

Abstract
Sensitization of resistant bacteria to existing antibiotics depends on the identification of candidate targets whose activities contribute to resistance. Using a transposon insertion library in an Escherichia coli mutant that was 2,000 times less susceptible to ciprofloxacin than its parent and the relative fitness scores, we identified 19 genes that contributed to the acquired ciprofloxacin resistance and mapped the shortest genetic path that increased the antibiotic susceptibility of the resistant bacteria back to a near wild-type level.
TEXT
Discovery of new antibacterials can be facilitated by the systematic identification of mutations that suppress antibiotic resistance phenotypes, which can be used to establish specific and prioritized lists of targets for drug development. Genetic suppressors of antibiotic resistance can be identified by using ordered libraries of mutants (e.g., references 1–4). Growth differences among mutants in response to antibiotics can be amplified by the competitive nature of the assays, which are based on pooled libraries (e.g., references 5, 6). Such comparative fitness assays may increase the sensitivity of the antibiotic suppressor screens, but they do so at the expense of very low specificity (5). However, in the absence of reference lists of genes whose inactivation is expected to result in increased antibiotic susceptibility, it is impossible to assess the sensitivity of such methods. Furthermore, there may not be a discernible relationship between the relative fitness under the antibiotic selective pressure and changes in the MIC of the antibiotic.
To independently evaluate these issues, we designed a comparative fitness study that used a laboratory strain of Escherichia coli that was resistant to clinically relevant concentrations of the fluoroquinolone antibiotic ciprofloxacin (Cipro) (7–10) and took advantage of existing information about the gene modulators of the parental, or intrinsic, quinolone resistance (4, 11–14).
Using a double-controlled experimental design (Fig. 1), we found that the true discovery rate of the screen was higher than 35%, with 90% sensitivity, assuming that the transposon insertions are functionally equivalent to the loss-of-function mutations. Additionally, we identified several new genes that contribute to ciprofloxacin resistance, which allowed us to develop a minimal functional classification of genes whose loss of function sensitizes E. coli to the fluoroquinolone, and demonstrated that ciprofloxacin resistance can be almost completely suppressed by a combination of three nonepistatic mutations with the largest individual fitness effects.
FIG 1.
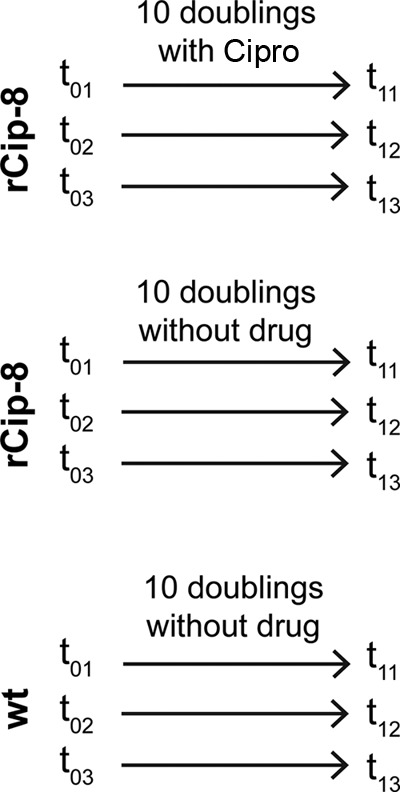
Experimental design. The schema shows all 18 samples used in the analysis, which have been procured from 9 growth experiments. The first subscript, 0 or 1, refers to the start and endpoint, respectively. The second subscript, 1, 2, or 3, refers to the independent sample drawn from the library mixture. Identity of the second subscripts (for one strain) implies that the same sample was used to seed the growth experiment. wt, Wild type.
Quantifiable mutant alleles were generated by saturation transposon mutagenesis using a modified mariner transposon (15, 16) in an E. coli K-12 wild-type strain and its ciprofloxacin resistant derivative, rCip-8 (see text in the supplemental material). The mutant was obtained in 4 selection steps, had a final MIC of 28 ± 4 μg/ml, and carried mutations known to be associated with quinolone resistance: gyrA (D87N), acrR (deletion of 1 bp in coding, 73/648 nt), marR (deletion of 1 bp in coding, 399/435 nt), and parC (S80I).
Following verification of the transposon libraries (see Table S1 and the text in the supplemental material), the rCip-8 mutant library (Cipro MIC, 30 μg/ml) was exposed to ciprofloxacin at 4 μg/ml for 10 generations. We observed that the abundance of insertions in 310 out of 4,516 genes significantly decreased (α = 0.05; P value adjusted for multiplicity of measurements, ≪0.05). However, insertion mutants in 253 of those 310 genes were also significantly and comparably depleted without the drug selective pressure in rCip-8, the wild-type sample, or both. That left 57 candidate genes whose insertions were likely to sensitize the resistant strain to the antibiotic. We were able to construct 52 out of 57 in-frame deletions (17, 18) in the rCip-8 background and test their effect on the MIC of Cipro. Nineteen of the 52 deletions sensitized the drug-resistant strain to Cipro. The 52 deletion mutants could be divided into the following two classes (see Table S2 in the supplemental material): class 1 where insertion alleles were significantly depleted only upon exposure of the culture to the drug and class 2 where insertion alleles were also reproducibly depleted in at least one antibiotic-free selection experiment but to a significantly lesser extent than under antibiotic pressure. Despite having the same number of genes, the first class contained more than 4 times as many correct predictions. The overall rate (for classes 1 and 2) of true positives was higher than 35%.
An earlier study identified 29 gene knockouts that suppressed ciprofloxacin resistance (4). Only 13 of those were among the 19 suppressors identified in the present screen in the Cipro-resistant strain (see Table S3 in the supplemental material), leaving 16 suppressors as potential false negatives. To directly evaluate the sensitivity of the screen, we tested 15 deletion mutants out of the missing 16 (one construction failed). We found that 10 of them affected the susceptibility of the wild-type strain but not the resistant strain. Therefore, the sensitivity of the screen was about 90% (1 − the number of false negatives [5 or 6]/the total number inferred and tested [52]).
We correlated the fold decrease in the MIC values of the deletion mutants relative to the resistant parent with the fold decrease in the abundance of transposon insertions between t0 and t1 samples (Fig. 2A). The extent of the depletion of transposon insertions explained more than 95% of the variance in the MIC values. Changes in the abundance of individual mutant alleles in a population can be used to evaluate the relative contribution of individual genes and gene classes to antibiotic resistance (Fig. 2B). The resistance of rCip-8 was primarily modulated by the activity of efflux pumps, followed by DNA repair and then regulators of the stress response and of the genes whose products are involved in lipopolysaccharide biosynthesis.
FIG 2.
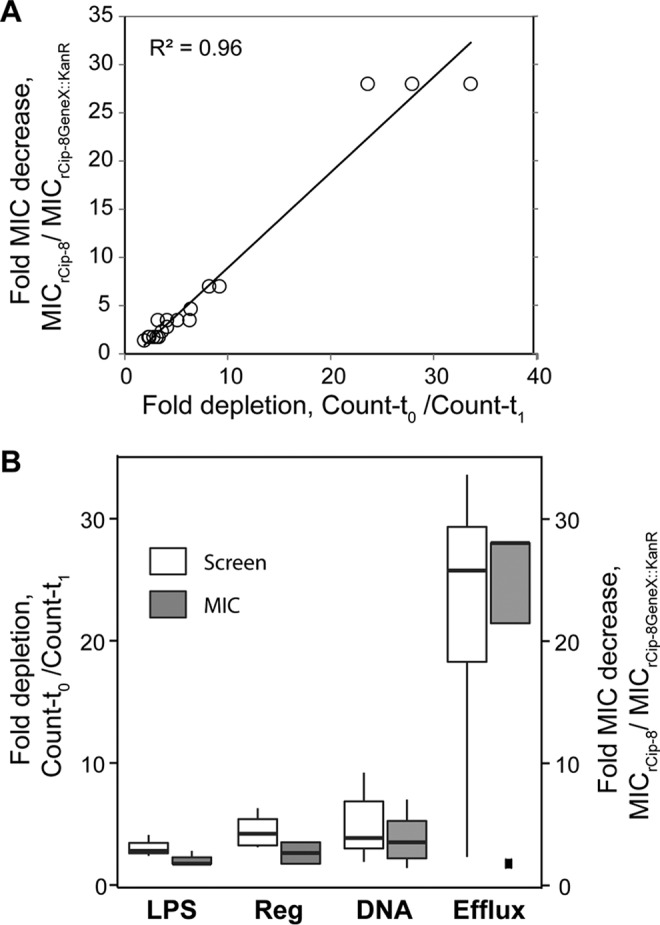
Correlation between the screen and antibiotic sensitivity metrics. The screen metric is a ratio of insertion allele abundances before and after selection. The antibiotic sensitivity metric is a ratio of the MIC of the resistant parent to the MIC of a corresponding knockout strain. (A) Scatterplot of two metrics obtained for each of the 19 genes detected in the screen and verified by individual knockouts. (B) Boxplot of corresponding gene classes broadly defined as follows. LSP, lipopolysaccharide biosynthesis and transport, yciM, yciS, and lpxM; Reg, regulation of transcription and/or translation, hfq, dksA, marA, and miaA; DNA, DNA metabolism, repair, and organization, hofO, recG, recB, recA, recC, xseA, recN, and uvrD; Efflux, tolC, acrA, acrB, and ygaH.
Deletions of 5 genes, hofO, lpxM, miaA, yciS, and ygaH, whose activity has not been previously associated with quinolone resistance, lowered the MIC of the resistant strain 1.4 to 2.8 times (Table 1). Three of them increased the ciprofloxacin susceptibility of the parental as well as the resistant strain, with a comparable reduction of the MIC (Table 1).
TABLE 1.
Effects of novel suppressors on ciprofloxacin MIC
| Additional suppressor allelea | Fitness ratiob | Genetic background MIC (μg/ml) |
||
|---|---|---|---|---|
| rCip-8 | Wild type | rCip-8ΔacrB | ||
| None | 1 | 28 | 0.015 | 1 |
| ΔhofO | 0.53 | 20 | 0.015 | 1 |
| ΔygaH | 0.43 | 16 | 0.015 | 1 |
| ΔlpxM | 0.36 | 16 | 0.007 | 0.5 |
| ΔmiaA | 0.3 | 16 | 0.007 | 1 |
| ΔyciS | 0.24 | 10 | 0.007 | 1 |
All suppressor alleles are gene knockouts, tested with the Kanr cassette present and removed.
The fitness ratio corresponds to the relative abundance of alleles carrying transposon insertions in their respective genes.
If the suppressor effects are additive and independent, then the shortest genetic path to sensitization of resistant bacteria below a breakpoint MIC can be a combination of suppressors with the largest sensitization effects. Although deletion of the acrB gene increased the susceptibility of the resistant strain by 28 times, the resulting MIC (1 μg/ml) was still above the clinical breakpoint concentration (19). We found that the deletions of 4 genes, predicted from the screen, can further sensitize rCip-8ΔacrB more than 2-fold: recB, recC, recG, and uvrD (Table 2). A combination of three deletions, acrB, recB, and uvrD, brought the MIC of the resistant strain within 2-fold of the sensitive wild-type strain, almost fully negating the effect of the quinolone resistance mutations in the DNA gyrase and topoisomerase IV genes.
TABLE 2.
Significant suppression of ciprofloxacin resistance by combinations of deletion alleles
| Additional suppressor allele | rCip-8 background MIC (μg/ml) |
|
|---|---|---|
| ΔacrB | ΔacrBΔuvrD | |
| None | 1 | 0.25 |
| ΔrecB | 0.15 | 0.03 |
| ΔrecC | 0.15 | NDa |
| ΔrecG | 0.2 | 0.06 |
| ΔuvrD | 0.25 | NAb |
ND, not determined.
NA, not applicable.
Supplementary Material
ACKNOWLEDGMENTS
We thank two anonymous reviewers for suggestions and criticism. We thank Evan Brutinel, Justin Courcelle, Jean-Marc Ghigo, Jeff Gralnick, Russell Monds, and Patrick Higgins for reagents and protocols and Deirdre Manion-Fischer for editing the manuscript.
Thu Tran was supported by an ASM undergraduate fellowship.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00889-16.
REFERENCES
- 1.Breidenstein EB, Khaira BK, Wiegand I, Overhage J, Hancock RE. 2008. Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob Agents Chemother 52:4486–4491. doi: 10.1128/AAC.00222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fajardo A, Martinez-Martin N, Mercadillo M, Galan JC, Ghysels B, Matthijs S, Cornelis P, Wiehlmann L, Tummler B, Baquero F, Martinez JL. 2008. The neglected intrinsic resistome of bacterial pathogens. PLoS One 3:e1619. doi: 10.1371/journal.pone.0001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomez MJ, Neyfakh AA. 2006. Genes involved in intrinsic antibiotic resistance of Acinetobacter baylyi. Antimicrob Agents Chemother 50:3562–3567. doi: 10.1128/AAC.00579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamae C, Liu A, Kim K, Sitz D, Hong J, Becket E, Bui A, Solaimani P, Tran KP, Yang H, Miller JH. 2008. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J Bacteriol 190:5981–5988. doi: 10.1128/JB.01982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Girgis HS, Hottes AK, Tavazoie S. 2009. Genetic architecture of intrinsic antibiotic susceptibility. PLoS One 4:e5629. doi: 10.1371/journal.pone.0005629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray JL, Kwon T, Marcotte EM, Whiteley M. 2015. Intrinsic antimicrobial resistance determinants in the superbug Pseudomonas aeruginosa. mBio 6:e01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becnel Boyd L, Maynard MJ, Morgan-Linnell SK, Horton LB, Sucgang R, Hamill RJ, Jimenez JR, Versalovic J, Steffen D, Zechiedrich L. 2009. Relationships among ciprofloxacin, gatifloxacin, levofloxacin, and norfloxacin MICs for fluoroquinolone-resistant Escherichia coli clinical isolates. Antimicrob Agents Chemother 53:229–234. doi: 10.1128/AAC.00722-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colpan A, Johnston B, Porter S, Clabots C, Anway R, Thao L, Kuskowski MA, Tchesnokova V, Sokurenko EV, Johnson JR. 2013. Escherichia coli sequence type 131 (ST131) subclone H30 as an emergent multidrug-resistant pathogen among US veterans. Clin Infect Dis 57:1256–1265. doi: 10.1093/cid/cit503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komp Lindgren P, Karlsson A, Hughes D. 2003. Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob Agents Chemother 47:3222–3232. doi: 10.1128/AAC.47.10.3222-3232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooper DC, Jacoby GA. 2015. Mechanisms of drug resistance: quinolone resistance. Ann N Y Acad Sci 1354:12–31. doi: 10.1111/nyas.12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper DL, Boyle DC, Lovett ST. 2015. Genetic analysis of Escherichia coli RadA: functional motifs and genetic interactions. Mol Microbiol 95:769–779. doi: 10.1111/mmi.12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khodursky AB, Cozzarelli NR. 1998. The mechanism of inhibition of topoisomerase IV by quinolone antibacterials. J Biol Chem 273:27668–27677. doi: 10.1074/jbc.273.42.27668. [DOI] [PubMed] [Google Scholar]
- 13.McDaniel LS, Rogers LH, Hill WE. 1978. Survival of recombination-deficient mutants of Escherichia coli during incubation with nalidixic acid. J Bacteriol 134:1195–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. 2009. Quinolones: action and resistance updated. Curr Top Med Chem 9:981–998. doi: 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouhenni R, Gehrke A, Saffarini D. 2005. Identification of genes involved in cytochrome c biogenesis in Shewanella oneidensis, using a modified mariner transposon. Appl Environ Microbiol 71:4935–4937. doi: 10.1128/AEM.71.8.4935-4937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brutinel ED, Gralnick JA. 2012. Anomalies of the anaerobic tricarboxylic acid cycle in Shewanella oneidensis revealed by Tn-seq. Mol Microbiol 86:273–283. doi: 10.1111/j.1365-2958.2012.08196.x. [DOI] [PubMed] [Google Scholar]
- 17.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U.S. Food and Drug Administration. 2011. Cipro I.V. U.S. Food and Drug Administration, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.