

Abstract
The threat posed by the chromosomally encoded class D β-lactamase of Acinetobacter baumannii (OXA-51/66) has been unclear, in part because of its relatively low affinity and turnover rate for carbapenems. Several hundred clinical variants of OXA-51/66 have been reported, many with substitutions of active-site residues. We determined the kinetic properties of OXA-66 and five clinical variants with respect to a wide variety of β-lactam substrates. The five variants displayed enhanced activity against carbapenems and in some cases against penicillins, late-generation cephalosporins, and the monobactam aztreonam. Molecular dynamics simulations show that in OXA-66, P130 inhibits the side-chain rotation of I129 and thereby prevents doripenem binding because of steric clash. A single amino acid substitution at this position (P130Q) in the variant OXA-109 greatly enhances the mobility of both I129 and a key active-site tryptophan (W222), thereby facilitating carbapenem binding. This expansion of substrate specificity represents a very worrisome development for the efficacy of β-lactams against this troublesome pathogen.
INTRODUCTION
Acinetobacter baumannii is a Gram-negative coccobacillus that is responsible for an increasing number of difficult-to-treat nosocomial infections (1). This species has been labeled a “serious threat” by the Centers for Disease Control and Prevention, in large part because of rising levels of antimicrobial resistance associated with it. A. baumannii is largely resistant to penicillins, cephalosporins, quinolones, aminoglycosides, and tetracyclines, and the only remaining sustainable treatment option is the carbapenem class of β-lactams (e.g., imipenem, doripenem, and meropenem). Now, growing levels of carbapenem resistance portend the potential for extensively drug-resistant A. baumannii strains that are resistant to almost all antibiotics (2–4). Carbapenem resistance has been attributed to the loss of outer membrane permeability upon porin deletion (5), overexpression of efflux pumps (6), and modification of the target transpeptidases (7), but the most notable contribution comes from the expression of β-lactamases. A wide variety of β-lactamases from all four classes (A to D) have been shown to be present in A. baumannii (8), but class D carbapenemases have received the most attention in recent years (9). Most class D carbapenemases are characterized by very high affinity for carbapenem substrates, coupled to relatively weak catalytic turnover rates (10). Five subfamilies of class D carbapenemases have been identified in A. baumannii, namely, OXA-23, OXA-24/40, OXA-51, OXA-58, and OXA-143 (1).
The general structural features and mechanism of class D β-lactamases have been elucidated, with high-resolution crystal structures solved for OXA-1 (11), OXA-10 (12), OXA-23 (13), OXA-24/40 (14), OXA-46 (15), OXA-48 (16), OXA-51 (17), and OXA-58 (18) (along with several variants) (13, 19, 20). For most of these enzymes, structures have been determined with substrates or inhibitors bound in the active site (21–26). Like class A and class C β-lactamases, members of class D make use of a serine-nucleophile acylation/deacylation double-displacement mechanism to hydrolyze the β-lactam ring (10). Two features are unique to class D enzymes, however. Their active sites are generally hydrophobic, with conserved residues such as V130, L168, and W167 (OXA-24 numbering) playing key roles in stabilizing active-site architecture and substrate binding (25, 27, 28). Second, carboxylation of an active-site lysine results in the formation of a carbamate general base that deprotonates the serine nucleophile and the deacylating water (29).
Surrounding the active site are three loops, sometimes referred to as the omega loop (12), the P loop (30), and a loop connecting two β-strands (β5 and β6 in OXA-48 and β8 and β9 in OXA-51) (16, 17) (Fig. 1). Many sequence modifications in these loops that enhance or alter the specificity or hydrolytic efficiency of class D β-lactamases have been characterized. Substitutions at positions in or near the omega loop of OXA-10 subfamily members give rise to increased activity against ceftazidime and are thus classified as extended-spectrum β-lactamases (ESBL) (31–33). Structural analysis of one such substitution revealed an altered omega loop that opened the active site for the binding of the bulky oxyimino side chain of ceftazidime (19). A single amino acid duplication (A220dup) in the β5β6 loop of OXA-23 (13) alters the structure of that loop in a way that allows bulky cephalosporins and the monobactam aztreonam to enter the active site, where they can be hydrolyzed. A similar substrate expansion is observed in OXA-23 or OXA-24 clinical variants with a proline-to-serine substitution in the same loop (20). The sequence and length of the β5β6 loop have been implicated in carbapenem targeting as well, as replacement of the β5β6 loop from the narrow-spectrum OXA-10 with the loop from OXA-48 imparts carbapenemase activity to the former (34). Other investigations into the mechanism by which class D β-lactamases hydrolyze carbapenems have focused on a highly conserved leucine (L168 in OXA-24/40) and how its side chain affects the ability of a water molecule to enter the active site and adopt the correct orientation for efficient deacylation of the carbapenem acyl-intermediate (16, 23). Some class D carbapenemases (OXA-23, OXA-24/40, and variants) contain a pair of residues (Y112 and M223 in OXA-24/40) that extend across the top of the active site from the P loop and the β5β6 loop (13, 14). This so-called “hydrophobic bridge” gives rise to a tunnel-like active site and is thought to be responsible for the very high affinity of these enzymes for doripenem and meropenem (14, 22).
FIG 1.
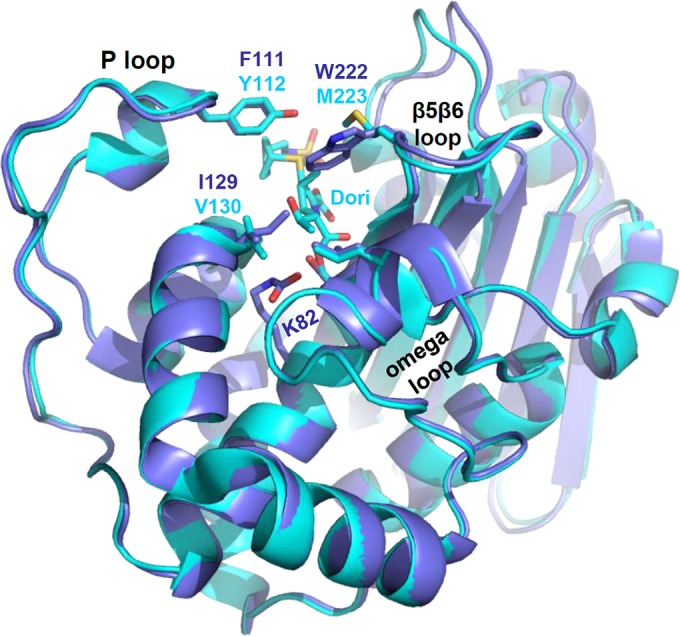
OXA-51 aligned to OXA-24/40/doripenem. The overall main-chain structure of OXA-51 (blue) matches very closely that of OXA-24/40 (cyan). The structure of doripenem (cyan, center top) indicates the position of the active site. Both structures contain three loops surrounding the active site: the P loop and the β5β6 loop (β8β9 in OXA-51) contribute large hydrophobic residues that cap the top of the active site, and the omega loop forms the front wall.
There are >500 unique β-lactamase enzymes that are officially recognized as part of class D, as indicated by their OXA identification numbers. Of these, ∼125 are members of the OXA-51-like subfamily, a group named after an enzyme that was first isolated from an Acinetobacter baumannii clinical sample from Argentina in 1996 (35). In contrast to plasmid-borne carbapenemases such as OXA-23 and OXA-24, the genes that encode OXA-51 and its closely related variants are usually chromosomal. This observation gave rise to the suggestion that OXA-51 may be a ubiquitous, native carbapenemase of A. baumannii (36). Phylogenetic evidence, however, suggests that OXA-65 or OXA-66 (variants of OXA-51 with 5 and 6 substitutions, respectively) might be closer to the ancestral sequence of the subfamily (37). Despite the abundance of variants from clinical A. baumannii isolates, the presence of an OXA-51-like gene does not always lead to carbapenem resistance. The lack of a resistance phenotype may be caused in part by generally low levels of OXA-51 expression and weaker binding affinity for carbapenems than those of OXA-23 and OXA-24/40. Their potential role in resistance was recognized upon the discovery that promoter-like elements such as ISAba1 could be inserted in front of the coding sequence to drive overexpression of the enzyme (38). Additionally, variants with single or multiple substitutions in the OXA-51 or OXA-66 background have been connected to antibiotic resistance phenotypes (39–42) and increased hydrolytic activity for carbapenems (43).
Our understanding of why OXA-51 has such weak carbapenemase activity was greatly enhanced by the elucidation of its structure by Smith and colleagues (17). While the overall fold of OXA-51 matches quite closely that of the potent carbapenemases OXA-23 and OXA-24/40, two key active-site differences help explain the relatively weak binding of carbapenems to OXA-51. First, a tryptophan (W222) occupies the position of the methionine of the hydrophobic bridge found in OXA-23 and OXA-24/40. Instead of forming a bridge across the top of the active site, however, the indole side chain of W222 projects into the binding cavity, where it would be expected to clash with the C-1 methyl group of meropenem or doripenem (Fig. 1). In order for those drugs to bind, the W222 side chain would need to flip ∼180° away from the binding site (17). Second, an active-site isoleucine (I129) projects its δ carbon into the active site, where it would likely clash with the carbapenem's hydroxyethyl group. This position is occupied by valine in nearly all other non-OXA-51-like class D β-lactamases, and structural studies show a very tight fit between the valine of those enzymes and bound carbapenem substrates (21–24).
Because OXA-51-like genes are nearly ubiquitous in A. baumannii, any evolutionary adaptation in this family which increases activity against carbapenems (either binding affinity or turnover) is a serious threat. In this study, we provide evidence that these changes are indeed emerging rapidly.
MATERIALS AND METHODS
Site-directed mutagenesis.
The gene for OXA-66 (codons 26 to 274) was purchased from GenScript BioTech Corporation (Piscataway Township, NJ) and cloned into a pET24a(+) vector using the NdeI and XhoI restriction sites. Substitutions at sites P130, I129, W222, and P226 were introduced by overlap extension PCR (44) using primers purchased from Integrated DNA Technologies (Coralville, IA) and Phusion High-Fidelity polymerase (ThermoScientific, Inc.). DNA sequences were confirmed by Sanger dideoxy sequencing at the University of Michigan Sequencing Core. Final plasmid preparations were used to transform BL21(DE3) Escherichia coli cells.
Expression and purification of variant enzymes.
Expression and purification of OXA-66 and all other variants were carried out by methods identical to those used previously for OXA-24 (20). Each enzyme was purified to >95% purity using carboxy-methyl cellulose CM23 (Whatman, United Kingdom), concentrated to 1 to 3 mg/ml, snap-frozen in liquid nitrogen, and stored at −80°C. The concentration of each enzyme preparation was determined using UV spectroscopy (280 nm) in the presence of 6 M guanidium HCl, with extinction coefficient values calculated by the method of Gill and von Hippel (45).
Determination of steady-state kinetic parameters.
Kinetic assays were performed by combining enzyme variants with β-lactam substrates in 50 mM NaH2PO4 plus 25 mM NaHCO3, pH 7.4, at room temperature. Mixtures were placed in quartz cuvettes (path length, 1.0 cm or 0.2 cm), and absorbance was monitored over time. Changes in absorbance were converted to velocity using the change in extinction coefficient values specific for each substrate (ampicillin, Δε = −900 M−1 · cm−1; doripenem, 297 nm, Δε = −11,460 M−1 · cm−1; imipenem, 300 nm, Δε = −9,000 M−1 · cm−1; cefotaxime, 260 nm, Δε = −7,500 M−1 · cm−1; ceftriaxone, 255 nm, Δε = −7,800 M−1 · cm−1; ceftazidime, 260 nm, Δε = −8,660 M−1 · cm−1; aztreonam, 320 nm, Δε = −700 M−1 · cm−1). For enzyme/substrate combinations that yielded very low Km values, Ks values were measured by using the substrate as a competitor with ampicillin acting as a reporter, as described previously (20).
Molecular dynamics simulations.
OXA-66 and its P130Q variant (OXA-109) were constructed for two parallel simulations using the OXA-51 apo structure (residues 38 to 274) (Protein Data Bank [PDB] accession number 5KZH), MMTSB (46), CHARMM (47) and CHARMM-GUI (48) script libraries, and the VMD package (49). For the OXA-66 model, the V48, Q107, P194, and D225 residues of OXA-51 sequence were replaced with A, K, Q, and N residues, respectively, using an MMTSB mutate script. For OXA-109, the P130 residue was replaced with glutamine, in addition to the other substitutions. OXA enzymes possess the sequence motif PASTFK (residues 78 to 83) containing a carboxylated lysine that acts as a general base. Force-field parameters for the carboxylated lysine are provided in our previous study (20). Both proteins were solvated with TIP3P (50) water molecules in a cubic box with a dimension of 74 Å. Potassium and chloride ions were added to neutralize the net charge of the system and to obtain the concentration of 0.15 M (37 anions and 35 cations). The system size was 41,235 atoms/ions for OXA-66 and 41,250 atoms/ions for OXA-109. The CHARMM27 protein force field (47) and the CHARMM, version 37b2, simulation package were used. Geometries of both systems were optimized using the Steepest Descent algorithm for 1,000 steps and then using the adopted basis Newton-Raphson (ABNR) method (51) until the gradient threshold of 0.01 kcal/mol/Å was achieved. During the minimization and simulations, periodic boundary conditions were applied, and electrostatic interactions were calculated with the particle mesh Ewald method (52). A switching function was applied to calculate the van der Waals interactions with a nonbonded cutoff of 12 Å. The SHAKE algorithm (53) was used to constrain the bond lengths involving hydrogen atoms. After minimization, the systems were heated gradually from 10 K to 300 K for 60 ps (with a 1-fs time step) and then simulated under NPT (constant number of atoms, pressure, and temperature) conditions for 200 ps total (with a 1-fs time step) to equilibrate the water molecules. Harmonic constraints of 10 kcal/mol/Å2 were initially placed on all of the proteins' heavy atoms and were removed during the last 170 ps of NPT simulations. The unconstrained systems were then simulated for 100 ps under NVT (constant volume) with a 1-fs time step, after which the time step was increased to 2 fs. The initial 10 ns was considered the equilibration period, and the data were collected over 189.6 ns of each productive trajectory. The Cα root mean square deviation (RMSD) profiles of core secondary elements for OXA-66 and OXA-109 are shown in Fig. S1 in the supplemental material.
Accession number.
The OXA-51 structure has been deposited in PDB under accession number 5KZH.
RESULTS AND DISCUSSION
In order to observe the patterns of amino acid substitutions that occur in the subfamily of OXA-51-like proteins, we carried out a BLAST search of the NCBI protein database using OXA-51 or OXA-66 as a query sequence. Searches were performed with cutoffs of 90% query coverage, 90% sequence identity, and the default BLOSUM62 matrix and were limited to Acinetobacter baumannii and Acinetobacter calcoaceticus. Analysis of the aligned structures revealed 192 unique sequences that met the criteria when either sequence was used to search. The alignments revealed that the highest number of substitutions were located near three sequence motifs which are highly conserved in class D β-lactamases and are known to be in or near the active site (Fig. 1 and 2). The area containing the sequence motif SAI (residues 127 to 129) revealed that substitutions for I129 (L/V/M) and P130 (S/L/Q/A/T/R) were very common, along with one sequence that had a substitution at V131 (I). The omega loop contains the WL motif (residues 166 and 167), which is unique to class D β-lactamases. This region was dominated by substitutions at L167 (V/Q); but three variants had substitutions at V168 (M/A), and one had a variation at W166 (L). The β8β9 loop (analogous to the β5β6 loop of OXA-48) showed common substitutions in many unique sequences, most notably W222 (L/G) and D225 (N). While it is possible that the prevalence of some of these substitutions in a large number of unique enzymes represents an early mutation event that was maintained as other substitutions accumulated, a phylogenetic analysis by Evans et al. suggests that substitutions at I129, P130, L167, and D225 have arisen independently several times (37). Building on our previous work that showed gain-of-function activity for I129L and L167V (43), we chose to analyze five new OXA-66 variants: OXA-109 (P130Q), OXA-234 (W222L), OXA-PQ/WL (an unnumbered class D β-lactamase with the double substitution P130Q/W222L [54]), OXA-172 (I129V/W222L), and OXA-173 (I129V/W222L/P226L). The sequences of these variants are shown in Fig. 2, with common motifs and substitutions highlighted.
FIG 2.

Alignment of OXA-66 and eight clinical variants. OXA-66 and several variants containing one, two, or three substitutions were aligned using ClustalW. The red bar between residues 25 and 26 represents the site of proteolytic digestion for removal of the export sequence. Key active-site residues are highlighted with blue, and the substitutions are highlighted with green. OXA-PQ/WL is a variant without an OXA number and is the P130Q/W222L double mutant of OXA-66 (NCBI RefSeq number WP_001022759).
The OXA-66 coding sequence (codons 26 to 274) was purchased as a synthetic gene in the pET24a expression vector, and the substitutions described above were introduced by overlap extension PCR (44). Each variant was expressed and purified using carboxymethyl cellulose ion exchange chromatography. The kinetic parameters kcat and Km for each variant were determined using UV spectroscopy. A wide variety of β-lactam substrates were used, including the penicillin ampicillin, the carbapenems imipenem and doripenem, the cephalosporins cefotaxime, ceftazidime, and ceftriaxone, and the monobactam aztreonam. The kcat and Km values that we measured for OXA-66 closely match those for OXA-51 (43), suggesting that the six amino acid differences between these two proteins (T5A, E36V, V48A, Q107K, P194Q, and D225N) likely have little effect on the hydrolytic efficacy of the proteins.
All variants displayed parameters that differed markedly from those of the parental enzyme OXA-66 although the patterns were unique for each variant and each class of drug (Table 1). The substitution of a glutamine for P130 results in an overall increase of at least 10-fold in kcat/Km values for both doripenem and imipenem over the value observed for OXA-66. In the case of imipenem, the gain in overall efficiency is derived almost entirely from a lower Km value, suggesting that the substitution results in a higher affinity for that drug. For doripenem, a modest decrease in Km (>2.5-fold) was accompanied by an approximately 4-fold increase in kcat, indicating that there may be gains in both affinity and turnover efficiency. The decrease in Km for doripenem was enough to put the value below the level at which it can be reliably quantified using UV spectroscopy, so Ks values were determined indirectly by competition using ampicillin as a reporter substrate. The Ks of doripenem (0.56 μM) indicated that the P130Q substitution did, in fact, lead to increased affinity for carbapenems of at least 5-fold. That said, the binding does not appear to be quite as tight as that observed for OXA-24, OXA-23, or even the I129L mutation of OXA-51, all of which display Ks values of <0.15 nM (13, 20, 43). The gains afforded by the P130Q substitution were largely limited to carbapenems as there was only a slight decrease in the very high Km for the penicillin ampicillin and no detectable hydrolytic activity for the advanced-generation cephalosporins cefotaxime, ceftazidime, and ceftriaxone or for the monobactam aztreonam.
TABLE 1.
Kinetic parameters of OXA-66 and five variants
| Enzyme and drug | Km (μM) | Ks (μM)a | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|---|
| OXA-66 | ||||
| Ampicillin | >20,000 | >180 | NA | |
| Doripenem | 5.4 ± 0.4 | >2.7 | 0.052 ± 0.001 | 0.0096 ± 0.0007 |
| Imipenem | 71 ± 1 | >47 | 0.69 ± 0.01 | 0.0097 ± 0.0002 |
| Cefotaxime | NAb | <0.02 | NA | |
| Ceftriaxone | NA | <0.02 | NA | |
| Ceftazidime | NA | <0.02 | NA | |
| Aztreonam | NA | <0.05 | NA | |
| OXA-66 P130Q (OXA-109) | ||||
| Ampicillin | 9,100 ± 1,500 | 510 ± 30 | 0.056 ± 0.010 | |
| Doripenem | <2.0 | 0.56 ± 0.03 | 0.20 ± 0.01 | >0.1 |
| Imipenem | 5.9 ± 0.6 | 3.6 ± 0.6 | 0.55 ± 0.01 | 0.093 ± 0.010 |
| Cefotaxime | NA | <0.02 | NA | |
| Ceftriaxone | NA | <0.02 | NA | |
| Ceftazidime | NA | <0.02 | NA | |
| Aztreonam | NA | <0.05 | NA | |
| OXA-66 W222L (OXA-234) | ||||
| Ampicillin | 2,200 ± 200 | 370 ± 10 | 0.17 ± 0.01 | |
| Doripenem | <2.0 | 0.35 ± 0.05 | 0.034 ± 0.001 | >0.017 |
| Imipenem | 5.6 ± 0.3 | 2.7 ± 0.2 | 0.14 ± 0.01 | 0.025 ± 0.002 |
| Cefotaxime | NA | <0.02 | NA | |
| Ceftriaxone | NA | <0.02 | NA | |
| Ceftazidime | NA | <0.02 | NA | |
| Aztreonam | NA | <0.05 | NA | |
| OXA-66 P130Q/W222L | ||||
| Ampicillin | 870 ± 90 | 340 ± 10 | 0.39 ± 0.04 | |
| Doripenem | <2.0 | 0.16 ± 0.02 | 0.13 ± 0.01 | >0.065 |
| Imipenem | 2.2 ± 0.2 | 0.54 ± 0.07 | 0.25 ± 0.01 | 0.11 ± 0.01 |
| Cefotaxime | NA | <0.02 | NA | |
| Ceftriaxone | NA | <0.02 | NA | |
| Ceftazidime | NA | <0.02 | NA | |
| Aztreonam | NA | <0.05 | NA | |
| OXA-66 I129V/W222L (OXA-172) | ||||
| Ampicillin | 2,400 ± 300 | 770 ± 50 | 0.32 ± 0.04 | |
| Doripenem | <2.0 | 0.011 ± 0.001 | 0.022 ± 0.001 | >0.011 |
| Imipenem | <2.0 | 0.049 ± 0.009 | 0.14 ± 0.01 | >0.07 |
| Cefotaxime | NA | <0.02 | NA | |
| Ceftriaxone | NA | <0.02 | NA | |
| Ceftazidime | NA | <0.02 | NA | |
| Aztreonam | NA | <0.05 | NA | |
| OXA-66 I129V/W222L/P226L (OXA-173) | ||||
| Ampicillin | 36 ± 7 | 130 ± 10 | 3.6 ± 0.8 | |
| Doripenem | <2.0 | 0.020 ± 0.005 | 0.012 ± 0.001 | >0.006 |
| Imipenem | <2.0 | 0.041 ± 0.009 | 0.022 ± 0.002 | >0.011 |
| Cefotaxime | 100 ± 4 | 0.52 ± 0.01 | 0.0052 ± 0.0002 | |
| Ceftriaxone | 5.7 ± 1.0 | 2.3 ± 0.5 | 0.055 ± 0.003 | 0.010 ± 0.002 |
| Ceftazidime | >1,000 | >0.25 | NA | |
| Aztreonam | 170 ± 10 | 0.34 ± 0.01 | 0.0020 ± 0.0001 |
Ks values were determined by competition kinetics with ampicillin as a reporter substrate.
NA, not applicable.
As noted above, the P130Q substitution has appeared clinically in combination with other mutations in the active site, most notably W222L. A laboratory mutant (W222M) in the OXA-51 background has been shown to increase affinity and overall hydrolytic efficiency against carbapenems (17). The position of W222 at the rim of the active site, with its bulky indole side chain projecting into the active site, is predicted to cause steric occlusion of carbapenem substrates (17). We tested variants containing the W222L substitution alone (OXA-234) and in combination with P130Q (OXA-66 PQ/WL). Similar to OXA-51 W222M, OXA-234 displayed lower Km and Ks values for doripenem and imipenem as well as for the penicillin ampicillin. Unlike the situation with the P130Q substitution, however, the W222L variant led to slightly lower kcat values for carbapenem substrates, thereby moderating the overall gain of hydrolytic efficiency observed. The double substitution of P130Q and W222L resulted in even lower Ks values for carbapenems, suggesting that the two substitutions can work together to improve affinity. Such high carbapenem affinities precluded us from measuring an exact Km, but even lower limits of the kcat/Km ratios indicate minimum 6-fold and 12-fold increases for overall efficiency of doripenem and imipenem hydrolysis, respectively. This suggests that if either of these substitutions appeared on its own, the addition of the other might confer a selective advantage against carbapenem therapy.
W222L is also found in combination with I129V in OXA-172, a variant that was isolated from carbapenem-resistant A. baumannii in Taiwan (39). The I129V substitution in OXA-172 is highly similar to the previously described I129L variant and is sequentially adjacent to P130. This variant displays carbapenem Ks values as low as those observed for OXA-23 and OXA-24 (<50 nM) and only modestly reduces kcat values compared to those of wild-type OXA-66. The OXA-172 (OXA-66 I129V/W222L) Km for ampicillin, however, was unchanged compared to that of OXA-234 (OXA-66 W222L), and all three variants containing W222L (OXA-234, OXA-66 PQWL, and OXA-172) showed no detectable turnover of advanced-generation cephalosporins or aztreonam.
Perhaps the most interesting OXA-66 variant we tested was OXA-173, another enzyme that was first identified in Taiwan. OXA-173 contains the same two substitutions found in OXA-172 (I129V and W222L) but has the additional substitution P226L. The proline at this position is highly conserved in class D carbapenemases, and substitutions for it have been shown to result in expanded substrate profiles in both the OXA-23 and OXA-24 subfamilies (20). Our analysis of OXA-173 showed a similar expansion of specificity, with a greatly reduced Km for the penicillin ampicillin and, for the first time in an OXA-51-like enzyme, hydrolytic activity against advanced-generation cephalosporins and the monobactam aztreonam. Along with these gains, we did not observe any major loss of carbapenemase activity for OXA-173. These results indicate that this variant is capable of binding and hydrolyzing a breadth of substrate classes that is unusual for class D β-lactamases.
Having demonstrated that all five variants of OXA-66 display reduced Km/Ks values for a variety of β-lactam substrates, we sought to explain how each substitution might alter the active site. The mechanism by which an I129V substitution in OXA-66 results in tighter binding of carbapenems can be easily rationalized. The potential for a steric clash between the side chain of I129 and the hydroxyethyl group of carbapenem substrates has been noted (17, 43). Substitution of a valine for an isoleucine at this position eliminates the δ carbon and its potential hindrance of the hydroxyethyl moiety. The homologous position in both OXA-23 and OXA-24 is occupied by valine, and both of these enzymes possess very low Km values for carbapenems. Similarly, the gain-of-function activities observed for the variants containing W222L can be rationalized from structural analysis. Smith et al. note that for a carbapenem to bind in OXA-51, the side-chain indole group of W222 must flip ∼180° away from the expected position of carbapenem binding, and that flip would appear to be energetically unfavorable (17). The side chain of leucine is much smaller than that of tryptophan, so the steric clash between doripenem and the far end of the indole side chain (particularly Cη1) would likely be eliminated with the W222L substitution. To illustrate these potential effects, we prepared a model of OXA-172 (OXA-66 I129V/W222L) and superposed it with a model of OXA-66 and OXA-24/40 in complex with doripenem (OXA-24/40 K84D/doripenem) (Fig. 3A). First, we used the mutagenesis function of PyMol to change OXA-51 (PDB code 4ZDX) to OXA-66 by introducing five substitutions (E36V, A48V, Q107K, P194Q, and D225N). We then created OXA-172 by introducing the additional substitutions I129V and W222L. These two PDB models were superposed on the structure of OXA-24/40 K84D/doripenem (PDB code 3PAE) to show how the substitutions potentially affect the interaction with doripenem. Figure 3A strongly supports the hypothesis that the I129V and W222L substitutions eliminate the steric clash with doripenem that would occur with the OXA-66 wild type.
FIG 3.
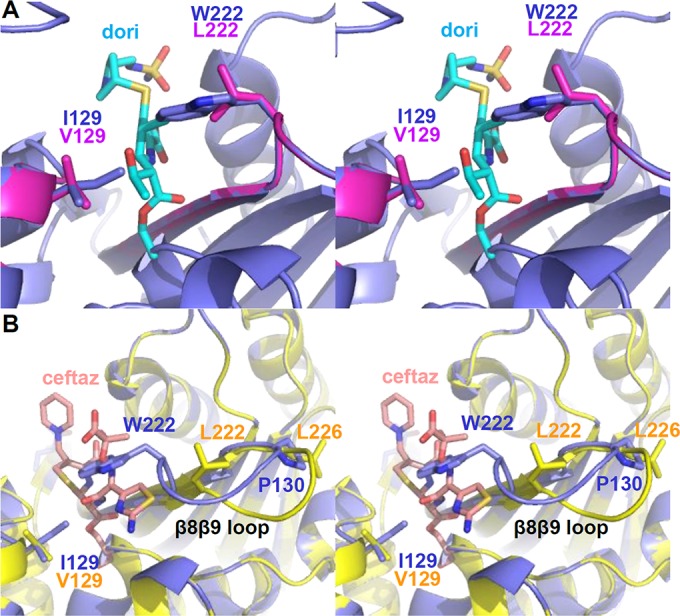
Models of OXA-172 and OXA-173. To help explain the potential structural effects of the substitutions that resulted in enhancement of hydrolytic activities, we constructed models of variants that contained those substitutions. (A) Model of OXA-66 I129V/W222L (OXA-172; magenta) superposed with a model of OXA-66 wild type (blue) and OXA-24/40/doripenem (PDB code 3PAE) (cyan; ligand only). (B) Model of OXA-66 I129V/W222L/P226L (OXA-173; yellow) superposed with a model of OXA-66 wild type (blue) and OXA-225/ceftazidime (PDB code 4X55) (salmon; ligand only). dori, doripenem; ceftaz, ceftazidime.
Previous studies involving the P227S substitution of OXA-225 and OXA-160 provide a possible explanation for the greatly expanded specificity provided by the substitution at the structurally homologous P226 position in OXA-173. No matter what residue is substituted for P226, the loss of the proline's rigid side-chain ring will allow much greater freedom of the Φ torsion angle. In OXA-160, this enhanced flexibility allows the β5β6 loop to adopt an alternative conformation that makes room for the bulky oxyimino side chains of advanced-generation cephalosporins and aztreonam (20). To illustrate this, we built a model of OXA-173 (OXA-66 I129V/W222L/P226L) by adding the P226L substitution to the OXA-172 model described above. This model was aligned with the structure of OXA-160 (OXA-24/40 P227S; PDB code 4X53) in the Coot program (55). The β8β9 loop of the OXA-173 model was then altered to match the backbone trace of the homologous loop from OXA-160. To show how a cephalosporin substrate might bind in the active sites of OXA-66 and OXA-173, these two models were superposed on the structure of OXA-225/ceftazidime (PDB code 4X55). Figure 3B shows that if the P226L substitution in OXA-173 has the same effect that P227S has on OXA-160, the β8β9 loop will move away from the active site and create a much larger area for binding. This explains how OXA-173 can bind large cephalosporin substrates, including those with bulky side chains like ceftazidime.
While simple structural comparisons can explain the effects of most of the substitutions we tested, the cause of significantly reduced carbapenem Km values observed for the P130Q substitution is not as readily apparent. The fact that this residue is sequentially adjacent to I129, along with the observation that substitutions at these two positions never appear in the same variant, suggests that they may be acting through the same mechanism. We wondered if the change in flexibility that would accompany the loss of the rigid proline residue at position 130 may affect the geometric constraints of the neighboring I129 and thus relieve the potential steric clash between that residue and the hydroxyethyl moiety of carbapenem substrates. To test this, we carried out 200-ns molecular dynamics simulations of fully hydrated OXA-66 wild type and its P130Q variant (OXA-109). The starting structures of OXA-66 and OXA-66 P130Q (OXA-109) were prepared by mutation of OXA-51 (PDB code 5KZH) (17).
In order to relieve the steric clash with carbapenems, the side chain of isoleucine would need to rotate in a way that would move its δ carbon away from the predicted location of the drug's hydroxyethyl group. Isoleucine can adopt three rotamers, gauche (−), gauche (+), and trans, with gauche (−) being the form that is observed in the wild-type OXA-51 structure (Fig. 4). Throughout the 190 ns of productive trajectory of wild-type OXA-66, the side chain of I129 adopts the gauche (−) rotamer 96% of the time, maintaining its δ carbon in a position unfavorable to carbapenem binding. The majority of the remaining 4% of the time is represented by a single continuous period in which I129 adopts the trans rotamer, with the δ carbon rotated toward the side chain of L167. The introduction of a P130Q substitution into the OXA-66 background alters the I129 rotamer distribution strikingly (Fig. 4). The time spent in the carbapenem-unfavorable gauche (−) rotamer is reduced to 35%, while the trans form rises to 42% and the gauche (+) is 24%. It should be noted that the trans form is analogous to the rotamer form of V130 observed in the carbapenem-bound form of OXA-24/40 (22). Thus, even in the absence of bound carbapenem, the P130Q substitution induces a large increase in the population of the I129 rotamer that is most compatible with binding that class of drug.
FIG 4.

Rotamer position of I129 in OXA-66 and OXA-109. The χ1 torsion angle for I129 was plotted as a function of time for OXA-66 (blue) and OXA-109 (green). Rotamers (approximate structures shown at right) were binned as follows: gauche (−), −120° < χ1 <0°; gauche (+), 0° < χ1 <120°; trans, −180° < χ1 < −120° and 120° < χ1 <180°. For purposes of clarity, χ1 values between −120° and −180° were shifted to the top of the graph by adding 360°.
We considered two possible explanations for this effect. First, the loss of the rigid ring structure connecting the side chain of P130 to its backbone nitrogen may facilitate an increase in main-chain flexibility and thereby give the side chain of I129 more freedom to overcome the energetic barrier to rotation. Arguing against this, the trajectories reveal that in both OXA-66 and OXA-109, the I129 side chain is able to sample all three states. The difference was thus not in the ability of I129 to access the other rotamers but, rather, in the longer duration during which it populated the trans and gauche (+) positions in OXA-109. Additionally, the main-chain torsion angles for both positions 129 and 130 are essentially unchanged from OXA-66 to OXA-109 (Table 2). Observation of the behavior of I129 during the OXA-66 trajectory reveals a second possibility. The rotation of I129 away from its favored gauche (−) position requires one of its two γ carbons to come into close contact with the δ carbon of the neighboring P130. To switch to the trans rotamer, I129L Cγ2 must approach uncomfortably close to P130 Cδ (∼3.5 Å). Rotating the other way to the gauche (+) rotamer results in a similar steric clash for γ1. Thus, the gauche (−) position for I129 is the most thermodynamically stable position because the alternative rotamers are crowded by the δ carbon of the neighboring P130. If this hypothesis is true, substitution of any other residue (not just glutamine) for proline at position 130 would result in relief of these steric influences on rotamer selection, more readily allowing the forms favorable to carbapenem binding. In support of this, analysis of position 130 in OXA-51/66 clinical variants reveals examples of every amino acid substitution that can be achieved with a single nucleotide switch (alanine, serine, leucine, glutamine, threonine, and arginine).
TABLE 2.
Average main-chain torsion angles for residues at position 129 and 130
| Enzyme | Φ angle (°) | Ψ angle (°) |
|---|---|---|
| OXA-66 I129 | −57.3 ± 7.9 | −44.8 ± 5.8 |
| OXA-109 I129 | −65.2 ± 9.3 | −37.4 ± 8.7 |
| OXA-66 P130 | −58.9 ± 10.6 | −32.1 ± 9.8 |
| OXA-109 Q130 | −61.6 ± 7.5 | −37.0 ± 7.9 |
This explanation is further supported by the increased binding affinity for carbapenems that is observed when an I129L substitution is introduced into OXA-51 (20). The lack of β-branching in the leucine would be predicted to allow its rotation away from the hydroxyethyl without the unfavorable interactions with P130. But if it is the β-branched structure of I129 that induces an energetic cost upon rotation to the carbapenem-favorable trans rotamer, why does the similarly β-branched valine present in OXA-23 and OXA-24 not result in weaker carbapenem binding? The answer may lie in the sole difference between valine and isoleucine, that is, the δ carbon of the latter. In the case of OXA-23 with meropenem bound, the valine maintains the gauche (−) rotamer and therefore does not clash with P130. Its lack of a δ carbon, however, presents much less steric challenge to the meropenem hydroxyethyl (23). In OXA-24/40 with doripenem bound, V130 rotates into the trans rotamer where one of its γ carbons is forced into close proximity with the δ carbon of the neighboring proline (22). The absence of the terminal δ carbon on the valine in this case may allow more room for that side chain to move toward leucine 168 (on the omega loop) and possibly reduce any clash with the neighboring proline (P131). Further examination of the packing of bound carbapenem, I129, P130, and L167 by more extensive molecular dynamics or structural techniques may identify other subtle differences between OXA-66 and OXA-24/40 that could influence the suitability of a β-branched residue in the trans rotamer at residue 129.
We examined the trajectories to see if the P130Q substitution had any influence on W222, whose indole ring presents the other major steric challenge for carbapenem binding. We saw unexpectedly large effects of the P130Q substitution on the rotational fluctuations of the two side-chain torsion angles (χ1 and χ2) for W222 (data not shown). We illustrate this effect by plotting the distance between the carbon at the far end of the indole ring (carbon Cη2) and the expected position of the C-1 methyl carbon of doripenem over the productive period of the simulation trajectory (Fig. 5). The position of doripenem was modeled through alignment of core secondary element Cα atoms of OXA-66 or OXA-109 to the OXA-24/40/doripenem structure (PDB code 3PAE). As Fig. 5 shows, this distance remains close to 2 Å throughout the OXA-66 trajectory (1.96 ± 1.01 Å), indicating that W222 projects into the active site in a position incompatible with doripenem binding for most of the simulation time. For the OXA-109 trajectory, however, the side chain of W222 regularly flips out of the active site, resulting in much greater average distances (6.13 ± 2.61 Å). Thus, the presence of a single substitution (P130Q) has effects on both I129 and W222 that are predicted to increase the binding affinity of carbapenem substrates. A representative frame of OXA-109 in which these two residues simultaneously adopt positions that are favorable to doripenem binding is shown superimposed on an OXA-66 frame showing these residues in the position they adopt in the OXA-51 crystal structure (Fig. 6).
FIG 5.

Comparison of the distance between the side chain of W222 and the expected position of doripenem in OXA-66 and OXA-109. The distance between the C-1 methyl carbon of the pyrroline ring of doripenem and the Cη2 of W222 was measured for each frame of the OXA-66 trajectory (blue) and the OXA-109 trajectory (green). To estimate the position of doripenem in the active sites of OXA-66 and OXA-109, the structures were aligned with the structure of OXA-24/40/doripenem (PDB code 3PAE). Representative snapshots of the position of W222 pointing away from the binding site (OXA-109; green) or toward the binding site (OXA-66; blue) are shown at right.
FIG 6.
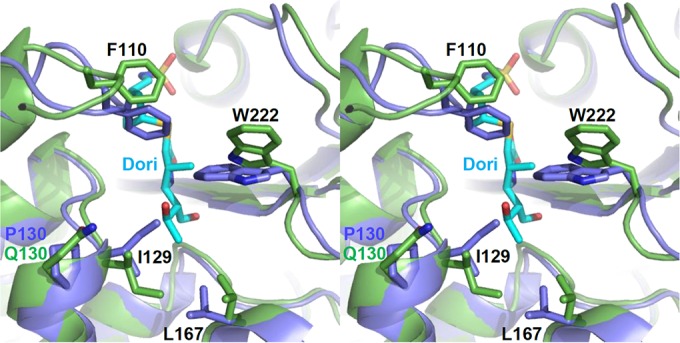
Stereo view of the superposition of frames from the molecular dynamics simulation of OXA-66 (blue) and OXA-109 (green) with acyl-doripenem from the crystal structure of OXA-24/40 (PDB 3PAE) (cyan; OXA-24/40 protein not shown). In OXA-66, I129 and W222 predominantly occupy positions that would be expected to clash extensively with the hydroxyethyl (I129) and pyrroline C-1 methyl (W222) groups of doripenem. The introduction of the P130Q substitution in OXA-109 leads to alternative conformations of both residues that greatly alleviate the steric clash.
Given that the P130Q substitution is on the opposite side of the active site from W222, how could the former induce such a large conformational effect on the latter? As Smith et al. point out, the orientation of W222 is dependent on the compensating stabilizing forces for its alternative positions (directed toward or away from the binding site) (17). In the crystal structure they present, the indole ring directly contacts the δ carbon of I129 (3.8 Å and 4.0 Å to the Cζ2 and Cη2 carbons of W222, respectively). The rotation of I129 away from the gauche (−) rotamer would eliminate these stabilizing contacts and might thus increase W222 side-chain mobility. W222 contacts a large number of residues, and thus more extensive networks of interactions may be involved.
The results we present here show that substitutions in the active site of OXA-66 lead to higher catalytic efficacy for the hydrolysis of carbapenems and, in some cases, other classes of β-lactam antibiotics. The ease with which single substitutions yield catalytic gains raises an important question: if the presence of an isoleucine at position 129 and a tryptophan at position 222 depresses activity so much, why do most OXA-51/66 enzymes have them? One possible answer is that these two residues are part of a hydrophobic packing unit (along with L167, L110, F111, and W220) that significantly stabilizes the overall structure. Further studies that investigate the effect of these substitutions on protein stability may reveal that there is some trade-off between stability and activity similar to what has been observed for other β-lactamases (20, 56, 57). The fact that these substitutions have appeared in clinical A. baumannii isolates so often and have many times been shown to increase carbapenem resistance in those isolates suggests that any such trade-offs do not diminish the adaptive potential that they provide.
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert Bonomo for providing doripenem and for helpful discussion.
This research was supported by National Institutes of Health grant 1R15AI082416 (David A. Leonard) and the Grand Valley State University Ott-Stiner Fellowship (Emma C. Schroder). Simulations were conducted with the high-performance computer cluster at Grand Valley State University supported by National Science Foundation grant number CNS-1228291 (Agnieszka Szarecka).
Emma C. Schroder and Cynthia M. June performed the molecular biology. Emma C. Schroder, Aysegul Saral, and Kyle A. Sugg performed the kinetics. Emma C. Schroder, Zachary L. Klamer, Troy Wymore, and Agnieszka Szarecka executed and analyzed the molecular dynamics simulations. Emma C. Schroder, Agnieszka Szarecka, Cynthia M. June, and David A. Leonard wrote the manuscript.
We declare that we have no competing financial interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01277-16.
REFERENCES
- 1.Evans BA, Hamouda A, Amyes SG. 2013. The rise of carbapenem-resistant Acinetobacter baumannii. Curr Pharm Des 19:223–238. doi: 10.2174/138161213804070285. [DOI] [PubMed] [Google Scholar]
- 2.Gould K. 2016. Antibiotics: from prehistory to the present day. J Antimicrob Chemother 71:572–575. doi: 10.1093/jac/dkv484. [DOI] [PubMed] [Google Scholar]
- 3.Falagas ME, Bliziotis IA. 2007. Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int J Antimicrob Agents 29:630–636. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Doi Y, Husain S, Potoski BA, McCurry KR, Paterson DL. 2009. Extensively drug-resistant Acinetobacter baumannii. Emerg Infect Dis 15:980–982. doi: 10.3201/eid1506.081006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Limansky AS, Mussi MA, Viale AM. 2002. Loss of a 29-kilodalton outer membrane protein in Acinetobacter baumannii is associated with imipenem resistance. J Clin Microbiol 40:4776–4778. doi: 10.1128/JCM.40.12.4776-4778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coyne S, Guigon G, Courvalin P, Périchon B. 2010. Screening and quantification of the expression of antibiotic resistance genes in Acinetobacter baumannii with a microarray. Antimicrob Agents Chemother 54:333–340. doi: 10.1128/AAC.01037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gehrlein M, Leying H, Cullmann W, Wendt S, Opferkuch W. 1991. Imipenem resistance in Acinetobacter baumannii is due to altered penicillin-binding proteins. Chemotherapy 37:405–412. doi: 10.1159/000238887. [DOI] [PubMed] [Google Scholar]
- 8.Hujer KM, Hujer AM, Hulten EA, Bajaksouzian S, Adams JM, Donskey CJ, Ecker DJ, Massire C, Eshoo MW, Sampath R, Thomson JM, Rather PN, Craft DW, Fishbain JT, Ewell AJ, Jacobs MR, Paterson DL, Bonomo RA. 2006. Analysis of antibiotic resistance genes in multidrug-resistant Acinetobacter sp. isolates from military and civilian patients treated at the Walter Reed Army Medical Center. Antimicrob Agents Chemother 50:4114–4123. doi: 10.1128/AAC.00778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans BA, Amyes SG. 2014. OXA β-lactamases. Clin Microbiol Rev 27:241–263. doi: 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leonard DA, Bonomo RA, Powers RA. 2013. Class D β-lactamases: a reappraisal after five decades. Acc Chem Res 46:2407–2415. doi: 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun T, Nukaga M, Mayama K, Braswell EH, Knox JR. 2003. Comparison of β-lactamases of classes A and D: 1.5-Å crystallographic structure of the class D OXA-1 oxacillinase. Protein Sci 12:82–91. doi: 10.1110/ps.0224303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maveyraud L, Golemi D, Kotra LP, Tranier S, Vakulenko S, Mobashery S, Samama JP. 2000. Insights into class D β-lactamases are revealed by the crystal structure of the OXA10 enzyme from Pseudomonas aeruginosa. Structure 8:1289–1298. doi: 10.1016/S0969-2126(00)00534-7. [DOI] [PubMed] [Google Scholar]
- 13.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. 2013. Structures of the class D Carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob Agents Chemother 57:4848–4855. doi: 10.1128/AAC.00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santillana E, Beceiro A, Bou G, Romero A. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci U S A 104:5354–5359. doi: 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. 2010. Crystal structure of the narrow-spectrum OXA-46 class D β-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob Agents Chemother 54:2167–2174. doi: 10.1128/AAC.01517-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Docquier JD, Calderone V, De Luca F, Benvenuti M, Giuliani F, Bellucci L, Tafi A, Nordmann P, Botta M, Rossolini GM, Mangani S. 2009. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem Biol 16:540–547. doi: 10.1016/j.chembiol.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Smith CA, Antunes NT, Stewart NK, Frase H, Toth M, Kantardjieff KA, Vakulenko S. 2015. Structural basis for enhancement of carbapenemase activity in the OXA-51 family of class D β-lactamases. ACS Chem Biol 10:1791–1796. doi: 10.1021/acschembio.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith CA, Antunes NT, Toth M, Vakulenko SB. 2014. The crystal structure of the carbapenemase OXA-58 from Acinetobacter baumannii. Antimicrob Agents Chemother 58:2135–2143. doi: 10.1128/AAC.01983-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meziane-Cherif D, Bonnet R, Haouz A, Courvalin P. 2016. Structural insights into the loss of penicillinase and the gain of ceftazidimase activities by OXA-145 β-lactamase in Pseudomonas aeruginosa. J Antimicrob Chemother 71:395–402. doi: 10.1093/jac/dkv375. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell JM, Clasman JR, June CM, Kaitany KC, LaFleur JR, Taracila MA, Klinger NV, Bonomo RA, Wymore T, Szarecka A, Powers RA, Leonard DA. 2015. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii. Biochemistry 54:1976–1987. doi: 10.1021/bi501547k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider KD, Karpen ME, Bonomo RA, Leonard DA, Powers RA. 2009. The 1.4 Å crystal structure of the class D β-lactamase OXA-1 complexed with doripenem. Biochemistry 48:11840–11847. doi: 10.1021/bi901690r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. 2011. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J Mol Biol 406:583–594. doi: 10.1016/j.jmb.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith CA, Antunes NT, Stewart NK, Toth M, Kumarasiri M, Chang M, Mobashery S, Vakulenko SB. 2013. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem Biol 20:1107–1115. doi: 10.1016/j.chembiol.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pernot L, Frénois F, Rybkine T, L'Hermite G, Petrella S, Delettré J, Jarlier V, Collatz E, Sougakoff W. 2001. Crystal structures of the class D β-lactamase OXA-13 in the native form and in complex with meropenem. J Mol Biol 310:859–874. doi: 10.1006/jmbi.2001.4805. [DOI] [PubMed] [Google Scholar]
- 25.Baurin S, Vercheval L, Bouillenne F, Falzone C, Brans A, Jacquamet L, Ferrer JL, Sauvage E, Dehareng D, Frère JM, Charlier P, Galleni M, Kerff F. 2009. Critical role of tryptophan 154 for the activity and stability of class D β-lactamases. Biochemistry 48:11252–11263. doi: 10.1021/bi901548c. [DOI] [PubMed] [Google Scholar]
- 26.Bou G, Santillana E, Sheri A, Beceiro A, Sampson JM, Kalp M, Bethel CR, Distler AM, Drawz SM, Pagadala SR, van den Akker F, Bonomo RA, Romero A, Buynak JD. 2010. Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J Am Chem Soc 132:13320–13331. doi: 10.1021/ja104092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vercheval L, Bauvois C, di Paolo A, Borel F, Ferrer JL, Sauvage E, Matagne A, Frère JM, Charlier P, Galleni M, Kerff F. 2010. Three factors that modulate the activity of class D β-lactamases and interfere with the post-translational carboxylation of Lys70. Biochem J 432:495–504. doi: 10.1042/BJ20101122. [DOI] [PubMed] [Google Scholar]
- 28.Buchman JS, Schneider KD, Lloyd AR, Pavlish SL, Leonard DA. 2012. Site-saturation mutagenesis of position V117 in OXA-1 β-lactamase: effect of side chain polarity on enzyme carboxylation and substrate turnover. Biochemistry 51:3143–3150. doi: 10.1021/bi201896k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci U S A 98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szarecka A, Lesnock KR, Ramirez-Mondragon CA, Nicholas HB, Wymore T. 2011. The class D β-lactamase family: residues governing the maintenance and diversity of function. Protein Eng Des Sel 24:801–809. doi: 10.1093/protein/gzr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hocquet D, Colomb M, Dehecq B, Belmonte O, Courvalin P, Plésiat P, Meziane-Cherif D. 2011. Ceftazidime-hydrolysing β-lactamase OXA-145 with impaired hydrolysis of penicillins in Pseudomonas aeruginosa. J Antimicrob Chemother 66:1745–1750. doi: 10.1093/jac/dkr187. [DOI] [PubMed] [Google Scholar]
- 32.Danel F, Hall LM, Livermore DM. 1999. Laboratory mutants of OXA-10 β-lactamase giving ceftazidime resistance in Pseudomonas aeruginosa. J Antimicrob Chemother 43:339–344. doi: 10.1093/jac/43.3.339. [DOI] [PubMed] [Google Scholar]
- 33.Danel F, Hall LM, Gur D, Livermore DM. 1995. OXA-14, another extended-spectrum variant of OXA-10 (PSE-2) β-lactamase from Pseudomonas aeruginosa. Antimicrob Agents Chemother 39:1881–1884. doi: 10.1128/AAC.39.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Luca F, Benvenuti M, Carboni F, Pozzi C, Rossolini GM, Mangani S, Docquier JD. 2011. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc Natl Acad Sci U S A 108:18424–18429. doi: 10.1073/pnas.1110530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown S, Young HK, Amyes SG. 2005. Characterisation of OXA-51, a novel class D carbapenemase found in genetically unrelated clinical strains of Acinetobacter baumannii from Argentina. Clin Microbiol Infect 11:15–23. doi: 10.1111/j.1469-0691.2004.01016.x. [DOI] [PubMed] [Google Scholar]
- 36.Merkier AK, Centrón D. 2006. blaOXA-51-type β-lactamase genes are ubiquitous and vary within a strain in Acinetobacter baumannii. Int J Antimicrob Agents 28:110–113. doi: 10.1016/j.ijantimicag.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 37.Evans BA, Hamouda A, Towner KJ, Amyes SG. 2008. OXA-51-like β-lactamases and their association with particular epidemic lineages of Acinetobacter baumannii. Clin Microbiol Infect 14:268–275. doi: 10.1111/j.1469-0691.2007.01919.x. [DOI] [PubMed] [Google Scholar]
- 38.Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM, Pitt TL. 2006. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol Lett 258:72–77. doi: 10.1111/j.1574-6968.2006.00195.x. [DOI] [PubMed] [Google Scholar]
- 39.Chen TL, Lee YT, Kuo SC, Hsueh PR, Chang FY, Siu LK, Ko WC, Fung CP. 2010. Emergence and distribution of plasmids bearing the blaOXA-51-like gene with an upstream ISAba1 in carbapenem-resistant Acinetobacter baumannii isolates in Taiwan. Antimicrob Agents Chemother 54:4575–4581. doi: 10.1128/AAC.00764-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zander E, Chmielarczyk A, Heczko P, Seifert H, Higgins PG. 2013. Conversion of OXA-66 into OXA-82 in clinical Acinetobacter baumannii isolates and association with altered carbapenem susceptibility. J Antimicrob Chemother 68:308–311. doi: 10.1093/jac/dks382. [DOI] [PubMed] [Google Scholar]
- 41.Hu WS, Yao SM, Fung CP, Hsieh YP, Liu CP, Lin JF. 2007. An OXA-66/OXA-51-like carbapenemase and possibly an efflux pump are associated with resistance to imipenem in Acinetobacter baumannii. Antimicrob Agents Chemother 51:3844–3852. doi: 10.1128/AAC.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YT, Turton JF, Chen TL, Wu RC, Chang WC, Fung CP, Chen CP, Cho WL, Huang LY, Siu LK. 2009. First identification of blaOXA-51-like in non-baumannii Acinetobacter spp. J Chemother 21:514–520. doi: 10.1179/joc.2009.21.5.514. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell JM, Leonard DA. 2014. Common clinical substitutions enhance the carbapenemase activity of OXA-51-like class D β-lactamases from Acinetobacter spp. Antimicrob Agents Chemother 58:7015–7016. doi: 10.1128/AAC.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill SC, von Hippel PH. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 46.Feig M, Karanicolas J, Brooks CL. 2004. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model 22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 47.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jo S, Kim T, Iyer VG, Im W. 2008. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 49.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 51.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 52.Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N · log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 53.Ryckaert J-P, Ciccotti G, Berendsen HJ. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 54.Sahl JW, Gillece JD, Schupp JM, Waddell VG, Driebe EM, Engelthaler DM, Keim P. 2013. Evolution of a pathogen: a comparative genomics analysis identifies a genetic pathway to pathogenesis in Acinetobacter. PLoS One 8:e54287. doi: 10.1371/journal.pone.0054287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 56.Beadle BM, Shoichet BK. 2002. Structural bases of stability-function tradeoffs in enzymes. J Mol Biol 321:285–296. doi: 10.1016/S0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Minasov G, Shoichet BK. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol 320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.