT4 (Fig. 8D to J). Collectively, these data suggested that deacetylation of MTPα by SIRT4 is an important target of SIRT4 and contributes to SIRT4-mediated hepatic steatosis in mice fed an HF/HS diet.
FIG 8.

Deacetylation of MTPα by SIRT4 contributes to SIRT4-mediated hepatic steatosis in HF/HS diet-fed mice. (A and B) Male C57BL/6J mice fed an HF/HS diet for 2 months were injected once a week with adenovirus encoding SIRT4, together with or without adenovirus encoding WT MTPα or 3K mutant MTPα, through the tail vein. Measurements were performed at 15 days postinfection. The mRNA levels and protein levels of SIRT4 and MTPα in the livers were determined by RT-qPCR (A) and Western blotting (B). (C) MTPα-3K acetylation levels were compared to MTPα protein levels. (D) Plasma β-hydroxybutyrate levels. (E) Organ weights. (F) Representative images of H&E staining and Oil Red O staining of liver sections (magnification, ×100). (G) Triglyceride and cholesterol levels in livers. (H) Levels of plasma AST and ALT. (I) Expression levels of inflammatory genes in livers. (J) GTT (left) and ITT (right) results. *, P < 0.05 versus LacZ; #, P < 0.05 versus SIRT4 plus LacZ; †, P < 0.05 versus SIRT4 plus MTPα-WT. n = 6 to 10/group. The error bars indicate SD.
Acetylation of MTPα is upregulated in the livers of fasted mice while it is downregulated in the livers of both mice and patients with NAFLD.
Our data have shown that MTPα-3K acetylation promotes its protein stability to facilitate fatty acid oxidation. Then, we asked whether MTPα-3K acetylation is regulated under fasting and refeeding states. After 16 h of fasting, the MTPα protein level was elevated, while the SIRT4 protein level was decreased, in the livers of lean mice, which was restored by 8 h of refeeding (Fig. 9A). To determine the MTPα-3K acetylation level, we examined MTPα-3K acetylation by normalizing it to MTPα protein levels. As expected, MTPα-3K acetylation increased upon fasting and was restored to a normal level after refeeding (Fig. 9B). Thus, these results demonstrated that SIRT4-regulated MTPα-3K acetylation is linked to the nutritional status in mice. Next, the MTPα protein level and its 3K acetylation in the livers of obese mice with fatty liver disease and patients with NAFLD were investigated. As shown in Fig. 9C, compared to the control groups of mice, the total protein level of MTPα was significantly decreased in the livers of obese mice (HF/HS diet-fed mice, ob/ob mice, and db/db mice). Moreover, liver samples from obese mice showed lower levels of 3K acetylation than those from the control groups of mice (Fig. 9D). SIRT4, the deacetylase of MTPα, was significantly upregulated in the livers of obese mice (Fig. 9C), which is consistent with the previous study (24). We then determined the levels of MTPα, MTPα-3K, and SIRT4 by performing immunohistochemistry in paraffin-embedded human liver tissues to expand our study. In agreement with the results in mouse liver samples, we observed that both the levels of total MTPα and the levels of MTPα-3K were lower, whereas the levels of SIRT4 were higher, in the livers of NAFLD patients than in the livers of healthy human subjects (Fig. 9E and F). These data support the notion that dysregulated acetylation of MTPα is involved in the pathogenesis of NAFLD.
FIG 9.

Acetylation of MTPα is upregulated in livers of fasted mice, while it is downregulated in livers of obese mice with fatty liver disease and patients with NAFLD. (A and B) Livers were harvested from lean mice under fed, fasted (16 h), and refed (8 h) states, lysed, and subjected to Western blotting. (A) The MTPα and SIRT4 protein levels were compared to those of β-actin. (B) The MTPα-3K acetylation levels were compared to MTPα protein levels. (C and D) Livers from obese mice (HF/HS diet-fed mice, ob/ob mice, and db/db mice) and control mice were lysed and subjected to Western blotting, similar to panels A and B. (E and F) Immunohistochemical staining of total MTPα, 3K-acetylated MTPα, and total SIRT4 proteins in human NAFLD and normal liver tissues. One example is shown in panel E (magnification, ×100), and statistical analysis of all the samples is shown in panel F. (G) Proposed model for the role of dysregulated MTPα-3K acetylation in the development of NAFLD. The expression of SIRT4 in the liver is upregulated after overnutrition, thereby decreasing the acetylation level of MTPα on lysine residues 350, 383, and 406. Decreased acetylation leads to increased ubiquitylation of MTPα on the same three lysine residues and facilitates the degradation of MTPα by the proteasome. This effect contributes to the impaired fatty acid β-oxidation and promotes the development of NAFLD. Further studies are needed to reveal the mechanism by which overnutrition leads to the upregulation of SITR4 in the liver.
DISCUSSION
Dysregulated lipid metabolism plays an important role in the pathogenesis of NAFLD (8). In this study, we demonstrated that acetylation promotes the protein stability of the lipid metabolic enzyme MTPα and that derangement of the enzymes' acetylation could play a role in the development of NAFLD (Fig. 9G).
MTPα catalyzes the second and third steps in the physiologically important β-oxidation pathway of fatty acid metabolism. Although impairment of mitochondrial β-oxidation of fatty acid has been considered to account for hepatic steatosis (8), the expression level and the potential role of MTPα in the pathogenesis of NAFLD have not been fully elucidated. Our data showed that downregulation of MTPα promotes hepatic steatosis both in FFA-treated AML12 cells and primary hepatocytes and in the livers of HF/HS diet-fed mice (Fig. 5 and 6, respectively). Importantly, a decrease in the MTPα protein level was detected in the livers of obese mice with fatty liver disease and in the liver samples from patients with simple steatosis (Fig. 9). These data confirm that MTPα downregulation contributes to the development of NAFLD.
In this study, we report a novel mechanism by which the MTPα level is regulated at the posttranscriptional level. It was found that acetylation stabilizes MTPα and prevents its ubiquitylation. Moreover, our data indicate that acetylation and ubiquitylation in MTPα could compete with each other by targeting the same lysine residues at K350, K383, and K406. Similar to this model, different modifications of the same lysine residues have been reported in the regulation of other proteins (26, 27). It is proposed that acetylation and ubiquitylation have opposing effects on the regulation of MTPα by competitively modifying the same lysine residues. Thus, the results showing that the acetylation-deficient MTPα-3KR and the acetylation-mimetic MTPα-3KQ mutants have similar biochemical and functional behaviors could be explained by the fact that the mutations on the three residues disrupt lysine ubiquitylation that primarily occurs on the same residues. Importantly, we found that the levels of both MTPα and MTPα-3K are decreased while the level of MTPα deacetylase SIRT4 is increased in NAFLD liver tissues compared to normal liver tissues (Fig. 9). These observations not only confirm MTPα acetylation in vivo but also suggest that MTPα-3K acetylation may play a role in the development of NAFLD.
The ubiquitin-proteasome system (UPS) plays an important role in controlling the levels of various cellular proteins and regulates the processes in many aspects of cell function. Increasing evidence shows that the UPS is implicated in the degradation of mitochondrial proteins. Ubiquitin-conjugated proteins in purified mitochondria were detected by mass spectrometry (23). Moreover, cells treated with proteasome inhibitors exhibit elevated levels of ubiquitylated mitochondrial proteins, suggesting the potentially important roles of the proteasome in mitochondrial protein degradation (28). Consistently, many studies have identified mitochondrial substrates of the UPS. The outer mitochondrial membrane proteins Mfn1/Mfn2 are polyubiquitylated and degraded by the proteasome (29). The proteasome plays a role in degradation of the intermembrane space protein endonuclease G (30). Uncoupling protein 2 (UCP2), which is an inner mitochondrial membrane protein, can be polyubiquitylated in the mitochondria and then retrotranslocated from the mitochondria to the cytoplasm and degraded by the proteasome (31). In this study, we showed that the mitochondrial protein MTPα can be polyubiquitylated and that inactivation of the proteasome prevents its turnover, indicating that MTPα is also a target of the UPS. Taken together, the evidence is compelling that the UPS participates in protein quality control in the mitochondrial compartments.
The sirtuin family consists of seven family members (SIRT1 to -7), each containing a conserved catalytic core domain with NAD+-dependent deacetylase, deacylase, and/or ADP-ribosyltransferase activities (32, 33). Three of the mammalian sirtuins (SIRT3, SIRT4, and SIRT5) are located in the mitochondria and may play roles as sensors of energy status in the organelle (34). SIRT4 is one of the least-characterized mitochondrial sirtuins. Recent studies have suggested that SIRT4 uses NAD to ADP-ribosylate and inactivate glutamate dehydrogenase, thereby suppressing insulin secretion from pancreatic beta cells (35). No deacetylase activity was reported for SIRT4 using a histone or BSA substrate (36). However, a recent study indicated that SIRT4 could directly bind, deacetylate, and repress malonyl CoA deacarboxylase (MCD) (20), which suggests that the absence of deacetylase activity for SIRT4 may have been a reflection of the lack of an appropriate substrate. Here, we show that SIRT4 is a deacetylase for MTPα, further supporting the existence of SIRT4 deacetylase activity. In the future, it would be interesting to assess whether SIRT4 has other deacetylation substrates.
As a mitochondrial sirtuin, SIRT4 has been shown to repress fatty acid oxidation (24, 25). However, how SIRT4 inhibits fatty acid oxidation and the connection of SIRT4 to NAFLD is not fully understood. In this study, it was found that SIRT4-mediated deacetylation destabilizes MTPα to inhibit lipid catabolism. Importantly, our data demonstrated that knockdown of SIRT4 in the liver increased MTPα acetylation and reduced hepatic steatosis and its associated pathology and metabolic disorders in HF/HS diet-fed mice (Fig. 7). Moreover, SIRT4 is upregulated in the livers of obese mice with fatty liver disease and in the liver samples from patients with simple steatosis (Fig. 9), which is consistent with previous studies (24, 25). These data, together with previous studies (24, 25), reveal the potential mechanism of SIRT4-mediated inhibition of fatty acid oxidation and highlight an important role of SIRT4 in NAFLD. Thus, it can be speculated that modulating SIRT4 activities could be used as a novel therapeutic approach to obesity-related metabolic disorders, such as NAFLD and insulin resistance. It should be noted, however, that little is known about how SIRT4 is upregulated in NAFLD, which merits further investigation.
It is the conventional belief that acetylation is a negative regulator of a metabolic enzyme's function. For example, SIRT5-mediated deacetylation of carbanoyl phosphate synthetase 1 (CPS1) upregulates its activity to increase amino acid catabolism (37). Acetylation of lactate dehydrogenase A (LDH-A) on lysine residue 5 (K5) inhibits its enzyme activity and promotes its degradation via chaperone-mediated autophagy (CMA) (22). Under high-glucose conditions, the gluconeogenic rate-limiting enzyme phosphoenolpyruvate carboxykinase (PEPCK1) is hyperacetylated, which promotes its ubiquitylation and degradation (19). However, there is growing evidence of a positive role of acetylation in regulating the functions of metabolic enzymes. For example, acetylation activates the enzyme activity of malate dehydrogenase (MDH) in the tricarboxylic acid (TCA) cycle and of enoyl-coenzyme A hydratase/3-hydroxyacyl-coenzyme A dehydrogenase (EHHADH) in fatty acid oxidation (38). In this study, we demonstrated that acetylation could stabilize MTPα to promote fat oxidation. Thus, acetylation can regulate metabolic enzyme functions in a variety of manners, leading to different effects.
In summary, our work has uncovered the role of acetylation-mediated MTPα protein stability in regulating liver lipid homeostasis and suggests that dysregulation of the enzyme's acetylation could play a part in the pathogenesis of NAFLD. These findings provide evidence that drugs modulating the acetylation of MTPα may merit exploration as therapeutic agents for NAFLD.
ACKNOWLEDGMENTS
This research was partially supported by National Key Basic Research Project Grants 2011CB910201 and 2013CB530601 and National Natural Science Foundation of China grants 81390350 and 31571471 (to Q.-Q.T.), Major State Basic Research Development Program of China Grant 2012CB524906 (to X.G.), and National Natural Science Foundation of China Grant 31370027 (to L.G.). The department is supported by Shanghai Leading Academic Discipline Project B110 and 985 Project 985 III-YFX0302.
REFERENCES
- 1.Dietrich P, Hellerbrand C. 2014. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol 28:637–653. doi: 10.1016/j.bpg.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Saito T, Misawa K, Kawata S. 2007. Fatty liver and non-alcoholic steatohepatitis. Intern Med 46:101–103. doi: 10.2169/internalmedicine.46.1784. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. 2004. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 4.Neuschwander-Tetri BA, Caldwell SH. 2003. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology 37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 5.Marchesini G, Babini M. 2006. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol 54:229–239. [PubMed] [Google Scholar]
- 6.Day CP, James OF. 1998. Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 7.Koppe SW. 2014. Obesity and the liver: nonalcoholic fatty liver disease. Transl Res 164:312–322. doi: 10.1016/j.trsl.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Koo SH. 2013. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol 19:210–215. doi: 10.3350/cmh.2013.19.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M, Hirasawa T, Itoh Y, Okanoue T. 2006. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int 26:613–620. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- 10.Ibdah JA, Paul H, Zhao Y, Binford S, Salleng K, Cline M, Matern D, Bennett MJ, Rinaldo P, Strauss AW. 2001. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest 107:1403–1409. doi: 10.1172/JCI12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW. 1999. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med 340:1723–1731. doi: 10.1056/NEJM199906033402204. [DOI] [PubMed] [Google Scholar]
- 12.Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, Matern D, Rinaldo P, Cline JM. 2005. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 128:1381–1390. doi: 10.1053/j.gastro.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Rector RS, Morris EM, Ridenhour S, Meers GM, Hsu FF, Turk J, Ibdah JA. 2013. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 57:2213–2223. doi: 10.1002/hep.26285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. 2014. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 15.Guan KL, Xiong Y. 2011. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci 36:108–116. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, Jiang Y, Zhou X, Li TT, Guan KL, Lei QY, Xiong Y. 2013. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell 52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao D, Mo Y, Li MT, Zou SW, Cheng ZL, Sun YP, Xiong Y, Guan KL, Lei QY. 2014. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest 124:5453–5465. doi: 10.1172/JCI76611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, Castro RE, Rodrigues CM. 2015. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci 129:721–739. doi: 10.1042/CS20140732. [DOI] [PubMed] [Google Scholar]
- 19.Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL, Zhao S. 2011. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 43:33–44. doi: 10.1016/j.molcel.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, Lovitch SB, Sharpe AH, Kurland IJ, Steegborn C, Gygi SP, Muoio DM, Ruderman NB, Haigis MC. 2013. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell 50:686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang ZC, Liu Y, Xiao LL, Li SF, Jiang JH, Zhao Y, Qian SW, Tang QQ, Li X. 2015. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J Hepatol 63:1466–1475. doi: 10.1016/j.jhep.2015.07.037. [DOI] [PubMed] [Google Scholar]
- 22.Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, Guan KL. 2013. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 23:464–476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeon HB, Choi ES, Yoon JH, Hwang JH, Chang JW, Lee EK, Choi HW, Park ZY, Yoo YJ. 2007. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem Biophys Res Commun 357:731–736. doi: 10.1016/j.bbrc.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, Ren X, Wu Z, Streeper RS, Bordone L. 2010. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem 285:31995–32002. doi: 10.1074/jbc.M110.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laurent G, de Boer VC, Finley LW, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, Haigis MC. 2013. SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol Cell Biol 33:4552–4561. doi: 10.1128/MCB.00087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gronroos E, Hellman U, Heldin CH, Ericsson J. 2002. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10:483–493. doi: 10.1016/S1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, Kracht M, Schmitz ML, Burstein E. 2012. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene 31:611–623. doi: 10.1038/onc.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. 2007. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One 2:e1066. doi: 10.1371/journal.pone.0001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. 2010. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radke S, Chander H, Schafer P, Meiss G, Kruger R, Schulz JB, Germain D. 2008. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem 283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azzu V, Brand MD. 2010. Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci 123:578–585. doi: 10.1242/jcs.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houtkooper RH, Pirinen E, Auwerx J. 2012. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13:225–238. doi: 10.1038/nrn3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finkel T, Deng CX, Mostoslavsky R. 2009. Recent progress in the biology and physiology of sirtuins. Nature 460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sebastian C, Satterstrom FK, Haigis MC, Mostoslavsky R. 2012. From sirtuin biology to human diseases: an update. J Biol Chem 287:42444–42452. doi: 10.1074/jbc.R112.402768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. 2006. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 36.Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E. 2007. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem 282:33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. 2009. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. 2010. Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]